

Abstract
A critical regulator of the apoptotic machinery is the Bcl-2 family proteins whose over expression confers a protective effect on malignant cells against death signals of apoptosis. Cancer cells that are resistant to various anti-cancer drugs and treatment regimen are found to over express these Bcl-2 proteins such as Bcl-2, Bcl-XL, Mcl-1, Bcl-w and A1/Bfl1. In recent years there has been an exponential growth in the identification as well as synthesis of non-peptidic cell permeable small-molecule inhibitors (SMIs) of protein-protein interaction. The focus of this article is on inhibitors of anti-apoptotic protein Bcl-2. This review summarizes an up to date knowledge of the available SMIs, their mode of action as well as their current status in preclinical as well as clinical development.
Keywords: SMI, Bcl-2 family proteins, Apoptosis
REVIEW
Apoptosis is a natural process through which multicellular organisms eliminate ageing or damaged cells. This is a very coordinated mechanism involving activation of numerous affecter proteins such as caspases that, in turn, promote a cascade of events leading to cell death. Bcl-2 and its related family of proteins (Bcl-XL, Bcl-w, A1 and Mcl-1) are the key negative regulators of the apoptotic switch (Cory and Adams 2002). On the contrary the structurally similar Bax and Bak proteins promote cell death. This switch between life and death is also governed by related proteins such as Bim, Bad, puma and noxa that share a signature pro-death domain BH3 (Bcl-2 homology region 3). On their release the BH3-only proteins are simulated by diverse intracellular stress signals which cause the BH3 domain to dock at the extended hydrophobic groove on the pro-life Bcl-2-like proteins, thereby neutralizing them (Liu et al, 2003; Sattler et al., 1997). This in turn leads to the accumulation of pro-apoptotic Bax and Bak on the endoplasmic reticulum and mitochondrial membranes, resulting in the release of apoptogenic proteins such as cytochrome c to trigger activation of caspases and ultimately leading to cell death. In cancer the anti-apoptotic proteins especially Bcl-2, are found to be over expressed. Furthermore the signaling through BH3 domain is also found to be impaired in most if not all tumor types (Villunger et al 2003). Surprisingly, all cancers harbor complete core apoptotic machinery making it ideal for targeted therapies using anticancer agents (Cory and Adams 2002).
An overview of the Bcl-2 family of proteins
Bcl-2 derives its name from B-cell lymphoma 2, as it is the second member of a range of proteins initially described as a reciprocal gene translocation in chromosomes 14 and 18 in follicular lymphomas. Proteins in the Bcl-2 family govern the mitochondrial outer membrane permeablization (MOMP) and thus play a key role in regulating the apoptotic process (Adams and Cory 1998; Green and Reed 1998; Gross et al., 1998). The Bcl-2 family proteins include anti-apoptotic protein as well as pro-apoptotic members (see Table 1) that share homology in up to 4 conserved regions named Bcl-2 homology (BH) 1-4 domains BH1, BH2, BH3 and BH4 (Chittenden et al., 1995; Geneste et al.,2006; Wang et al., 1996; Letai et al., 2002; Kelekar et al 1998; Huang et al., 2000 and Huang et al., 1998). All 4 homology regions are present in Bcl-2 and Bcl-XL (Kelekar et al 1998). The Bcl-2 family has been divided into 3 major subclasses; 1) the anti-apoptotic proteins, which include Bcl-2 and Bcl-XL; 2) the pro-apoptotic proteins, which was further subdivided to include multidomain proteins, like Bax and Bak, which possess sequence homology in BH1-3 domains, and 3) the “BH3-domain only” proteins that share sequence homology within the amphipathic α-helical BH3 region (Chittenden et al., 1995 and Wang et al., 1996). The BH3-only proteins are pro-apoptotic and include Bid, Bik, Bim, Bad, Puma and Noxa (Letai et al., 2002 and Kelekar et al 1998).
Table 1.
Classification of Bcl-2-family proteins
| APOPTOTIC PROPERTY |
SUBCLASS | PROTEIN NOMENCLATURE |
DOMAINS |
|---|---|---|---|
| Anti-apoptotic | Multidomain |
Bcl-2, Bcl-Xl, Mcl- 1, A1, Bcl-w, Bcl-B |
BH1, BH2, BH3, BH4 |
| Proapoptotic | Multidomain | Bax, Bak, Bok | BH1, BH2, BH3 |
| BH3 only (Direct Activator) |
Bid, Bim |
BH3 |
|
| BH3 (Derepressor) |
Bad, Bik, Bnip3, Puma, Noxa, Bmf, Hrk, others |
BH3 |
|
A balance between members of the Bcl-2 family is believed to determine whether mitochondria remain intact or permeabilized and release proteins that promote cell death (Oltvai et al., 1993; Korsmeyer et al., 1993; Sedlak et al., 1995 and Yang et al., 1995). One of these released proteins is cytochrome c, forms a 7-fold-symmetric supercomplex (“the apoptosome”) with Apaf-1, which activates caspase-9 (Acehand et al., 2002 and Zhou et al 1999). Caspase-9, in turn activates caspase-3, the protease that cleaves the majority of caspase substrates during apoptosis. The intrinsic apoptotic pathway is controlled by Bcl-2 family proteins near or embedded in the endoplasmic reticulum (ER) and the outer mitochondrial membrane (Chao et al 1998; Reed 1997a and Wei et al., 2001). Results of studies in Bax/Bak double knockout (DKO) mouse fibroblasts reveal that binding of BH3 domain from Bim, Bad, Noxa and Bid to either Bax or Bak is essential for BH3-domain-induced apoptosis (Letai et al., 2002; Reed 1997a ;Cheng et al.,2001 and Zhong et al.,2001).
The new class of drugs we describe in this review function by perturbing the Bcl-2 system, which functions as gatekeeper in the intrinsic pathway of apoptosis. As suggested more than a decade ago by Stan Korsmeyer and other workers (Oltvai et al.,1993; Korsmeyer et al.,1993; Sedlak et al., 1995 and Yang et al., 1995), much of the stability of the Bcl-2 system arises through protein-protein interaction between pro-apoptotic and anti-apoptotic members, listed in Table 1. These proteins feature a long amphipathic α-helix in their 3-dimensional structure, whose sequence is partially conserved between family members and during evolution. The length and position of the BH3 domain in the folded structure, however, is highly-conserved, because it fits into a hydrophobic groove found on the surface of anti-apoptotic proteins such as Bcl-XL, Bcl-2, Mcl-1 and others (Figure 1.
Figure 1. Hydrophobic Groove in the Anti-Apoptotic Bcl-2 Family Proteins is the Specific Binding Sites for SMIs like ABT-737 and TW-37.
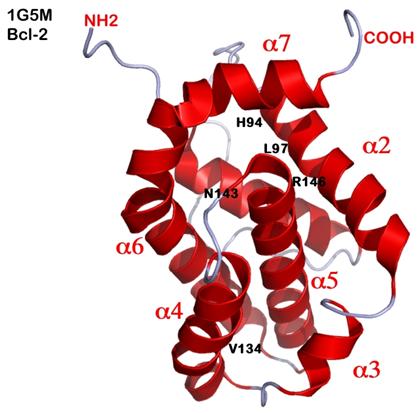
BH3-mimetic SMIs mimic the amphipathic α-helix of the BH3-only pro-apoptotic proteins, which bind to a hydrophobic cleft formed by helices α2, α4, and α5 in the folded structure shown for Bcl-2, taken from PDB file 1G5M. Critical residues in Bcl-2 include R146 in α5 and N143 located on the loop between α4 and α5. Other residues in Bcl-2 key to drug binding are the hydrophobic sidechains of residues H94 and L97 in helix α2. The chemical interactions between Bcl-2 and drug are thus chiefly hydrophobic (van der Waals), except for one ionic interaction between R146 and a nucleophile (often negatively charged) in the drug. In TW-37, the polyphenolic ring interacts closely with R146 and N143. Derivatization of all 3 hydroxyl groups in the polyphenolic ring to hydroxyl methyl creates TW-37A (the inactive congener).
Members of the Bcl-2 proteins are promising targets for cancer therapy
Over expression of Bcl-2, Mcl-1, Bcl-XL and other members contribute to cancer progression and confer resistance to apoptosis induced by standard anti-cancer therapies (Garcia et al., 2007). Most of the currently available cancer chemotherapeutic agents target cellular DNA integrity or replication, and indirectly trigger apoptosis in tumor cells (Borner., 2003). Tumors expressing high levels of Bcl-2, Mcl-1, or Bcl-XL, are often found to be resistant to chemotherapeutic agents or radiation therapy (Lee et al., 1999; Simonian et al., 1997 and Reed et al., 1996). Thus, inhibition of the function of the anti-apoptotic members represents a novel and promising strategy for designing new class of anticancer drugs that can overcome the resistance of cancer cells to chemotherapy or radiation.
Various approaches of targeting Bcl-2
Several approaches have been developed to target Bcl-2 including an approach to inhibit Bcl-2 expression levels. This formed the basis of the concept behind Bcl-2 anti-sense therapy (Cotter 1996; Morris et al., 2002 and Mohammad et al., 2002). A promising Bcl-2 anti-sense oligonucleotide G3139 was in several clinical trials for treatment of various forms of cancer. It has been found that robust intracellular concentrations could be achieved in vivo in bone marrow (range, 3.4-40.6 pmol/mg protein) and peripheral blood mononuclear cells (range, 0.47-19.4 pmol/mg protein) from acute myeloid leukemia patients treated with G3139. Further, these intracellular concentrations were related to Bcl-2 mRNA down regulation (Dai et al., 2005). The second approach is to block the activity of Bcl-2 using an antibody against Bcl-2. An intracellular anti-Bcl-2 single-chain antibody has been shown to increase drug-induced cytotoxicity in the MCF-7 breast cancer cell lines as well as other cancers (Piche et al., 1998). The third approach is to use a ribozyme against Bcl-2. More recently, a synthetic, cell permeable Bak BH3 peptide that binds to Bcl-2 has been shown to induce apoptosis in vitro and have in vivo activity in human myeloid leukemia growth in severe combined immunodeficient mice (Wang et al., 2000). A chemical strategy has also been pursued by some researcher using hydrocarbon stabling to generate stapled BH3 peptide with increased pharmacological properties. (Walensky et al., 2004). The stapled peptides, called “stabilized alpha-helix of BCL-2 domains” (SAHBs), are helical, protease-resistant, and cell-permeable molecules that bind with increased affinity to multidomain BCL-2 member pockets. Such a SAHB of the BH3 domain from the BID protein was shown to specifically activate the apoptotic pathway to kill leukemia cells. Further, other stapled BID-BH3 peptides have also been synthesized that have shown to have better apoptotic potential than parent peptide alone (Walensky et al., 2006). Although partially successful, yet none of these approaches has proven useful in the clinic, and attention has thus focused on newer agents with better clinical outcome such as non-peptidic small molecule inhibitor, which is the theme of this review.
Targeting the BH3 binding site by SMIs
SMIs are organic molecules of low molecular weight (usually less than 750 daltons). Their small size makes their use in vivo even more practical, and possibly more cost-efficient, compared to oligonucleotides or other small peptides. The anti-apoptotic function of Bcl-2 is attributed, at least in part, to the ability to heterodimerize with pro-apoptotic members such as Bim and Bid. Three dimensional structures of Bcl-XL from X-ray and NMR studies reveal that a hydrophobic groove (the BH3 binding pocket) into which the Bim or Bid BH3 domain is able to bind (Muchmore et al., 1996). This binding pocket in Bcl-2 is essential for its anti-apoptotic function. It has been hypothesized that SMIs that bind to this BH3 binding site in Bcl-2 may be capable of blocking the heterodimerization of Bcl-2 with pro-apoptotic members in the Bcl-2 protein family, such as Bid and Bim. Drug occupation of the hydrophobic groove is thus thought to disarm the anti-apoptotic function of Bcl-2 (and others) and induce apoptosis. This decade has witnessed a tremendous enthusiasm in the area of SMI design and development, about half dozen groups have taken up the challenge to develop SMIs targeting the “elongated hydrophobic cleft.” These SMIs thus act as BH3-mimetics (see Table 2). The following paragraphs summarize the currently studied SMIs, their mode of action as well as a brief description of their success in the clinic.
Table 2.
List of BH-3 non-peptidic small molecule inhibitors
| Compound | Preclinical Use | Clinical Trials |
Molecular Targets* |
|---|---|---|---|
ABT-737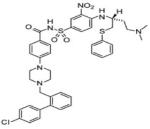
|
Multiple myeloma; acute myeloid leukemia; small cell lung cancer; lymphoma |
Phase I/II | Bcl-2 and Bcl-Xl (low nM affinity) |
|
ABT-263 (structure is proprietary) |
Multiple myeloma; small cell lung cancer; non-Hodgkin Lymphoma; chronic lymphocytic leukemia |
Phase I/IIa | Bcl-Xl, Bcl-2, Bcl-w, Bcl-B |
|
Gossypol (BL-193, AT-101) 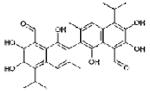
|
Head and neck tumors, malignant gliomas |
Phase II/III; | Mcl-1, Bcl-2, Bcl-Xl (highest to lowest affinity) |
|
Obatoclax (GX015-070) 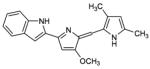
|
Myeloma; mantle cell lymphoma |
Phase I/II | Bcl-2, Bcl-Xl, Bcl-w, Mcl-1 |
TW-37
|
Non-Hodgkins Lymphoma, Pancreatic, Lung |
Preclinical | Bcl-w, Bcl-Xl, A1, Mcl-1, Bcl-2 (highest to lowest affinity) |
|
Apogossypolone (ApoG2) 
|
Non-Hodgkins Lymphoma, Lymphoma |
Preclinical | Bcl-2, Mcl-1, Bcl-Xl (highest to lowest affinity) |
HA14-1
|
Leukemia and others | Preclinical | Bcl-2, Bcl-Xl, Bcl-w |
Tetrocarcin A
|
Leukemia and others | Preclincal | Bcl-2 and Bcl-Xl |
|
Chelerythrine Chloride 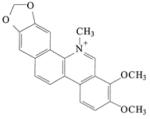
|
Squamous cell carcinoma and others |
Preclincal | Bcl-2 and Bcl-Xl |
Antimycin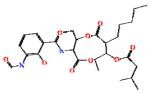
|
Various types of Cancer | Preclinical and Phase I |
Bcl-2 and Bcl-Xl |
BHI-1 derivatives
|
Under test/trials | Preclinical | Bcl-2, Bcl-Xl, Bcl-w |
(1) Gossypol
Gossypol, also known as BL-193, was the first compound (Table 1) to reach the clinic, but its mechanism of action at the time was not known (Bushunow et al., 1999; Stein et al., 1992). Gossypol is a natural polyphenol product isolated from cottonseeds and roots. It has been studied since the 1980s for its contraceptive and anticancer effects. There are three isoforms of gossypol, which include (−)-BL-193, (+)-BL-193, and (±)-BL-193. The (−)-BL-193 has been shown to be more potent than its either isoforms in its growth-inhibitory effects. Multidimensional nuclear magnetic resonance methods have shown (−)-BL-193 binds the hydrophobic groove of Bcl-2 and Bcl-XL (Wang et al., 2006). Although this compound has been primarily studied as an inhibitor of Bcl-2, its other mechanisms of action have also been proposed. It has been shown earlier that gossypol could induce oxidative DNA breakage in vitro in the presence of metal ions such as copper (Zaidi and Hadi 1992a and b). In a recent report it has been shown that gossypol induces apoptosis in chronic lymphocytic leukemia through generation of ROS which in turn mediate the release of cytochrome c and thus causing apoptosis (Balakrishnan et al., 2008). Gossypol is currently in preclinical testing.
(2) TW-37
TW-37 is a benzenesulfonyl derivative that was first synthesized by researchers at University of Michigan. The drug has both pro-apoptotic (Mohammad et al., 2007) and antiangiogenic effects (Zeitlin et al., 2006). It was originally designed to target the BH3-binding groove in the Bcl-XL, and has shown to have high affinities for Bcl-2, Bcl-XL, and, unlike most SMIs, also Mcl-1 (Mohammad et al., 2007). Zeitlin et al. (2006) have shown the antiangiogenic effects occur by inducing apoptosis in endothelial cells. These researchers also found that low concentrations of TW-37 result in inhibition of migration and capillary sprouting assays. Our laboratory has extensively studied this SMI for its apoptotic action not just on leukemia or lymphoma (Mohammad et al., 2007) but also on pancreatic cancer (Wang et al., 2008 and Azmi et al., 2008.). The observed effect of TW-37 is described in the forthcoming passage. The drug is still in the preclinical phase of testing.
(3) Apogossypolone (ApoG2)
ApoG2 is a derivative of gossypol that was designed by Ascenta in order to reduce the non-specific reactivity and toxicity of gossypol and is currently in the preclinical phase of testing This modification involved the removal of two reactive aldehyde groups on the polyphenolic rings of gossypol. Current research shows ApoG2 is a potent inhibitor of Mcl-1 and Bcl-2 proteins (Zhang et al., 2006). Studies from our laboratory have shown ApoG2 blocks binding of Bim and Bcl-2 and induces apoptosis in lymphoma cell lines with minimal toxicity (Mohammad et al., 2008). Further it has also been shown that Apog2 induces apoptosis in follicular Small Cleaved Cell Lymphoma model pre-B-acute lymphoblastic leukemia, mantle cell lymphoma, marginal zone lymphoma, as well as chronic lymphocytic leukemia. Therefore, ApoG2 could potentially be a more effective drug in the lymphoma clinic spanning a greater array of patients. (Arnold et al., 2008).
(4) ABT-737
ABT-737 was made by collaboration from IDUN laboratories and Abbott laboratories. It has been shown to inhibit the anti-apoptotic proteins Bcl-2, Bcl-XL and Bcl-w, but not other anti-apoptotic proteins such as Bcl-B, Mcl-1 and A1 (Oltersdorf et al., 2005). The drug's inability to target Mcl-1 causes many cell types to be refractory to its effects. Experiments by van Delft et al. demonstrate downregulation of Mcl-1 resulted in increased sensitivity to ABT-737 (Van Delft et al., 2006). These researchers also found Mcl-1 overexpression resulted in increased drug resistance. This drug is currently in phase 2 of clinical testing.
(5) ABT-263
ABT-263 an orally available drug that is structurally related to ABT-737 and has shown potent cytotoxicity against numerous human tumor cell lines including many lymphoid malignancies (Lock et al., 2007). It is considered a second generation inhibitor of Bcl-2 family proteins with high affinity to the anti-apoptotic proteins Bcl-XL, Bcl-2, Bcl-w and Bcl-B (Wilson et al., 2007). Recent preclinical studies demonstrated in vitro activity of ABT-263 against ALL cell lines but showed limited single agent activity against solid tumor cell lines (Lock et al 2007). As with ABT-737 it does not have a high affinity for Mcl-1. It is currently in phase I/II clinical testing.
(6) Obatoclax (GX-015-070)
Obatoclax is an indole bipyrrole compound that was developed by Gemin X. It has been shown by Trudel et al. to not only induce apoptosis by inhibiting the interaction between pro-apoptotic and anti-apoptotic proteins, but also to upregulate the pro-apoptotic protein Bim (Trudel et al., 2007). Specifically, these researchers found Obatoclax inhibits binding of Bak to Mcl-1 and the up-regulation of Bim induces cytochrome c release and activation of caspase-3. It is currently being assessed in phase I and phase II clinical trials for solid tumors and hematological malignancies (Borthakur et al., 2006). In a recent report it has been shown that obatoclax could synergize with ABT-737 to induce apoptosis. These findings suggest that this agent may not only augment the clinical activity of traditional chemotherapy, but it can potentiate the activity of other BH3 mimetics with different binding affinities/patterns (Konopleva et al., 2008). Bcl-2 phosphorylation due to activation of MEK/ERK pathway is an important physiological phenomena that results in decreased sensitivity towards obatoclax. In an interesting study by Perez-Galan and co-workers (2008), it was shown that sensitivity to obatoclax could be increased by synergizing it with ERK inhibitors giving a new window of opportunity towards treating resistant cells. Unfortunately, this compound may need to be redesigned for pharmacokinetic reasons; Trudel et al. recently report that treatment of mice with bolus injection of GX-015-070 fails to reach pharmacologically-effective levels in the blood; and dose escalation is limited by “significant neurologic toxicity”(Trudel et al., 2007).
Other SMIs currently under study
Apart from the above mentioned SMIs there are a number of small-molecule inhibitors with diverse chemical structure under study. For example HA14-1 which was derived through the DISCOVER program using three dimensional database (San Leandro, CA). HA14-1 has high binding affinity to Bcl-2. HA141 once bound to Bcl-2 inhibits its interaction with Bak peptide. Due to its instability and redox activity a newer and stable version was generated (sHA14-1) with better in vitro activity against cancer cells (Tian et al., 2008). Another naturally occurring SMI is tetrocarcin A (TC-A) which is produced by actinomycetes. In an interesting report it was found that TC-A and bcl-2 antisense oligonucleotides reduce radioresistance of tumor cells overexpressing Bcl-2. Therefore, it was suggested that combination of radiotherapy and Bcl-2 inhibitors may prove to be a useful therapeutic approach for treating tumors that overexpress Bcl-2 (Hara et al., 2005). Chelerythrine chloride is another SMI produced by natural benzophenanthridine alkaloid extracted from the stems of Bocconia vulcanica. It was reported that Chelerythrine induces apoptosis in SQ-20B cells by inhibiting protein kinase C (Chmura et al., 2000). Chelerythrine chloride was identified as an inhibitor of the interaction between the Bak BH3 domain and Bcl-XL (Chan et al., 2003). Recently, it was discovered that chelerythrine binds at a distinct site, different from the classic BH3 binding cleft (Zhang et al., 2006). It was found that in contrast to other the previously established pathway, chelerythrine appears to also induce alternative Bax/Bak-independent apoptotic mechanism that involves cyclosporine A-sensitive mitochondrial membrane permeability (Wan et al., 2008). Antimycin is yet another BH3 mimetic from natural source (Streptomyces). Antimycin A has been shown to compete with the Bak BH3 for binding to the hydrophobic pocket of Bcl-2 and Bcl-XL proteins. It can induce apoptosis in Bcl-XL overexpressing TAMH murine hepatocyte cells (Tzung et al., 2001). Apart from its binding specificity towards Bcl-2, antimycin has been shown to affect the apoptotic machinery. Piskernik and colleagues (2008) using radical spin trap techniques have shown that antimycin A could release superoxide radicals (O2.−) from mitochondria to cytoplasm which is important for the induction of those processes leading to cell dysfunction and cell death.
Pan Bcl-2 SMIs
Over the last couple of years our group has extensively studied two new compounds (TW-37 and ApoG2), which seem to be close to the goal of becoming broad-spectrum or pan-Bcl-2 SMIs (Mohammad et al., 2007, Wang et al., 2008 and Azmi et al., 2008). For a drug to be successful in clinic it has to fulfill two key parameters beyond pharmacokinetic and drug formulation. The first parameter is selectivity and specificity. There is no doubt that the new compounds such as our (−)-gossypol, TW-37, ApoG2 and Abbott's ABT-737 and GX-015-070 are promising from the point of view of nanomolar Ki (determined in cell-free experiments) and nanomolar IC50 (determined on select cultured cells) (Wang et al., 2008 and Azmi et al., 2008). The parameter which is not universal to all pharmaceuticals is a parameter of critical importance to cancer. Ordinarily, a new drug must be selective and specific enough to hit intended targets and not too many side-targets, but in cancer, a new drug must not inherently induce drug-resistance (Frantz et al., 2005).
Since there are 6 genes encoding 6 key targets of BH3-mimetics, and cells express a narrow or broad subset of these; a valuable drug must hit them all, “Pan Bcl-2 drug”. It is not clear yet what determines the IC50 or biological efficacy of a BH3-mimetic SMI. Does the IC50 simply reflect the spectrum of targets in the treated cell type? It does not seem to be so. For example, the Abbott drug ABT-737 binds to Bcl-2 with a KD of 1 nM, and Bcl-XL with a respectable affinity as well; neverless, some cells treated with ABT-737 show an IC50 around 500 nM and others (OPM1, H929, and U266) even higher, 8000-15000 nM (Trudel et al., 2007).
SMIs TW-37 as anti-lymphoma agent
One of the most promising aspects of SMIs in treating cancer is that their targets and mechanisms of action are different from those of cytotoxic drugs and radiation. This makes it feasible to combine SMIs with other treatments, creating a synergistic therapy, without likely development of cross-resistance or increased toxicity. In an in vivo study it was found that TW-37 synergizes with CHOP. For example, the MTD of the TW-37+CHOP combination was determined to be 40 mg/kg (iv via tail vein, divided in 3 injections for 3 days) plus CHOP at its MTD (cyclophosphamide “C”, 40 mg/kg, iv; doxorubicin “H”, 3.3 mg/kg, iv; vincristine “O”, 0.5 mg/kg, iv; and prednisone “P”, 0.2 mg/kg, orally every day for 5 days). Animals at this dose experienced weight loss of <5% and had scruffy fur, however, with full recovery 48 to 72 h after completion of treatment. However, daily injections of 40 mg/kg for four consecutive days was toxic, as shown by a loss of >20% body weight. In addition, 60 mg/kg per injection, i.v. injected daily for 3 days was toxic. shows the tumor weight of mice treated with TW-37, CHOP, and their combination, compared with control. Mice in all treatment groups developed s.c. tumors. Tumor weights in the TW-37 + CHOP combination decreased significantly (P < 0.01) compared with either TW-37 or CHOP group, an impressive log10 reduction of 1.8. Antitumor activity of TW-37 alone, CHOP alone, or TW-37 + CHOP combination against WSU-DLCL2-bearing SCID mice as measured by T/C, T-C, and log10 kill were 57%, 19%, and 11%; 4, 8, and 12 days; and 0.6, 1.2, and 1.8, respectively (Figure 2). T/C values are used to determine tumor response. CHOP alone and TW-37 + CHOP were considered active against WSU-DLCL2 tumor (T/C < 42%). The dose and schedule of TW-37 alone and in combination with CHOP against WSU-DLCL2 xenograft tumor merits refinement, planned for future work.
Figure 2.
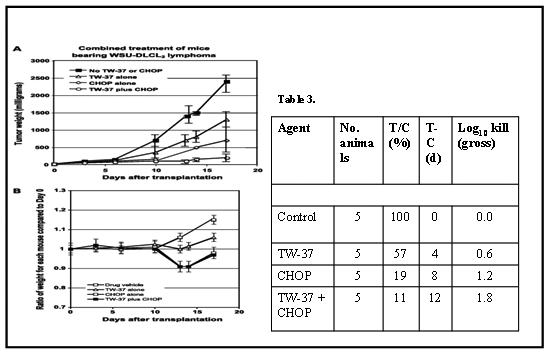
(A)Tumor reduction resulting from the four-drug regimen CHOP in combination with the BH3 mimetic SMI TW-37 in a mouse xenograft model of diffuse lymphoma. A, tumor weight (mg) of WSU-DLCL2-bearing SCID in control (diluent), TW-37, CHOP, and TW-37/CHOP. Points, mean; bars, SD. The TW-37 treatment group was 20 mg/kg given i.v. for three consecutive d. The CHOP treatment group was at its MTD. The combined treatment group was at the MTD of CHOP + 20 mg TW-37 per kg (i.v.) for three consecutive d. Tumor weight decreased significantly in mice that received TW-37/CHOP combination compared with either treatment group alone (TW-37 alone or CHOP alone), with P < 0.01. All animals were treated 5 d after tumor transplantation. Together, combination treatment with CHOP and TW-37 lead to a log10 1.8 reduction of tumor mass during the treatment period (Table 3). B, we weighed the mice over 17 d of treatment using the same treatment dose and scheduling as in (A). After 12 d, mice treated with CHOP alone or the TW-37 + CHOP combination lost ∼9% of their body weight compared with initial weight at day 0.
(B) The mice were weighed over 17 days of treatment using the same treatment dose and scheduling as in Fig 2 A After 12 days, mice treated with CHOP lost ∼9% of their body weight compared with initial weight; the curve for CHOP alone overlaps the curve for the combination, showing that addition of TW-37 to CHOP did not cause any additional toxicity.
(C) Table 3. Antitumor activity of TW-37, CHOP, and their combination in WSU-DLCL2–bearing SCID mice
Other Pharmacological targets of SMIs ApoG2 and TW-37
Studies over the last couple of years in our laboratory have revealed that apart from Bcl-2 family proteins, several other cellular targets have been found to be modulated by the above mentioned SMIs. In an interesting study in pancreatic cancer cells it was shown that at nanomolar concentrations cell growth inhibiton by TW-37 was accompanied by increased apoptosis and concomitant attenuation of NF-kB. Further, downregulation of NF-kB downstream genes such as matrix metalloproteases (MMP-9) and vascular endothelial growth factor (VEGF), resulting in the inhibition of pancreatic cancer cell migration, invasion and angiogenesis in vitro (Wang et al., 2008). We also tested TW-37 against Colo-357 in a SCID xenograft model. Our results show that TW-37 was effective in decreasing tumor weight significantly compared to untreated animals. Most importantly, we have done the combination of TW-37 with gemcitabine on cell growth and apoptosis assay in BxPC-3 and Colo-357 cell lines. We found that TW-37 sensitizes these 2 cell lines to gemcitabine-induced growth inhibition and apoptosis (Wang et al., 2008). Therefore, Bcl-2 inhibitor could be a novel agent for designing innovative approaches for demonstrating their antitumor activity against PC and, as such, could also be useful in enhancing the antitumor activity of conventional therapeutic agents for the treatment of PC patients. These results suggest that SMIs like TW-37 also play a role in regulating critical genes such as NF-kB and its downstream pathways involved in angiogenesis.
Current studies are being pursued in order to identify the effect of TW-37 on other apoptotic genes such as prostate apoptosis response 4 (PAR-4) which is a leucine zipper protein originally identified in prostate cancer cells undergoing apoptosis (Sells et al., 1997). PAR-4 is ubiquitously expressed in normal tissues and cell types and is found primarily in the cytoplasm. In contrast, PAR-4 localizes both to the cytoplasm and the nucleus in many, but not all, cancer cells and cancer tissues where it sensitizes cells to the action of diverse apoptotic stimuli and causes tumor regression. Our preliminary data reveals that TW-37 as well as its parent SMI, Apog2, both induce nuclear localization of PAR-4 and thus sensitize pancreatic cancer cells to apoptosis (Azmi et al., 2008). In combination studies with gemcitabine, pre-treatment with Apog2 or TW-37 lead to sensitization of Colo-357 cells to the growth inhibitory and apoptotic action of a therapeutic drug, gemcitabine. In an in vivo setting, the maximum tolerated dose of TW-37 in xenograft of severe combined immunodeficient (SCID) mice (40 mg/kg for three i.v. injections) led to significant tumor inhibition (Figure 3 A). In order to identify the clinical relevance of our in vitro results, an initial pilot experiment was performed using a xenograft animal model of pancreatic cancer. Immunohistohemical analysis of Colo-357 xenograft animal tissue stained with PAR-4 antibody revealed some interesting results. In the untreated control tumor tissues, we did not find any significant presence of PAR-4 and correspondingly negligible apoptosis or necrosis (Figure 3 B left panel). In contrast, in the TW-37 treated tumors, we found extensive PAR-4 staining as well as high amount of necrotic cells (Figure 3 B right panel). These observations provide evidence in support of the “proof-of-principle” for targeting PAR-4 by SMIs, which could be an important and new area in the treatment of pancreatic cancer. Our results suggest that the observed anti-tumor activity of SMIs is mediated through a novel pathway involving induction PAR-4. Further in-depth analysis is underway to identify other key regulators affected in the PAR-4 mediated apoptosis pathway.
Figure 3. TW-37 inhibits tumor growth and induces PAR-4 expression in cancer tissue.
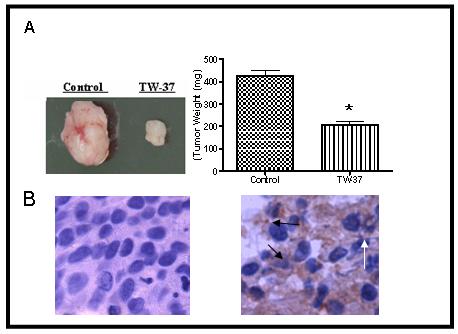
Colo-357 xenografts were inoculated s.c. in SCID mice. Once transplanted, fragments developed into palpable tumors (about 80 mg), and groups of nine animals were removed randomly and assigned to different treatment groups. Mice were injected with TW-37 at 20 mg/kg iv x3 days, for two cycles. The control group received vehicle only. (A) TW-37 retards the growth of Colo-357 tumor xenografts in nude mice. (B) PAR-4 staining of tumor tissue showing PAR-4 induction by TW-37-treated animal tumors. Control tumors (Left panel) show insignificant PAR-4 staining. TW-37 treated tumors (Right Panel) show prominent PAR-4 staining (indicative of white arrows) as well as extensive necrosis (indicated by black arrows).
Current and Future Developmental Goals
Identifying a perfect SMI is indeed very challenging both for chemist as well as biologist. The ideal BH3-mimetic small molecule drug would be orally-available, reach and maintain pharmacologically-active levels in the bloodstream, target a broad class of pro-survival Bcl-2-related targets, and function to kill tumor cells by a canonical mechanism-based mode of action. Furthermore, the compound should have anti-tumor activity as a single agent, and show additive or synergistic anti-tumor action when combined with other approved agents. No single compound described in this review fulfills all the above requirements. For example, even high bolus injections of GX015-070 (Obatoclax) (Perez-Gelan et al., 2007) in human pharmacokinetic studies, shows that the drug demonstrates an achievable Cmax of 10 to 80 ng/mL, which is below the level indicated for in vitro activity on most tumor targets, consistent with its failure to reach levels in a mouse xenograft tumor model sufficient to disrupt Mcl-1/Bax protein-protein interaction (Trudel et al., 2007). Abbott's ABT-263 represents an effort to improve on its earlier ABT-737 by making the compound orally-available, enabling phase 1/2a clinical trials (Lock et al., 2007), but ABT-263 still misses Mcl-1 as a target, showing a KD in fluorescence polarization assays of ∼500 nM. By not binding well to the important target Mcl-1, tumors treated with ABT-737 are likely to develop Mcl-1-mediated drug resistance, and ABT-263 would not be expected to improve much over ABT-737 in this aspect (Shoemaker et al., 2007 and Tahir et al., 2007). Several of the compounds touted as BH3-mimetics such as chelerythrine (Chan et al., 2003) do not actually bind to the hydrophobic pocket in Bcl-XL where the BH3 helix of tBid or Bim normally bind, but bind instead to another distinct site in Bcl-XL (70); thus they do not fulfill the mechanistic requirements of action as a true BH3-mimetic. The same criticism extends to the oftcited BH3-mimetic HA14 (Wang et al., 2000), which binds in vitro to Bcl-2 family proteins in isolation, but in the cellular environment actually decomposes to generate reactive oxygen species, which may produce its apoptotic properties (Doshi et al., 2007).
Even if a compound fulfills the essential requirements of BH3-mimetic action in living cells, it may fail in the clinic, if the compound misses important pro-survival targets, as ABT-737 (and ABT-263) fail to bind to Mcl-1. Drug specificity and selectivity is hailed as the Holy Grail by pharmacologists, but in the cancer arena, specificity should be broad to cover secondary targets whose presence can lead to resistance to the initial compound. Such is the argument (Mohammad et al., 2007) for a “pan-Bcl-2” SMI—a compound which may not bind to all of its targets in the low nanomolar range, but binds to at least Bcl-2 and Mcl-1 to disarm the pro-survival capacities of these key targets. Such “dirty drugs” may prove useful in knocking out Bcl-2-family members as well as kinase family members (Frantz et al., 2005). Thus, the future may lie for the time being still in the organic chemistry lab, to come up with novel compounds or modify pre-existing compounds which fail the “pan-Bcl-2” test. In this regard, Xing and co-workers (Xing et al., 2007; Doshi et al., 2006; Doshi et al., 2007 and Tian et al., 2008), are providing an array of fresh organic compounds which have yet to be tested against a large array of cell lines, a necessary step before testing in animal models.
Summary
SMIs of Bcl-2 family proteins as a means of novel therapy are a very exciting research area. The development of drugs able to induce apoptosis in tumor cells, without damaging healthy normal cells in the vicinity of the tumor or anywhere else in the body, is an important challenge in the fight against cancer. Many chemotherapeutic drugs cannot distinguish between cancerous and normal healthy cells in patient's body, thus causing toxicity. There is substantial evidence to support the use of SMIs of Bcl-2 proteins to overcome apoptotic resistance in a variety of human cancers with minimal toxicity to normal cells. Our current research investigations indicate that SMIs can target other important molecules such as PAR-4, Akt, PDPK1, notch-1, NF-kB, survivin and other. However, much remains to be deciphered regarding the mechanism of inhibition of Bcl-2 family proteins.
SMIs may be able to restore the normal apoptotic pathway in malignant cells with high expression of Bcl-2 family proteins and make these cells more susceptible to conventional chemotherapy. Although initially designed against Bcl2, a number of SMIs (described in this review) show a vast spectrum of activities on a number of different pharmacological targets. Therefore, it is still to early to estimate the true value of these inhibitors ad as such these SMIs may well represent an important starting point for the development of an entirely new class of anti-cancer agents.
Acknowledgements
We acknowldedge the generous financial support by National Institutes of Health RO1 Grant (Leukemia Lymphoma CA 109389 and Pancreatic R01 CA109389 to R.M. Mohammad) and from the Leukemia and Lymphoma Society (6028-07, to R.M. Mohammad).
Contract Grant Sponsor: NIH; Contract Grant Number: RO1 Grant (CA 109389), to R.M. Mohammad
Contract Grant Sponsor: NIH; Contract Grant Number: R01CA109389 to (Mohammad RM)
Contract Grant Sponsor: Leukemia and Lymphoma Society Contract Grant Number 6028-07, to R.M. Mohammad.
REFERENCES
- Cory S, Adams JM. The Bcl-2 family: regulators of the cellular life-or-death switch. Nat Rev Can. 2002;2:647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science (New York, NY. 28. 1998;281(5381):1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science (New York, NY. 28. 1998;281(5381):1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. The EMBO journal. 15. 1998;17(14):3878–85. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geneste O, Hickman J, Bennett R, Rain J-C. inventors. 2006 WO2005014638. [Google Scholar]
- Dai G, Chan KK, Liu S, Hoyt D, Whitman S, Klisovic M, Shen T, Caliguir MA, Byrd J, Grever M, Marcucci G. Cellular uptake and intracellular levels of the antisense Bcl-2 antisense G3139 in cultured cells and treated patients with acute myeloid leukemia. Clin Can Res. 2005;11:2998–3008. doi: 10.1158/1078-0432.CCR-04-1505. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 12. 1996;86(1):147–57. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Chittenden T, Flemington C, Houghton AB, Ebb RG, Gallo GJ, Elangovan B, et al. A conserved domain in Bak, distinct from BH1 and BH2, mediates cell death and protein binding functions. The EMBO journal. 15. 1995;14(22):5589–96. doi: 10.1002/j.1460-2075.1995.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yin XM, Chao DT, Milliman CL, Korsmeyer SJ. BID: a novel BH3 domain-only death agonist. Genes & development. 15. 1996;10(22):2859–69. doi: 10.1101/gad.10.22.2859. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer cell. 2002;2(3):183–92. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Kelekar A, Thompson CB. Bcl-2-family proteins: the role of the BH3 domain in apoptosis. Trends in cell biology. 1998;8(8):324–30. doi: 10.1016/s0962-8924(98)01321-x. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 8. 2000;103(6):839–42. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Huang DC, Adams JM, Cory S. The conserved N-terminal BH4 domain of Bcl-2 homologues is essential for inhibition of apoptosis and interaction with CED-4. The EMBO journal. 16. 1998;17(4):1029–39. doi: 10.1093/emboj/17.4.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Seminars in cancer biology. 1993;4(6):327–32. [PubMed] [Google Scholar]
- Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, et al. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proceedings of the National Academy of Sciences of the United States of America. 15. 1995;92(17):7834–8. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 27. 1995;80(2):285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 27. 1993;74(4):609–19. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Seminars in cancer biology. 1993;4(6):327–32. [PubMed] [Google Scholar]
- Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, et al. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proceedings of the National Academy of Sciences of the United States of America. 15. 1995;92(17):7834–8. doi: 10.1073/pnas.92.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 27. 1995;80(2):285–91. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Molecular cell. 2002;9(2):423–32. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. The Journal of biological chemistry. 23. 1999;274(17):11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annual review of immunology. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- Reed JC. Bcl-2 family proteins. Oncogene. 1998;17(25):3225–36. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- Reed JC. Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Advances in pharmacology San Diego, Calif. 1997;41:501–32. doi: 10.1016/s1054-3589(08)61070-4. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science (New York, NY. 27. 2001;292(5517):727–30. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Molecular cell. 2001;8(3):705–11. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes & development. 2001 Jun 15;15(12):1481–6. doi: 10.1101/gad.897601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A, Cayla X, Rebollo A. Inventors. 2007 EP1788394. [Google Scholar]
- Borner C. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Molecular immunology. 2003;39(11):615–47. doi: 10.1016/s0161-5890(02)00252-3. [DOI] [PubMed] [Google Scholar]
- Lee JU, Hosotani R, Wada M, Doi R, Kosiba T, Fujimoto K, et al. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur J Cancer. 1999;35(9):1374–80. doi: 10.1016/s0959-8049(99)00134-3. [DOI] [PubMed] [Google Scholar]
- Simonian PL, Grillot DA, Nunez G. Bcl-2 and Bcl-XL can differentially block chemotherapy-induced cell death. Blood. 1. 1997;90(3):1208–16. [PubMed] [Google Scholar]
- Reed JC, Miyashita T, Takayama S, Wang HG, Sato T, Krajewski S, et al. BCL-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. Journal of cellular biochemistry. 1996;60(1):23–32. doi: 10.1002/(SICI)1097-4644(19960101)60:1%3C23::AID-JCB5%3E3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Geneste O, Hickman J, Bennett R, Rain J-C. Inventors. 2006 EP1651668. [Google Scholar]
- Cotter FE. Antisense therapy for lymphomas. Hematological oncology. 1997;15(1):3–11. doi: 10.1002/(sici)1099-1069(199702)15:1<3::aid-hon583>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Morris MJ, Tong WP, Cordon-Cardo C, Drobnjak M, Kelly WK, Slovin SF, et al. Phase I trial of BCL-2 antisense oligonucleotide (G3139) administered by continuous intravenous infusion in patients with advanced cancer. Clin Cancer Res. 2002;8(3):679–83. [PubMed] [Google Scholar]
- Mohammad R, Abubakr Y, Dan M, Aboukameel A, Chow C, Mohamed A, et al. Bcl-2 antisense oligonucleotides are effective against systemic but not central nervous system disease in severe combined immunodeficient mice bearing human t(14;18) follicular lymphoma. Clin Cancer Res. 2002;8(4):1277–83. [PubMed] [Google Scholar]
- Piche A, Grim J, Rancourt C, Gomez-Navarro J, Reed JC, Curiel DT. Modulation of Bcl-2 protein levels by an intracellular anti-Bcl-2 single-chain antibody increases drug-induced cytotoxicity in the breast cancer cell line MCF-7. Cancer research. 15. 1998;58(10):2134–40. [PubMed] [Google Scholar]
- Wang JL, Zhang ZJ, Choksi S, Shan S, Lu Z, Croce CM, et al. Cell permeable Bcl-2 binding peptides: a chemical approach to apoptosis induction in tumor cells. Cancer research. 15. 2000;60(6):1498–502. [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wardner G, Verdine GL, Korsmeyer JL. Activation of apoptosis in vitro by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Joon oh K, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, et al. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature. 23. 1996;381(6580):335–41. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2. 2005;435(7042):677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer cell. 2006;10(5):389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Carol H, Houghton PJ, Morton CL, Kolb EA, Gorlick R, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2007:17. doi: 10.1002/pbc.21433. [DOI] [PubMed] [Google Scholar]
- Wilson WH, Tulpule A, Levine AM, Dunleavy K, Krivoshik AP, Hagey AE, et al. A Phase 1/2a Study Evaluating the Safety, Pharmacokinetics, and Efficacy of ABT-263 in Subjects with Refractory or Relapsed Lymphoid Malignancies. Blood (ASH Annual Meeting Abstracts) 2007;110(11):1371. [Google Scholar]
- Bushunow P, Reidenberg MM, Wasenko J, Winfield J, Lorenzo B, Lemke S, et al. Gossypol treatment of recurrent adult malignant gliomas. Journal of neuro-oncology. 1999;43(1):79–86. doi: 10.1023/a:1006267902186. [DOI] [PubMed] [Google Scholar]
- Stein RC, Joseph AE, Matlin SA, Cunningham DC, Ford HT, Coombes RC. A preliminary clinical study of gossypol in advanced human cancer. Cancer chemotherapy and pharmacology. 1992;30(6):480–2. doi: 10.1007/BF00685601. [DOI] [PubMed] [Google Scholar]
- Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. Journal of medicinal chemistry. 19. 2006;49(21):6139–42. doi: 10.1021/jm060460o. [DOI] [PubMed] [Google Scholar]
- Zaidi R, Hadi SM. Complexes involving gossypol, DNA and Cu(II) Biochem Int. 1992;28:1135–1143. [PubMed] [Google Scholar]
- Zaidi R, Hadi SM. Strand scission in DNA by gossypol and Cu(II): role of Cu(I) and free radicals. 1992 doi: 10.1002/jbt.2570070404. [DOI] [PubMed] [Google Scholar]
- Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Zeev Estrov, Bornmann W, Kantargian H, Viallet J, Samudio I, Andreef M. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax) Can Res. 2008;68:3419–3420. doi: 10.1158/0008-5472.CAN-07-1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Galan P, Roue G, Guerra ML, Nquyen M, Villamor N, Montserrat E, Shore GC, Campo E, Colomer D. BCL-2 phosphorylation modulates sensitivity to the BH3 mimetic GX15-070 (Obatoclax) and reduces its synergistic interaction with bortezomib in chronic lymphocytic leukemia cells. Leukemia. 2008 July 3; doi: 10.1038/leu.2008.175. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood. 2007 Jun 15;109(12):5430–8. doi: 10.1182/blood-2006-10-047951. [DOI] [PubMed] [Google Scholar]
- Borthakur G, O'Brien S, Ravandi-Kashani F, Giles F, Schimmer AD, Viallet J, et al. A Phase I Trial of the Small Molecule Pan-Bcl-2 Family Inhibitor Obatoclax Mesylate (GX15-070) Administered by 24 Hour Infusion Every 2 Weeks to Patients with Myeloid Malignancies and Chronic Lymphocytic Leukemia (CLL), Poster Board #-Session: 832-II. Blood (ASH Annual Meeting Abstracts) 2006 Abstract 2654.108. [Google Scholar]
- Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 1. 2007;13(7):2226–35. doi: 10.1158/1078-0432.CCR-06-1574. [DOI] [PubMed] [Google Scholar]
- Zeitlin BD, Joo E, Dong Z, Warner K, Wang G, Nikolovska-Coleska Z, et al. Antiangiogenic Effect of TW37, a Small-Molecule Inhibitor of Bcl-2. Cancer research. 1. 2006;66(17):8698–706. doi: 10.1158/0008-5472.CAN-05-3691. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ling X, Zhang Y, Lin Y, Min P, Stoudemire J. A novel pan inhibitor of Bcl-2 and Mcl-1 Apogossypolone (ApoG2) with superior stability and improved activity against human leukemia and lymphoma cells. Ascenta Therapeutics, Inc.; San Diego, CA: 2006. [Google Scholar]
- Mohammad R, Young D, Chen B, Aboukameel A, Wang G, Chen J, et al. ApoG2, a potent, non-toxic small-molecule inhibitor of Bcl-2 family: A preclinical trial in lymphoma. University of Michigan; Ann Arbor, MI: [Google Scholar]
- Arnold AA, Aboukameel O, Chen J, Yang D, Wang S, Al-Katib S, Mohammad RM. Preclinical studies of Apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in Follicular Small Cleaved Cell Lymphoma model. Mol Can. 2008;7:20. doi: 10.1186/1476-4598-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, et al. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 20. 2000;97(13):7124–9. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Das SG, Doshi JM, Peng J, Lin J, Xing C. sHA 14-1, a stable and ROS-free antagonist against anti-apoptotic Bcl-2 proteins, bypasses drug resistances and synergizes cancer therapies in human leukemia cell. Can Lett. 2008;259:198–208. doi: 10.1016/j.canlet.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Nakashima T, Uosaki Y, Hara M, Ikeda S, Kanda Y. Synthesis of tetrocarcin derivatives with specific inhibitory activity towards Bcl-2 functions. Bioorganic & medicinal chemistry letters. 9. 2001;11(7):887–90. doi: 10.1016/s0960-894x(01)00094-4. [DOI] [PubMed] [Google Scholar]
- Wan KF, Chan SF, Sukumaran SK, Lee MC, Yu VC. Chelerythrine induces apoptosis through a Bax/Bak-independent mitochondrial mechanism. 2008;283:8423–8433. doi: 10.1074/jbc.M707687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Omura-Miniamisawa M, Chao C, Nakagami Y, Ito M, Inoue T. Bcl-2 inhibitors potentiate the cytotoxic effects of radiation in Bcl-2 overexpressing radioresistant tumor cells. Int J Radiat Biol Phys. 2005;61:517–528. doi: 10.1016/j.ijrobp.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Chmura SJ, Dolan ME, Cha A, Mauceri HJ, Kufe DW, Weichselbaum RR. In vitro and in vivo activity of protein kinase C inhibitor chelerythrine chloride induces tumor cell toxicity and growth delay in vivo. Clin Cancer Res. 2000;6(2):737–42. [PubMed] [Google Scholar]
- Chan SL, Lee MC, Tan KO, Yang LK, Lee AS, Flotow H, et al. Identification of chelerythrine as an inhibitor of BclXL function. The Journal of biological chemistry. 6. 2003;278(23):20453–6. doi: 10.1074/jbc.C300138200. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Bhunia A, Wan KF, Lee MC, Chan SL, Yu VC, et al. Chelerythrine and sanguinarine dock at distinct sites on BclXL that are not the classic BH3 binding cleft. Journal of molecular biology. 1. 2006;364(3):536–49. doi: 10.1016/j.jmb.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, et al. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nature cell biology. 2001;3(2):183–91. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- Piskernik C, Haindl S, Behling T, Gerald Z, Kehrer H, Redl H, Kozlov AZ. Antimycin A and lipopolysaccharide cause the leakage of superoxide radicals from rat liver mitochondria. Biochem Biophys Acta. 2008;1782:280–285. doi: 10.1016/j.bbadis.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Chen M, Xing D, Chen T, Zhang L. Bim L involvement in Bax activation during UV irradiation-induced apoptosis. Biochemical and biophysical research communications. 29. 2007;358(2):559–65. doi: 10.1016/j.bbrc.2007.04.167. [DOI] [PubMed] [Google Scholar]
- Xing C, Wang L, Tang X, Sham YY. Development of selective inhibitors for anti-apoptotic Bcl-2 proteins from BHI-1. Bioorganic & medicinal chemistry. 1. 2007;15(5):2167–76. doi: 10.1016/j.bmc.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz S. Drug discovery: playing dirty. Nature. 13. 2005;437(7061):942–3. doi: 10.1038/437942a. [DOI] [PubMed] [Google Scholar]
- Trudel S, Stewart AK, Li Z, Shu Y, Liang SB, Trieu Y, et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res. 15. 2007;13(2 Pt 1):621–9. doi: 10.1158/1078-0432.CCR-06-1526. [DOI] [PubMed] [Google Scholar]
- Mohammad RM, Wang S, Banerjee S, Wu X, Chen J, Sarkar FH. Nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-XL, (−)-Gossypol, enhances biological effect of genistein against BxPC-3 human pancreatic cancer cell line. Pancreas. 2005;31(4):317–24. doi: 10.1097/01.mpa.0000179731.46210.01. [DOI] [PubMed] [Google Scholar]
- Mohammad RM, Wang S, Aboukameel A, Chen B, Wu X, Chen J, et al. Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-X(L) ((−)-gossypol) against diffuse large cell lymphoma. Molecular cancer therapeutics. 2005;4(1):13–21. [PubMed] [Google Scholar]
- Wang Z, Song W, Aboulameel A, Mohammad M, Wang G, Banerjee S, Kong D, Wang S, Sarkar FH, Mohammad RM. TW-37, a small molecule inhibitor of Bcl-2, inhibits cell growth and invasion in pancreatic cancer. 2008;123(4):958–66. doi: 10.1002/ijc.23610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sells SF, Wood DP, Jr., Barve SSJ, et al. Commonality of the gene programs induced by effectors of apoptosis in androgen-dependent and -independent prostate cells. Cell Growth Differ. 1997;5:457–66. [PubMed] [Google Scholar]
- Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang S, Wang G, Chen S, Sarkar F, Mohammad RM. Critical role of PAR-4 in sensitizing pancreatic caner cells to SMI induced apoptosis. Mol Can Ther. 2008 doi: 10.1158/1535-7163.MCT-08-0438. accepted article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Galan P, Roue G, Villamor N, Campo E, Colomer D. The BH3-mimetic GX15-070 synergizes with bortezomib in mantle cell lymphoma by enhancing Noxa-mediated activation of Bak. Blood. 15. 2007;109(10):4441–9. doi: 10.1182/blood-2006-07-034173. [DOI] [PubMed] [Google Scholar]
- Shoemaker AR, Mitten MJ, Oleksijew A, O'Connor JM, Wang B, Ackler S, et al. The Bcl-2 Family Inhibitor ABT-263 Shows Significant Anti-Tumor Efficacy in Models of B Cell Non-Hodgkins Lymphoma. Blood (ASH Annual Meeting Abstracts) 2007 Abstract 825. [Google Scholar]
- Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer research. 1. 2007;67(3):1176–83. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- Doshi JM, Tian D, Xing C. Ethyl-2-amino-6-bromo-4-(1-cyano-2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (HA 14-1), a Prototype Small-Molecule Antagonist against Antiapoptotic Bcl-2 Proteins, Decomposes To Generate Reactive Oxygen Species That Induce Apoptosis. Molecular pharmaceutics. 2007;4(6):919–28. doi: 10.1021/mp7000846. [DOI] [PubMed] [Google Scholar]
- Doshi JM, Tian D, Xing C. Structure-activity relationship studies of ethyl 2-amino-6-bromo-4-(1-cyano-2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (HA 14-1), an antagonist for antiapoptotic Bcl-2 proteins to overcome drug resistance in cancer. Journal of medicinal chemistry. 28. 2006;49(26):7731–9. doi: 10.1021/jm060968r. [DOI] [PubMed] [Google Scholar]
- Tian D, Das SG, Doshi JM, Peng J, Lin J, Xing C. sHA 14-1, a stable and ROS-free antagonist against anti-apoptotic Bcl-2 proteins, bypasses drug resistances and synergizes cancer therapies in human leukemia cell. Cancer letters. 8. 2008;259(2):198–208. doi: 10.1016/j.canlet.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]