

Abstract
Walker-Warburg syndrome (WWS) is a genetically heterogeneous autosomal recessive disease characterized by congenital muscular dystrophy, cobblestone lissencephaly, and ocular malformations. Mutations in six genes involved in the glycosylation of α-dystroglycan (POMT1, POMT2, POMGNT1, FCMD, FKRP and LARGE) have been identified in WWS patients, but account for only a portion of WWS cases. To better understand the genetics of WWS and establish the frequency and distribution of mutations across WWS genes, we genotyped all known loci in a cohort of 43 WWS patients of varying geographical and ethnic origin. Surprisingly, we reached a molecular diagnosis for 40% of our patients and found mutations in POMT1, POMT2, FCMD and FKRP, many of which were novel alleles, but no mutations in POMGNT1 or LARGE. Notably, the FCMD gene was a more common cause of WWS than previously expected in the European/American subset of our cohort, including all Ashkenazi Jewish cases, who carried the same founder mutation.
Keywords: Walker-Warburg syndrome; congenital muscular dystrophy; alpha-dystroglycan; POMT1, POMT2, FCMD, FKRP
INTRODUCTION
Walker-Warburg syndrome (WWS; MIM# 236670) is an autosomal recessive disorder characterized by cobblestone (type II) lissencephaly, hydrocephalus, severe cerebellar and ocular malformations, and congenital muscular dystrophy (CMD) (Dobyns, et al., 1989; Cormand, et al., 2001; Vajsar and Schachter, 2006). It is the most severe form of CMD, leading to significant developmental delay and a life expectancy of less than 3 years.
While WWS presents with a relatively homogeneous phenotype, it is genetically heterogeneous (Beltran-Valero de Bernabe, et al., 2002; Currier, et al., 2005; Vajsar and Schachter, 2006). The most common known causes of WWS are mutations in two glycosyltransferases, protein O-mannosyltransferase 1 and 2 (POMT1; MIM# 607423, NM_007171; POMT2; MIM# 607439, NM_013382) (Beltran-Valero de Bernabe, et al., 2002; Currier, et al., 2005; van Reeuwijk, et al., 2005b). POMT1 and POMT2 attach the first sugar in the O-mannose-linked glycan moiety of the α-dystroglycan protein, a transmembrane glycoprotein expressed on the surface of muscle cells and neurons (Barresi and Campbell, 2006). α-dystroglycan interacts with several extracellular matrix components in the basal membrane, and disruption of its function is thought to underlie the severe defects in muscle, eye and brain development in WWS patients (Barresi and Campbell, 2006). However, POMT1 mutations have been reported in only 10-20% of WWS cases (Currier, et al., 2005; Vajsar and Schachter, 2006), and only three WWS patients and three fetuses affected by fetal cobblestone lissencephaly have been found to carry POMT2 mutations (van Reeuwijk, et al., 2005b; Bouchet, et al., 2007).
Disruption of α-dystroglycan glycosylation can have variable effects on muscle, eye and brain development, leading to other CMD syndromes that affect the nervous system less severely than WWS (van Reeuwijk, et al., 2005a). Additional genes in the α-dystroglycan glycosylation pathway are mutated in autosomal recessive disorders similar to WWS and grouped under the term α-dystroglycanopathies: POMGNT1 (MIM# 606822, NM_017739) in Muscle Eye Brain disease (MEB; MIM# 253280) (Yoshida, et al., 2001), FCMD (MIM# 607440, NM_001079802) in Fukuyama congenital muscular dystrophy (FCMD; MIM# 253800) (Kobayashi, et al., 1998), fukutin-related protein (FKRP; MIM# 606596, NM_024301) in Muscular Dystrophy Congenital 1C (MDC1C; MIM# 606612) (Brockington, et al., 2001), and LARGE (MIM# 603590, NM_004737) in MDC1D (MIM# 608840) (Longman, et al., 2003). Further genetic analyses have revealed that these α-dystroglycanopathy genes are associated with extremely variable phenotypes ranging from mild CMD to WWS (van Reeuwijk, et al., 2005a; Barresi and Campbell, 2006; Godfrey, et al., 2007). FCMD mutations have been identified in four classic cases of WWS each (de Bernabe, et al., 2003; Silan, et al., 2003; Cotarelo, et al., 2008), LARGE mutations in two WWS cases (Godfrey, et al., 2007; van Reeuwijk, et al., 2007) and FKRP and POMGNT1 mutations in one WWS case each (Beltran-Valero de Bernabe, et al., 2004, Godfrey, et al., 2007).
Such genetic heterogeneity of WWS and the variability in the genotype/phenotype correlation for each identified gene have greatly complicated the genetic diagnosis of WWS patients. Because of the rarity of the disease, a comprehensive overview of the frequency and distribution of mutations in all known genes in a carefully phenotyped and homogeneous cohort of patients had not previously been undertaken. To explore the genetic heterogeneity of WWS and provide some guidance in the choice of genetic tests in these patients, we collected a cohort of classic WWS cases of different ethnic origins and analyzed both common causes of WWS (POMT1, POMT2) and genes that are more rarely mutated in this disease, such as FCMD, FKRP, POMGNT1 and LARGE.
SUBJECTS AND METHODS
Subjects
All subjects were enrolled after informed consent was obtained, and research was conducted according to protocols approved by the Institutional Review Boards of Beth Israel Deaconess Medical Center and Children’s Hospital, Boston. All patients were diagnosed with classic WWS based on the following criteria: (1) brain malformation on neuroimaging characterized by cobblestone lissencephaly, severe ventriculomegaly, and cerebellar hypoplasia; (2) the presence of ocular malformations; and (3) a clinical diagnosis of congenital muscular dystrophy, usually based on severe hypotonia (Dobyns, et al., 1989; Cormand, et al., 2001). Because of the clinical severity and reduced lifespan associated with this condition, serum creatine kinase (CK) and histopathological confirmation of muscular dystrophy were not available in most cases, although CK was highly elevated in all patients tested.
Whole genome scans and sequencing analysis
Genomic DNA was prepared from peripheral blood samples from patients and available family members according to standard protocols. 600 ng of genomic DNA were used to hybridize Affymetrix 250K StyI SNP arrays at the Broad Institute of MIT and Harvard, Cambridge, MA. Genotyping data was analyzed using the loss of heterozygosity algorithm on dChip software (Lin, et al., 2004) to identify regions of identity by descent (IBD) at known WWS loci. Sequencing of POMT1 (NM_007171.3), POMT2 (NM_013382.4), POMGNT1 (NM_017739.2), LARGE (NM_0044737.3), FKRP (NM_001039885.1) and FCMD (NM_006731.2) coding regions was performed on PCR products after amplification of genomic DNA by SeqWright (Houston, TX). PCR primers were designed for each exon including at least 50 base pairs of flanking intronic sequences and are available upon request. DNA changes are described according to the nucleotide numbering reflects in the cDNA sequence, with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (www.hgvs.org/mutnomen). For protein changes the initiation codon is codon 1. Whenever a putative mutation was identified, its inheritance pattern was confirmed in all available family members, and at least 96 control DNAs (192 alleles) were tested to confirm it was not a benign polymorphic change. Unfortunately, no parents were available in the four compound heterozygous cases identified in this study.
Multiplex Ligation-dependent Probe Amplification (MLPA)
MLPA analysis was performed using the SALSA MLPA EK-1 kit (MRC-Holland, Amsterdam, The Netherlands) according to the manufacturer specifications. Custom probes for each exon of the POMT2 gene were designed following instructions from MRC-Holland and sequences are available upon request. MLPA-PCR products were run on an ABI-3130XL genetic analyzer (Applied Biosystems, Foster City, CA), and MLPA peaks were quantified on GeneMapper software (Applied Biosystems) and analyzed following protocols from MRC-Holland.
RESULTS
We collected a cohort of 43 affected individuals (from 40 families) of varying provenance. Sixteen patients from 14 families were Middle Eastern children born to parents who were first cousins (9 from Saudi Arabia, 3 from Israel, 1 from Jordan, 1 from Syria). Twenty-seven additional cases from the United States and Europe were mostly Caucasian (20, including 3 of Ashkenazi Jewish origin), with 6 Hispanic patients and 1 African-American patient.
All individuals (n=104) received genome-wide screens on Affymetrix 250K StyI SNP arrays at the Broad Institute of MIT and Harvard, Cambridge, MA. We analyzed both consanguineous and non-consanguineous families for the presence of stretches of homozygous SNPs spanning the known WWS loci (POMT1; POMT2; POMGNT1, FCMD; FKRP and LARGE), using the loss of heterozygosity algorithm in dChip software (Lin, et al., 2004). Stretches of homozygosity/identity by descent (IBD) of the order of several centiMorgans (cM) were expected in the consanguineous families in our cohort, with the mutation-bearing fragment usually larger than 5 cM for children whose parents are first cousins (Woods, et al., 2006). Smaller regions of IBD may also be present in affected children from non-consanguineous marriages subject to founder effects. Sequencing of the coding sequence of the corresponding gene was performed wherever IBD was observed. In the non-consanguineous cases, all six genes were sequenced in all affected.
In addition, the Affymetrix arrays allow for copy-number change analysis and we assessed all known WWS genes for changes, such as heterozygous or homozygous deletion and duplications, using the copy-number function in dChip (Lin, et al., 2004). No copy-number changes were identified in the six WWS loci analyzed in our cohort, however the average distance between SNPs in these arrays is approx. 10 kb and it is possible that smaller deletions/duplications may be present.
POMT1
Five Middle Eastern families and one Hispanic family showed IBD at the POMT1 locus and upon sequencing of the POMT1 gene all but one affected carried a homozygous mutation (Table 1, Families 1,2,4-6). All POMT1 mutations identified were predicted to severely disrupt POMT1 translation and function by either splicing disruption (c.280+1G>T, c.1892+1G>T), frameshift (c.1260_1261delCT, p.L421fs) or early termination (c.2163C>A, Y721X). The same mutation (c.280+1G>T) was found in two unrelated Saudi families (Table 1, Family 1, 2) carrying different haplotypes (not shown), suggesting that this mutation either originated twice in the population, or was an extremely remote founder mutation in the Saudi population.
Table 1.
Mutations in POMT1, POMT2, FCMD and FKRP
| Family | Origin | Consanguinity | Gene | Mutation |
|---|---|---|---|---|
| 1, 2 | Saudi Arabia | 1st cousin | POMT1 | homozygous c.280+1G>T (p.E94fs) |
| 3 | US (African) | No | POMT1 | c.1097G>A (p.C366Y), c.2167_2168insG (p.G722fs)* |
| 4 | Israeli Arab | 1st cousin | POMT1 | homozygous c.1260_1261delCT (p.L421fs)* |
| 5 | Bahrain | 1st cousin | POMT1 | homozygous c.1892+1G>T (p.Q630fs) |
| 6 | US (Hispanic) | 1st cousin | POMT1 | homozygous c.2163C>A (p.Y721X)* |
| 7 | Palestinian | 1st cousin | POMT2 | homozygous c.924-2A>C (p.K307fs) |
| 8 | US (Caucasian) | No | POMT2 | c.1006+5G>A**, c.1890A>G (splicing?) |
| 9 | Saudi Arabia | 1st cousin | POMT2 | homozygous c.1159delA (p.I387Sfs) |
| 10 | US (Caucasian) | No | FKRP | c.341C>G (p.A114G)*, c.437A>C (p.E146A) |
| 11 | Saudi Arabia | 1st cousin | FKRP | homozygous c.962_970dup9 (p.A321_R323dup) |
| 12 | US (Caucasian) | No | FCMD | c.385delA (p.I129X), c.1176C>A (p.Y392X) |
| 13 | US (Hispanic) | Probable | FCMD | homozygous c.557A>G (p.H186R) |
| 14-16 | Ashkenazi | No | FCMD | homozygous c.1167_1168insA (p.F390fs)* |
previously identified mutations,
novel mutations at same site of previously identified mutations
RefSeq accession number of sequences: POMT1 NM_007171.3; POMT2 NM_013382.4; FKRP NM_001039885.1; FCMD NM_006731.2
Additional sequencing identified only one other patient carrying a POMT1 mutation (Table 1, Patient 3). An African-American male carried two likely POMT1 mutations: a novel missense change at cysteine 366 (c.1097G>A, p.C366Y) and a previously reported frameshift mutation (c.2167_2168insG, p.G722fs, (Beltran-Valero de Bernabe, et al., 2002)). This patient was presumed to be a compound heterozygote, but parental samples were not available to confirm this.
POMT2
POMT2 forms a heterocomplex with POMT1 to initiate glycosylation (Akasaka-Manya, et al., 2006). POMT2 mutations have been reported in only a few WWS cases (van Reeuwijk, et al., 2005b) and more recently in at least six independent MEB cases (Godfrey, et al., 2007). Five Middle Eastern families showed IBD at the POMT2 locus, and two mutations were found in the coding sequence of the POMT2 gene, one in a Saudi family (c.1159delA) causing a frameshift and early termination (p.I387Sfs; Table1, Family 9) and one in a Palestinian family disrupting the splice acceptor in intron 7 (c.924-2A>C, p.K307fs, Table 1, Family 7). IBD in four families without a mutation upon sequencing was not unexpected, as the consanguineous families in our cohort often have only one affected child with multiple regions of IBD across the genome due to the high degree of consanguinity. The SNP haplotypes in the four families were all different, ruling out a common undentified mutation. We asked whether these cases carried copy-number changes, such as deletions or duplication in the POMT2 gene. We had ruled out larger changes by copy-number analysis of the SNP data, but small copy-number changes at the single exon level would not be detected. We excluded homozygous deletion, since all exon could be amplified in all patients during PCR-based sequencing, but could not rule out homozygous duplications. Therefore, we further analyzed these patients via Multiplex Ligation-dependent Probe Amplification (MLPA) at the POMT2 locus (Schouten, et al., 2002). No single exon deletions or duplications were identified (not shown). It is possible that non-coding mutations may cause the disorder in these four families or they may carry mutations in yet unidentified genes.
Sequencing in our non-consanguineous cohort identified one additional case carrying two heterozygous putative splicing changes in POMT2 (Table 1, Patient 8). The first is a transition at a highly conserved guanine 5 basepairs (bp) downstream of exon 8 (c.1006+5G>A). While less universally conserved than the splice donor site, this G in intronic position +5 is an important part of the snRNP binding site (Cartegni, et al., 2002) and identical mutations at the same position have been reported to affect mRNA splicing in other CMD cases (Thi Tran, et al., 2005; Bouchet, et al., 2007). The second putative mutation is a synonymous change (c.1890A>G) located at position −2 at the end of exon 18, a position that is also highly conserved and often involved in splicing defects (Cartegni, et al., 2002). The patient is deceased and no cell lines are available for this case, therefore direct analysis of possible splicing disruptions could not be perfomed. To confirm that these were not rare benign SNP’s, we doubled the number of control DNAs tested and did not find these changes in 188 control individuals (376 chromosomes).
LARGE and POMGNT1
The LARGE gene is usually found mutated in a form of CMD (MDC1D) and the POMGNT1 gene is responsible for Muscle-Eye-Brain disease (MEB). Recently both LARGE and POMGNT1 mutations have been identified in rare WWS patients (van Reeuwijk, et al., 2007; Godfrey, et al., 2007). From our consanguineous cohort one Saudi family showed IBD at the LARGE locus and two Hispanic families showed IBD at the POMGNT1 locus, but no mutation was identified upon sequence analysis. Sequencing of the LARGE and POMGNT1 genes in the non-consanguineous group did not identify any potential mutations.
FKRP
FKRP mutations are associated with a wide spectrum of congenital muscular dystrophies with or without cognitive defects and brain malformations (Mercuri, et al., 2003; Louhichi, et al., 2004; Mercuri, et al., 2006). Only one case of WWS and two cases of MEB are known to carry FKRP mutations (Beltran-Valero de Bernabe, et al., 2004; Mercuri, et al., 2006). By homozygosity mapping, one Saudi family on our cohort showed IBD at the FKRP locus and carried a 9 bp duplication (c.962_970dup9) predicted to duplicate three amino acids, alanine 321 to arginine 323 (Table 1, Family 11; Figure 1A). Interestingly, the only previously reported FKRP mutation causing WWS is a transition affecting cysteine 318, which is close to the change in Family 11 (Figure 1C; Beltran-Valero de Bernabe, et al., 2004). Mutations at this location in the protein sequence may be particularly deleterious for brain development.
Figure 1. FKRP mutations in classic WWS patients.
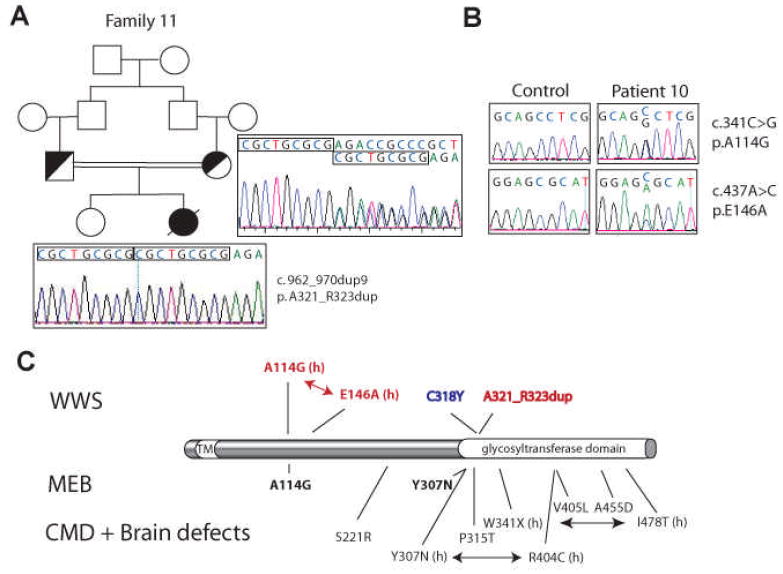
A. Patient 11 was born to first-cousin parents and carried a homozygous 9 basepair duplication starting at bp.962 in the FKRP gene. This mutation leads to a 3 amino acid duplicagtion. B. Two heterozygous missense mutations were identified in Patient 10. C. Schematic representation of the FKRP protein and mutation summary. The initiation codon is codon 1. Mutations listed above the protein cause WWS: previously reported mutations are in blue, mutations identified in this study are in red. Below the protein mutations causing MEB and CMD associated with variable brain malformations are shown. (h) means heterozygous mutations. Compound heterozygous mutations are connected by an arrow. FKRP mutations causing only CMD are not listed. TM: transmembrane domain.
One additional WWS case carrying heterozygous FKRP changes was identified in the European/American non-consanguineous pool (Table 1, Patient 10). A girl from the United States presented a known mutation (c.341C>G, p.A114G, Figure 1B-C (Mercuri, et al., 2006)), accompanied by a novel missense changes, c.437A>C (p.E146A). Both changes were tested in 188 control individuals (376 chromosomes). The A114G mutation has already been described in homozygosity in one MEB case (Mercuri, et al., 2006). A114 is another residue in the FKRP protein which, when mutated, may severely perturb brain development.
FCMD
Mutations in the putative glycotransferase FCMD were initially described in FCMD patients in Japan, where an ancestral retrotransposon insertion in the 3’ UTR of the gene is present in all affected individuals, usually in homozygosity or less commonly in heterozygosity in combination with mutations in the coding region (Figure 2C) (Kobayashi, et al., 1998; Kondo-Iida, et al., 1999). No homozygous mutations in the coding sequence were identified in FCMD patients, suggesting that such mutations may be embryonic lethal or cause a more severe disorder, such as WWS (Kondo-Iida, et al., 1999). The identification of homozygous coding mutations in the FCMD gene outside of Japan, in two Turkish families affected by WWS (de Bernabe, et al., 2003; Silan, et al., 2003) indicated that WWS may be allelic to FCMD. However, the relative contribution of FCMD mutations to the etiology of WWS has not been studied. Recently, two additional WWS patients were reported to carry FCMD mutations (Cotarelo, et al., 2008).
Figure 2. FCMD mutations in classic WWS patients.
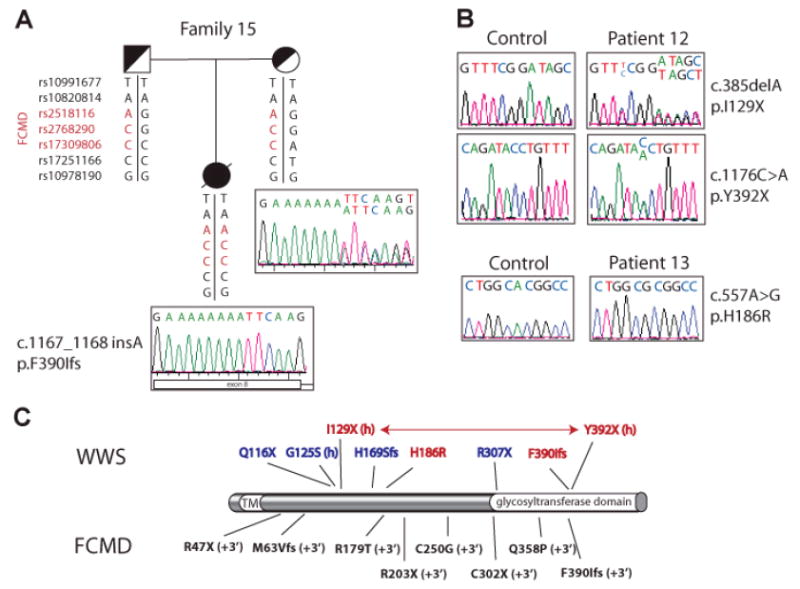
A. Patient 15 is of Ashkenazi Jewish origin. She carried a homozygous insertion in exon 8 of the FCMD gene, which was inherited from both parents. SNP genotyping results are indicated below each individual. The same mutation and the same SNP haplotype were found in two additional Ashkenazim affected in our cohort suggesting a founder effect in this population. B. Patient 12 carried two heterozygous nonsense mutations, while Patient 13 carried a homozygous missense mutation. C. Schematic representation of the fukutin protein and mutation summary. Mutations listed above the protein cause WWS: previously reported mutations are in blue (G125S was identified in heterozygosity with a small 3’ UTR deletion, which is different from the classic FCMD mutations (Cotarelo, et al., 2008)), mutations identified in this study are in red. (h) means heterozygous mutations. Compound heterozygous mutations are connected by an arrow. Below the protein coding mutations causing FCMD are shown; (+3’) next to each mutation indicates that the mutation is in heterozygosity and associated with the 3’ retransposon insertion characteristic of Japanese FCMD patients. TM: transmembrane domain.
No IBD was observed at the FCMD locus in our consanguineous families. Due to the high resolution of our SNP screen, we wondered whether small regions of IBD with identical haplotype could be used to identify a founder mutation in non-consanguineous families with the same ethnic origin. All three Ashkenazi Jewish families in our group carried the same homozygous haplotype at the FCMD locus (Figure 2A). Upon sequencing of the FCMD gene, we found the same homozygous insertion in all three affected children (c.1167_1168insA, Table 1, Family 14-16). This insertion had been described as a de novo heterozygous change coupled with the 3’ UTR insertion in a Japanese FCMD patient (Kondo-Iida, et al., 1999) and was concurrently identified by another group in Family 14, which was obtained from Coriell Cell Repositories, Camden, NJ (Cotarelo, et al., 2008). In our patient cohort, this mutation appears to represent an inherited founder mutation common to all Ashkenazi families (Figure 2A). We therefore analyzed 299 samples from the control Ashkenazi population in Israel to determine carrier frequency and found that 2/299 (0.7%) of the Israeli Ashkenazim carry this FCMD mutation in heterozygosity.
Sequencing in the remaining patients identified two additional cases carrying FCMD mutations. A Caucasian boy from the United States carried a deletion (c.381delA, p.I129X) and an early termination (c.1176C>A, p.Y392X) (Table 1, Patient 11; Figure 2B) and was presumed to be a compound heterozygote, but parental samples were not available to confirm this. A Mexican boy from the United States originally reported as being born from a non-consanguineous union showed large stretches of homozygosity in his genome scan, suggesting that the parents share a common ancestor. He carried a homozygous mutation in exon 4 (c.557A>G, p.H186R) (Table 1, Patient 12; Figure 2B).
DISCUSSION
To date, there has been no comprehensive analysis of the frequency and distribution of mutations in all known WWS genes in a large group of carefully phenotyped WWS patients. Previous reports indicated that only 10-20% of WWS cases could be explained by mutations in POMT1 and POMT2 (Currier, et al., 2005; Vajsar and Schachter, 2006). Mutations in FCMD, FKRP and LARGE were reported in a few WWS patients each (Silan, et al., 2003; Beltran-Valero de Bernabe, et al., 2003 and 2004; Godfrey, et al., 2007; van Reeuwijk, et al., 2007; Cotarelo, et al., 2008), and were presumed to be extremely rare. We assembled a large cohort of patients with typical WWS, drawn from Middle Eastern consanguineous families and from consanguineous and non-consanguineous families from Europe and the Americas. We found that 40% (16/40) of patients in our cohort carried mutations in the coding sequence of POMT1, POMT2, FKRP or FCMD with no POMGNT1 or LARGE mutations detected. FCMD and FKRP mutations in particular were much more common than previously suggested and were mostly identified in non-consanguineous patients of European descent (Table 2). All Ashkenazi Jewish patients in our group shared an identical haplotype at the FCMD locus and the same mutation suggesting a founder effect in this population (Table 2). We identified the carrier frequency of this mutation to be 0.7% in the Ashkenazi population in Israel, which will be extremely informative for genetic testing. A striking difference was observed in the geographic distribution of mutations, as Middle Eastern families were mostly carriers of POMT1 mutations, while the most common cause of European/American cases was FCMD mutations (Table 2).
Table 2.
Frequency of coding mutations according to origin
| POMT1 | POMT2 | FCMD | FKRP | |
|---|---|---|---|---|
| Middle East (14) | 35.7% (5/14) | 14.3% (2/14) | 0.0% | 7.1% (1/14) |
| Europe/Americas (27) | 7.4% (2/27) | 3.7% (1/27) | 18.5% (5/27) | 3.7% (1/27) |
| Ashkenazi (3) | 0.0% | 0.0% | 100.0% (3/3) | 0.0% |
| Hispanics (6) | 16.7% (1/6) | 0.0% | 16.7% (1/6) | 0.0% |
Our data support the hypothesis that WWS is an extremely genetically heterogeneous disease. Analysis of the medical records and neuroimaging data available for our cohort revealed that patients carrying POMT1, POMT2, FKRP and FCMD mutations are clinically and radiologically very similar (Table 3 and Figure 3). Moreover, they are indistinguishable from WWS patients not carrying mutations in any of these genes.
Table 3.
Summary of clinical findings in WWS patients carrying mutations in POMT1, POMT2, FKRP and FCMD
| Pat. | Gene | Brain findings
|
Eye findings
|
Muscle findings
|
Other
|
|||
|---|---|---|---|---|---|---|---|---|
| Cerebral Cortex | Ventricles | Cerebellum | Tone | CK (U/L) | ||||
| 1 | POMT1 | cobblestone | enlarged | small, dysmorphic | bilateral glaucoma and cataracts | low | 1545 | seizures, congenital hip dislocation |
| 2a | POMT1 | cobblestone | enlarged | small, dysmorphic | retinal dysplasia, glaucoma | low | 1565 | |
| 2b | POMT1 | cobblestone | enlarged | small hemispheres, no vermis | L microphthalmia, retinal dysplasia, AC malformation, glaucoma | low | 787 | seizures |
| 3 | POMT1 | lissencephaly, ctx too thin to assess | enlarged, encephalocele | small hemispheres, no vermis | microphthalmia | n/a | n/a | seizures, contractures, polydactyly |
| 4 | POMT1 | cobblestone | enlarged | small | n/a | n/a | n/a | contractures, bilateral kidney dysplasia, |
| 5 | POMT1 | lissencephaly, ctx too thin to assess | enlarged | small hemispheres, no vermis | shallow AC, retinal hypoplasia | low | n/a | L hydronephrosis |
| 6 | POMT1 | cobblestone | enlarged | small | cataracts, AC malformation | n/a | 5000 | |
| 7 | POMT2 | cobblestone | enlarged | small | cataracts | low | elevated | |
| 8 | POMT2 | cobblestone | enlarged | small hemispheres, no vermis | mild ON hypoplasia | n/a | 2000 | tower skull deformity |
| 9 | POMT2 | cobblestone | enlarged | small, dysmorphic | retinal dysplasia, ON hypoplasia | low | 1666 | hydronephrosis |
| 10 | FKRP | frontal PMG with cobblestone features | enlarged | small hemispheres, no vermis | detached retina | low | n/a | seizures |
| 11 | FKRP | cobblestone | enlarged | small, dysmorphic | bilateral cataracts | low | 5941 | seizures, arthrogryposis |
| 12 | FCMD | cobblestone | enlarged | small hemispheres, no vermis | ON hypoplasia | low | 2255 | seizures |
| 13 | FCMD | cobblestone | enlarged | small hemispheres, no vermis | ON hypoplasia | low | n/a | |
| 14 | FCMD | cobblestone | enlarged encephalocele | n/a | microphthalmia, microcornea, cataracts | elevated | ||
| 15 | FCMD | cobblestone | enlarged | small, dysmorphic | microphthalmia, retinal dysplasia | n/a | 39146 | mild hydronephrosis |
| 16 | FCMD | cobblestone | enlarged | small vermis | glaucoma | low | 9900 | seizures |
Abbreviations: R, right; L, left; AC, anterior chamber; ON, optic nerve; ctx, cortex; PMG, polymicrogyria
Figure 3. Brain imaging of children with WWS of varying genetic etiologies.
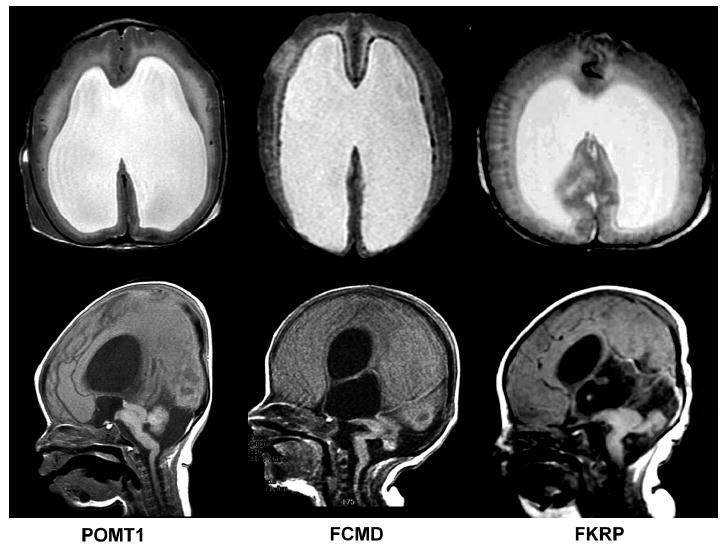
Magnetic resonance imaging (MRI) using T2-weighted axial (top row) and T1-weighted sagittal (bottom row) sequences demonstrates that affected individuals with mutations in the POMT1 (left), FCMD (middle), and FKRP (right) genes have indistinguishable radiological phenotypes. All demonstrate classic WWS findings including cobblestone lissencephaly with an absence of sulcation and an irregular gray-white junction, marked ventriculomegaly, and severe dysplasia of posterior fossa structures, including cerebellar hypoplasia and a “kinked” appearance of the brainstem.
Since genotype/phenotype correlation does not appear to allow for the prediction of the responsible mutated gene in this disorder, systematic mutation analysis and correlation with geographical and ethnic provenance may be useful to guide a genetic diagnosis. Our data suggests that POMT1 is more frequently mutated in Middle Eastern populations, while FCMD may be an additional candidate for testing in populations of European descent, in particular among Ashkenazi Jewish families. A reduced frequency of FCMD mutations in the Middle East may explain why FCMD was only identified twice by homozygosity mapping as a cause of WWS.
In addition, because the exact involvement of FCMD, FKRP and LARGE in α-dystroglycan glycosylation is still largely unknown, comprehensive mutation analysis in patients and genotype/phenotype correlation across the spectrum of disease caused by these genes may provide clues to gene function. Only one mutation in the putative glycosyltransferase FKRP has previously been identified as a cause of WWS (Beltran-Valero de Bernabe, et al., 2004). We have identified one known and two novel alleles: p.A114G was present in heterozygosity in one patient and it was previously found to cause MEB in homozygosity (Mercuri, et al., 2003), p.E146A is a novel change, and the 9bp duplication in Family 11 is a novel mutation near the site of the only previously described WWS mutation (C318Y) (de Bernabe, et al., 2003). While all other known FKRP mutations usually cause congenital muscular dystrophy with mild or no brain defects (Brockington, et al., 2001; Mercuri, et al., 2006), our results indicate that changes at certain residues in the protein are extremely deleterious for brain development. Functional analyses in animal models will determine how these mutations affect the FKRP protein.
Acknowledgments
The authors would like to thank all the families for their participation in this study, and all the collaborators who contributed samples, where no mutation was identified, Adria Bodell and Kira Apse for their help in coordinating collection of patient samples in the past, Seung-Yun Yoo, Eric Morrow and Christina Austin for insightful critical discussion. This work was supported by National Institutes of Health grant R37 NS35129-05 (CAW). M.C.M is supported by a Development Grant from the Muscular Dystrophy Association. B.S.C. is supported by the National Institutes of Health (K23 NS049159). CAW is an Investigator of the Howard Hughes Medical Institute.
References
- Akasaka-Manya K, Manya H, Nakajima A, Kawakita M, Endo T. Physical and functional association of human protein O-mannosyltransferases 1 and 2. J Biol Chem. 2006;281(28):19339–45. doi: 10.1074/jbc.M601091200. [DOI] [PubMed] [Google Scholar]
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, Celli J, van Beusekom E, van der Zwaag B, Kayserili H, Merlini L, Chitayat D, Dobyns WB, Cormand B, Lehesjoki AE, Cruces J, Voit T, Walsh CA, van Bokhoven H, Brunner HG. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71(5):1033–43. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, van Bokhoven H, van Beusekom E, Van den Akker W, Kant S, Dobyns WB, Cormand B, Currier S, Hamel B, Talim B, Topaloglu H, Brunner HG. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Genet. 2003;40(11):845–8. doi: 10.1136/jmg.40.11.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Voit T, Longman C, Steinbrecher A, Straub V, Yuva Y, Herrmann R, Sperner J, Korenke C, Diesen C, Dobyns WB, Brunner HG, van Bokhoven H, Brockington M, Muntoni F. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41(5):e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet C, Gonzales M, Vuillaumier-Barrot S, Devisme L, Lebizec C, Alanio E, Bazin A, Bessieres-Grattagliano B, Bigi N, Blanchet P, Bonneau D, Bonnieres M, Carles D, Delahaye S, Fallet-Bianco C, Figarella-Branger D, Gaillard D, Gasser B, Guimiot F, Joubert M, Laurent N, Liprandi A, Loget P, Marcorelles P, Martinovic J, Menez F, Patrier S, Pelluard-Nehme F, Perez MJ, Rouleau-Dubois C, Triau S, Laquerriere A, Encha-Razavi F, Seta N. Molecular heterogeneity in fetal forms of type II lissencephaly. Hum Mutat. 2007;28(10):1020–27. doi: 10.1002/humu.20561. [DOI] [PubMed] [Google Scholar]
- Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, Ponting CP, Estournet B, Romero NB, Mercuri E, Voit T, Sewry CA, Guicheney P, Muntoni F. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69(6):1198–209. doi: 10.1086/324412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3(4):285–98. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- Cormand B, Pihko H, Bayes M, Valanne L, Santavuori P, Talim B, Gershoni-Baruch R, Ahmad A, van Bokhoven H, Brunner HG, Voit T, Topaloglu H, Dobyns WB, Lehesjoki AE. Clinical and genetic distinction between Walker-Warburg syndrome and muscle-eye-brain disease. Neurology. 2001;56(8):1059–69. doi: 10.1212/wnl.56.8.1059. [DOI] [PubMed] [Google Scholar]
- Cotarelo RP, Valero MC, Prados B, Pena A, Rodriguez L, Fano O, Marco JJ, Martinez-Frias ML, Cruces J. Two new patients bearing mutations in the fukutin gene confirm the relevance of this gene in Walker-Warburg syndrome. Clin Genet. 2008;73(2):139–45. doi: 10.1111/j.1399-0004.2007.00936.x. [DOI] [PubMed] [Google Scholar]
- Currier SC, Lee CK, Chang BS, Bodell AL, Pai GS, Job L, Lagae LG, Al-Gazali LI, Eyaid WM, Enns G, Dobyns WB, Walsh CA. Mutations in POMT1 are found in a minority of patients with Walker-Warburg syndrome. Am J Med Genet A. 2005;133(1):53–7. doi: 10.1002/ajmg.a.30487. [DOI] [PubMed] [Google Scholar]
- Dobyns WB, Pagon RA, Armstrong D, Curry CJ, Greenberg F, Grix A, Holmes LB, Laxova R, Michels VV, Robinow M, Zimmerman RL. Diagnostic criteria for Walker-Warburg syndrome. Am J Med Genet. 1989;32(2):195–210. doi: 10.1002/ajmg.1320320213. [DOI] [PubMed] [Google Scholar]
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, Straub V, Robb S, Quinlivan R, Feng L, Jimenez-Mallebrera C, Mercuri E, Manzur AY, Kinali M, Torelli S, Brown SC, Sewry CA, Bushby K, Topaloglu H, North K, Abbs S, Muntoni F. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–35. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, Segawa M, Yoshioka M, Saito K, Osawa M, Hamano K, Sakakihara Y, Nonaka I, Nakagome Y, Kanazawa I, Nakamura Y, Tokunaga K, Toda T. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394(6691):388–92. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- Kondo-Iida E, Kobayashi K, Watanabe M, Sasaki J, Kumagai T, Koide H, Saito K, Osawa M, Nakamura Y, Toda T. Novel mutations and genotype-phenotype relationships in 107 families with Fukuyama-type congenital muscular dystrophy (FCMD) Hum Mol Genet. 1999;8(12):2303–9. doi: 10.1093/hmg/8.12.2303. [DOI] [PubMed] [Google Scholar]
- Lin M, Wei LJ, Sellers WR, Lieberfarb M, Wong WH, Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20(8):1233–40. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- Longman C, Brockington M, Torelli S, Jimenez-Mallebrera C, Kennedy C, Khalil N, Feng L, Saran RK, Voit T, Merlini L, Sewry CA, Brown SC, Muntoni F. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12(21):2853–61. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- Louhichi N, Triki C, Quijano-Roy S, Richard P, Makri S, Meziou M, Estournet B, Mrad S, Romero NB, Ayadi H, Guicheney P, Fakhfakh F. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics. 2004;5(1):27–34. doi: 10.1007/s10048-003-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri E, Brockington M, Straub V, Quijano-Roy S, Yuva Y, Herrmann R, Brown SC, Torelli S, Dubowitz V, Blake DJ, Romero NB, Estournet B, Sewry CA, Guicheney P, Voit T, Muntoni F. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53(4):537–42. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- Mercuri E, Topaloglu H, Brockington M, Berardinelli A, Pichiecchio A, Santorelli F, Rutherford M, Talim B, Ricci E, Voit T, Muntoni F. Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch Neurol. 2006;63(2):251–7. doi: 10.1001/archneur.63.2.251. [DOI] [PubMed] [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silan F, Yoshioka M, Kobayashi K, Simsek E, Tunc M, Alper M, Cam M, Guven A, Fukuda Y, Kinoshita M, Kocabay K, Toda T. A new mutation of the fukutin gene in a non-Japanese patient. Ann Neurol. 2003;53(3):392–6. doi: 10.1002/ana.10491. [DOI] [PubMed] [Google Scholar]
- Thi Tran HT, Takeshima Y, Surono A, Yagi M, Wada H, Matsuo M. A G-to-A transition at the fifth position of intron-32 of the dystrophin gene inactivates a splice-donor site both in vivo and in vitro. Mol Genet Metab. 2005;85(3):213–9. doi: 10.1016/j.ymgme.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Vajsar J, Schachter H. Walker-Warburg syndrome. Orphanet J Rare Dis. 2006;1:29. doi: 10.1186/1750-1172-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Reeuwijk J, Brunner HG, van Bokhoven H. Glyc-O-genetics of Walker-Warburg syndrome. Clin Genet. 2005a;67(4):281–9. doi: 10.1111/j.1399-0004.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabe D, Sabatelli P, Merlini L, Boon M, Scheffer H, Brockington M, Muntoni F, Huynen MA, Verrips A, Walsh CA, Barth PG, Brunner HG, van Bokhoven H. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet. 2005b;42(12):907–12. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Reeuwijk J, Grewal PK, Salih MA, Beltran-Valero de Bernabe D, McLaughlan JM, Michielse CB, Herrmann R, Hewitt JE, Steinbrecher A, Seidahmed MZ, Shaheed MM, Abomelha A, Brunner HG, van Bokhoven H, Voit T. Intragenic deletion in the LARGE gene causes Walker-Warburg syndrome. Hum Genet. 2007;121(6):385–90. doi: 10.1007/s00439-007-0362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG, Cox J, Springell K, Hampshire DJ, Mohamed MD, McKibbin M, Stern R, Raymond FL, Sandford R, Malik Sharif S, Karbani G, Ahmed M, Bond J, Clayton D, Inglehearn CF. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am J Hum Genet. 2006;78(5):889–96. doi: 10.1086/503875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, Inazu T, Mitsuhashi H, Takahashi S, Takeuchi M, Herrmann R, Straub V, Talim B, Voit T, Topaloglu H, Toda T, Endo T. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Developmental Cell. 2001;1(5):717–24. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]