

Abstract
Laser-scanning fluorescence microscopy for efficient acquisition of time-gated and spectrally resolved fluorescence images was developed based on line illumination of the laser beam and detection of the fluorescence image through a slit. In this optical arrangement, the fluorescence image was obtained by scanning only one axis perpendicular to the excitation line, and the acquisition time was significantly reduced compared with conventional laser-scanning confocal microscopy. A multidimensional fluorescence dataset consisting of fluorescence intensities as a function of x-position, y-position, fluorescence wavelength, and delay time after photoexcitation was analyzed and decomposed based on the parallel factor analysis model. The performance of the line-scanning microscopy was examined by applying it to the analysis of one of the plant defense responses, accumulation of antimicrobial compounds of phytoalexin in oat (Avena sativa), induced by the elicitor treatment.
Keywords: Fluorescence microscopy, Autofluorescence, PARAFAC, Phytoalexin
Introduction
Fluorescence microscopy is widely used to study biological phenomena in living cells and tissues. Recent progress in the development of laser-scanning microscopy and various fluorescent probes has demonstrated that a fluorescence-based method, which is basically free from background noise, is sensitive and selective [1–3]. However, fluorescence of native molecules existing in cells and tissues can be sometimes detected and becomes unwelcome background noise for fluorescence microscopy. When the wavelength of the excitation light is tuned to the near ultraviolet (UV) region, this “autofluorescence” is not negligible because many molecules show photoabsorption spectra in the near-UV region: aromatic amino acids (tryptophan, phenylalanine, tyrosine), the extracellular matrix (collagen, elastin), coenzymes relating to electron transfer systems (nicotinamide adenine dinucleotide, flavin adenine dinucleotide), and many kinds of secondary metabolites, especially in plants.
Since these molecules are, in general, physiologically important, their autofluorescence has been used to monitor the metabolic state of living cells and applied to tissue diagnostics [4–18]: Physiological alterations in tissues are detected as changes in the fluorescence properties of associated molecules, including an increase or decrease in the fluorescence intensity and a peak shift of the fluorescence spectrum. Most cells and tissues contain several autofluorescent molecules with broad and overlapping fluorescence spectra in the near-UV region. Therefore, it is essential to develop a technique for the decomposition of a mixture of unknown fluorescence components, and to trace the spectral change of a specific component. One of the straightforward and effective approaches is to acquire and analyze a multidimensional fluorescence dataset that includes, for example, fluorescence spectrum, excitation spectrum, time profile, anisotropy, and spatial localization. Two-dimensional fluorescence spectroscopy acquiring excitation and fluorescence spectra has been widely used at research and diagnostic levels because of the high selectivity and simple configuration of the measurement system [13–18]. A variety of multidimensional fluorescence microscopes have been developed based on unique optical configurations including Fourier or Hadamard transformations [19, 20], structured illumination [21], and line-scanning methods [22].
In this study, we developed a method of laser-scanning fluorescence microscopy obtaining time-gated and spectrally resolved fluorescence images, that is, fluorescence intensities as a function of x-position, y-position, fluorescence wavelength, and delay time after photoexcitation. Conventional laser-scanning confocal microscopy requires a long time to acquire a whole dataset of such multidimensional fluorescence intensities. In addition, the detector requires a relatively long exposure time because autofluorescence is generally very weak. Therefore, we developed a line-scanning technique, which is based on line illumination of the laser beam and detection of the fluorescence image through a slit instead of a pinhole [22–26]. In this optical arrangement, the fluorescence image was obtained by scanning only one axis perpendicular to the excitation line, and the acquisition time was significantly reduced compared with conventional laser-scanning confocal microscopy.
A multidimensional fluorescence dataset was analyzed and decomposed based on the parallel factor analysis (PARAFAC) model [27–29]. PARAFAC is one of several decomposition methods for a multidimensional dataset and is widely used in the field of chemometrics. A major advantage of the model is that data following this model can be uniquely decomposed into individual contributions. The fluorescence data are examples to which PARAFAC has been widely employed [30].
The performance of line-scanning microscopy, in which time-gated and spectrally resolved fluorescence images are obtained and decomposed based on the PARAFAC model, was examined by applying it to the analysis of one of the induced plant defense responses: the accumulation of antimicrobial compounds, which are generally known as phytoalexins, in oat (Avena sativa). Oat leaves produce avenanthramides such as phytoalexins when attacked by pathogens or treated with an elicitor [31–36]. Avenanthramides are substituted hydroxycinnamic acid conjugates, which demonstrate photoabsorption in the near-UV region. By using line-scanning microscopy, we have measured the autofluorescence of oat leaves and analyzed the accumulation of avenanthramides in response to the elicitor. The results demonstrate that our approach is powerful and effective for the analysis of complicated responses in living cells.
Materials and Methods
Line-scanning Microscopy
A schematic illustration of the line-scanning microscopy method is shown in Fig. 1. An amplified mode-locked Ti:sapphire laser (Coherent, RegA9000) was operated at a wavelength of 780 nm and a repetition rate of 200 kHz. The second harmonics (center wavelength of 390 nm, pulse duration of 150 fs) generated in a thin BBO crystal was used were an excitation light source. A line illumination pattern (in parallel with the y-direction in Fig. 1) was created by a cylindrical lens (f = 150 mm) and was focused on a sample with a 10X objective lens (Olympus, numerical aperture of 0.30). The excitation intensity was reduced to 10 pJ, which was measured in front of the objective lens.
Fig. 1.

A schematic illustration of line-scanning microscopy
The fluorescence image of the line illumination on the sample was relayed to the entrance slit of a polychromator (Acton, SpectraPro-150, 300 grooves per millimeter grating). The slit width was set at 70 μm, corresponding to 7 μm on the sample in this configuration. Fluorescence passing through the entrance slit was spectrally dispersed by the grating and detected by a charge-coupled device (CCD) camera (640 × 480 pixels). An image intensifier with a gate width of 200 ps (LaVision, PicoStar HR) was placed in front of the CCD to provide a time-resolved fluorescence image with a time resolution of 200 ps. A time delay between a laser pulse and a gating electronic pulse was changed by an electronic delay generator (Becker & Hickl GmbH, DEL-150). As a result, a single frame of the CCD provided information on the two-dimensional time-gated fluorescence data: The fluorescence image of the line illumination (y-direction) on the sample was vertically aligned on the two-dimensional data, while the fluorescence spectrum at each y-position in the excitation line was horizontally allocated. Hereafter, we refer to the two-dimensional time-gated data as a “y –λ map.” A y –λ map consists of 480-μm height (y) in length on the sample and 265-nm spectral bandwidth (λ). We obtained the sample images, which we call “x–y images,” with the time-gated fluorescence spectra by shifting the sample position along the x-direction (perpendicular to the excitation line) and reconstructing the dataset of y –λ maps acquired at each x-position. Furthermore, varying the gate timing of the in-/breaktensifier, we finally obtained multiple fluorescence images as a function of a wavelength (λ) and a delay time (τ). Since the scanning dimensions of a sample position can be reduced by combining line excitation and multichannel detection, this method acquires a whole dataset remarkably faster than conventional confocal microscopy. In this study, the typical exposure time of the detector was set at 200 ms. Under this condition, it takes about 10 min to acquire a dataset consisting of, for example, 640(x)× 480(y) × 640(λ) × 2 (τ).
PARAFAC Model
The three-dimensional PARAFAC model with F components can be written as
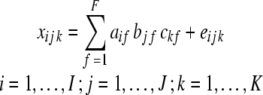 |
where xijk is the fluorescence intensity element of the three-dimensional dataset X (I ×J ×K). In our case, (a1f,...,aIf ), (b1f,...,bJf ), and (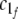 ,...,cKf ) correspond to a fluorescence spectrum, a time profile, and an x–y image of the f th component, respectively. The eijk is a residual. The PARAFAC model assumes that the fluorescence spectrum of each component is independent of the delay time and the position, whereas the relative contribution of each fluorescence component changes at the delay time and the position. This assumption is reasonable in the subnanosecond time resolution of our system.
,...,cKf ) correspond to a fluorescence spectrum, a time profile, and an x–y image of the f th component, respectively. The eijk is a residual. The PARAFAC model assumes that the fluorescence spectrum of each component is independent of the delay time and the position, whereas the relative contribution of each fluorescence component changes at the delay time and the position. This assumption is reasonable in the subnanosecond time resolution of our system.
For the PARAFAC calculations, we obtained y–λ maps by scanning the x-position (1-μm step, 640 positions in total). Two frames of different delay times (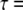 0.0 and 3.0 ns) were obtained at each x-position. As a result, a multidimensional fluorescence dataset which consisted of
0.0 and 3.0 ns) were obtained at each x-position. As a result, a multidimensional fluorescence dataset which consisted of 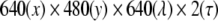 was obtained. This dataset was binned with 25-nm steps along the fluorescence wavelength dimension to reduce data size for calculations. The spatial dimensions of 640(x) × 480(y) were reshaped to a one-dimensional array (of size 307,200) in the PARAFAC calculation and then reshaped again to the spatial dimensions of 640(x) × 480(y) after the calculations were completed. Therefore, the dataset, which consisted of 10(λ) × 2 (τ) × 307,200(xy), was fitted by the PARAFAC model. In PARAFAC modeling, nonnegativity constraints were applied to all three dimensions. All the analyses were performed with the N-way toolbox for MATLAB [37], which is a set of MATLAB routines designed to perform multiway data analysis.
was obtained. This dataset was binned with 25-nm steps along the fluorescence wavelength dimension to reduce data size for calculations. The spatial dimensions of 640(x) × 480(y) were reshaped to a one-dimensional array (of size 307,200) in the PARAFAC calculation and then reshaped again to the spatial dimensions of 640(x) × 480(y) after the calculations were completed. Therefore, the dataset, which consisted of 10(λ) × 2 (τ) × 307,200(xy), was fitted by the PARAFAC model. In PARAFAC modeling, nonnegativity constraints were applied to all three dimensions. All the analyses were performed with the N-way toolbox for MATLAB [37], which is a set of MATLAB routines designed to perform multiway data analysis.
Sample Preparation
Oat seeds (A. sativa L., cv. Shokan 1) were soaked in distilled water for 24 h to facilitate germination, sown in wet vermiculite, and maintained at 20°C for 7 days under continuous artificial light exposure at a photosynthetic photon flux density of 50 μmol m − 2 s − 1 in the growth chamber.
Leaf segments were prepared from the primary leaves of 7-day-old oat seedlings. The lower epidermis was peeled away and the mesophyll cells were floated on 3 ml of the elicitor solution or distilled water in a Petri dish with the peeled surface in contact with the solution [38]. Penta-N-acetylchitopentaose solution (concentration of 1 mM) was used as an elicitor while distilled water was used as a control. There is no absorption band in the near-UV region in the penta-N-acetylchitopentaose solution. In addition, we confirmed that the solution does not emit fluorescence for the excitation light at 390 nm used in this study. All the experiments were performed in the leaf segments after incubation for 48 h at 20°C. Penta-N-acetylchitopentaose was purchased from Seikagaku Kogyo, Tokyo. Avenanthramide A (N-(4-hydroxycinnamoyl)-5-hydroxyanthranilic acid) is a gift from Dr. A. Ishihara (Kyoto University, Kyoto).
Results and Discussion
Characterization of Time-Resolved Fluorescence Spectra
A transmission image of mesophyll cells of an oat leaf treated with an elicitor is shown in Fig. 2a. Figure 2b shows a y –λ map observed at a delay time of 0.0 ns and at the position of x1 indicated by a dotted line in Fig. 2a. This kind of information on the time-gated fluorescence spectrum at each y-position can be obtained as a single frame of the CCD, demonstrating the unique and effective configuration of the line-scanning method. In Fig. 2b, in addition to the strong fluorescence component at a wavelength longer than 650 nm, weak fluorescence components can be recognized in the region of 450–650 nm: at least two components of a short-wavelength component centered at ~450 nm, and a middle-wavelength component centered at ~510 nm. As a result, three components with different fluorescence spectra were observed in the elicitor-treated cells.
Fig. 2.

a Transmission image of mesophyll cells of an oat leaf treated with an elicitor. b Time-gated y–λ map observed at the x1 position indicated by a dotted line in a. Delay time τ is 0.0 ns. c The same as b but τ is 3.0 ns. d–f The same as a–c, respectively, but the oat leaf was treated with distilled water as a control. All the time-gated y–λ maps are normalized by the fluorescence intensity of the short-wavelength components centered around 450 nm to focus on the weak fluorescence in 450–650 nm. Therefore, the intensity at a wavelength region longer than 650 nm is above the scale and is shown as a white area
Figure 2c shows a y –λ map observed at the same position of x1 but at a delay time of 3.0 ns, where fluorescence intensity was normalized by the short-wavelength component. The fluorescence pattern of the middle-wavelength component disappears and the fluorescence intensity of the long-wavelength component decreases, indicating that the fluorescence lifetimes of these components are shorter than that of the short-wavelength component.
Figure 2d is a transmission image of mesophyll cells of an oat leaf treated with distilled water as a control. In contrast to the three components observed in the elicitor-treated cells, only two components with different fluorescence spectra can be recognized in a y –λ map at a delay time of 0.0 ns and at the position of x1 (Fig. 2e): a short-wavelength component centered at ~450 nm and a long-wavelength component centered at a wavelength longer than 650 nm. In a y –λ map at a delay time of 3.0 ns (Fig. 2f), where the fluorescence intensity was normalized by the short-wavelength component, the intensity of the long-wavelength component decreases. This indicates that the fluorescence lifetime of the long-wavelength component, is shorter than that of the short-wavelength component as is the case with the elicitor-treated cells.
To study fluorescence properties of these components in more detail, time-integrated fluorescence spectra and time profiles of the fluorescence intensities, which were obtained by varying the delay time τ with a step of 0.02 ns and then averaging in each area of P1–P5 (see Figs. 2b, e), were plotted in Fig. 3a, b, respectively. Here, the fluorescence properties of the long-wavelength component P3 were in agreement with those of the component P5. Therefore, the fluorescence spectrum and the time profile of P3 were plotted as the only long-wavelength component in Fig. 3.
Fig. 3.

a Time-integrated fluorescence spectra and b time profiles of the fluorescence intensities. The lines are results experimentally observed and the circles are the components extracted by PARAFAC. The fluorescence spectra and the time profiles were drawn with appropriate shifts in a vertical direction for comparison
The sharp structure that appears in the fluorescence spectra around 455 nm is due to Raman scattering of water. Fluorescence properties of P1 and P4, which correspond to the short-wavelength components observed in the elicitor-treated and water-treated cells, respectively, are similar to each other. The middle-wavelength component (P2), which was observed only in the elicitor-treated cells, had a broad fluorescence spectrum. The fluorescence spectra of the long-wavelength components (P3 and P5) observed in the elicitor-treated and water-treated cells, respectively, have a narrow width and a peak of 677 nm.
The time profiles of these components shown in Fig. 3b can be fitted by two or three exponential decays convoluted with an instrumental response function. The parameters obtained by the fitting are listed in Table 1. The time profiles of P1 and P4 have the same time constants of <0.2, 1.2, and 3.0 ns but different ratios of the amplitudes. The time profile of P2 shows faster decay than those of P1 and P4. In the time profiles of P3 and P5, the long decay component of a few nanoseconds was not observed.
Table 1.
Decay times and their relative amplitudes obtained by fitting to the fluorescence time profiles in Fig. 3b
| Samples | Time constants, ns (amplitudes in percent) | ||
|---|---|---|---|
| P1 | <0.2 (45.5) | 1.2 (49.5) | 3.0 (5.0) |
| P2 | <0.2 (74.6) | 0.6 (23.3) | 2.0 (2.1) |
| P3 (P5) | <0.2 (56.5) | 0.6 (43.5) | |
| P4 | <0.2 (35.8) | 1.2 (52.6) | 3.0 (11.6) |
| Avenanthramide A | <0.2 (94.3) | 0.5 (5.4) | 3.5 (0.3) |
Spatial Localization of Fluorescence Components
To study the spatial localization of these florescence components, x–y images at different wavelengths are shown in Fig. 4. Here, x–y images of the elicitor-treated cells were constructed with the fluorescence at τ = 0.0 ns and averaged in a 25-nm bin size centered at λ = 442, 567, and 667 nm (Fig. 4a–c, respectively). Fluorescence at these selected wavelengths is mainly derived from the short-, middle-, and long-wavelength components, respectively. It is noticed that the minor contributions from other components vanish because of spectral overlap among different components.
Fig. 4.

a–c The x–y images of the elicitor-treated cells constructed with the fluorescence at τ = 0.0 ns and averaged in a 25-nm bin size centered at λ = 442, 567, and 667 nm, respectively. d–e The x–y images of the water-treated cells constructed with the fluorescence at τ = 0.0 ns and averaged in a 25-nm bin size centered at λ = 442 and 667 nm, respectively. f–h The x–y images of components I, II, and III, respectively, extracted by PARAFAC for the elicitor-treated cells. i–j The same as f and g, respectively, but PARAFAC was performed for a dataset at a wavelength range of up to 605 nm (see the text). The images in each row were scaled so that the highest intensity in the long-wavelength components (c, e, h) is 100
The short- and long-wavelength components have a similar spatial localization at this spatial resolution. Namely, the fluorescence of these components was observed in the same cell. On the other hand, the x–y image of the middle-wavelength component shows a complementary pattern to the other components. That is, the fluorescence intensity of the middle-wavelength component is high in the cell, whereas the fluorescence of the long-wavelength component is weak.
In the water-treated cells (control), two components with different fluorescence properties were recognized. Therefore, we show the x–y images constructed with fluorescence intensity centered at 442 and 667 nm (averaged in a 25-nm bin size), which correspond to the short- and long-wavelength components, respectively (Fig. 4d, e). The spatial patterns of these images are similar to each other. This is the same result as observed in the elicitor-treated cells.
PARAFAC Decomposition
To extract the major fluorescence components from the multidimensional dataset obtained in the elicitor-treated cells, we performed PARAFAC with three components. The size of the dataset used for the calculation was 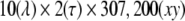 as described in Section 2.2. We named the three components extracted by PARAFAC as components I, II, and III, which corresponded to the short-, middle-, and long-wavelength components, respectively. The fluorescence spectra of these components are plotted in Fig. 3a. Component I is peak-shifted to a shorter wavelength than the short-wavelength component (P1) while component II is peak-shifted to a higher wavelength than the middle-wavelength component (P2). It is considered that the overlapping region between the two components was successfully separated by PARAFAC. Component III agrees well with the long-wavelength component (P3). The relative intensities of components I, II, and III at 0.0 and 3.0 ns are plotted in Fig. 3b. These were obtained in agreement with the time profiles of the short-, middle-, and long-wavelength components, respectively.
as described in Section 2.2. We named the three components extracted by PARAFAC as components I, II, and III, which corresponded to the short-, middle-, and long-wavelength components, respectively. The fluorescence spectra of these components are plotted in Fig. 3a. Component I is peak-shifted to a shorter wavelength than the short-wavelength component (P1) while component II is peak-shifted to a higher wavelength than the middle-wavelength component (P2). It is considered that the overlapping region between the two components was successfully separated by PARAFAC. Component III agrees well with the long-wavelength component (P3). The relative intensities of components I, II, and III at 0.0 and 3.0 ns are plotted in Fig. 3b. These were obtained in agreement with the time profiles of the short-, middle-, and long-wavelength components, respectively.
Figure 4f–h shows the spatial localization of components I, II, and III extracted by PARAFAC, respectively. Components I and III agree well with those of the short- and long-wavelength components, respectively. On the other hand, the spatial localization of component II is slightly different from that of the middle-wavelength component. The complementary relationship between components II and III is not clear compared with that between the middle- and long-wavelength components. In the fluorescence spectrum of component II (see Fig. 3a), a rise toward a longer wavelength exists at around 650 nm. This contribution is probably due to the cross talk of the long-wavelength component, which comes from very different fluorescence intensities between the middle- and long-wavelength components. It is suggested that the cross talk of the long-wavelength component results in different spatial patterns between the middle-wavelength component and component II.
To examine the spatial localizations of the short- and middle-wavelength components without the effect of cross talk, we performed PARAFAC with two components for a dataset at a wavelength range of up to 605 nm. We named the two components extracted by PARAFAC as components I’ and II’, respectively. The fluorescence spectra of components I’ and II’ are in agreement with those of components I and II, respectively, in a wavelength region lower than 605 nm. Further, the time profiles of components I’ and II’ correspond well to those of components I and II, respectively (data not shown). The spatial localization of components I’ and II’ are shown in Fig. 4i, j, respectively. The former component’s localization is the same as the spatial localization of component I (Fig. 4f) and was similar to that of the short-wavelength component (Fig. 4a). On the other hand, the latter component’s localization was different from the spatial localization of component II (Fig. 4g) but very similar to that of the middle-wavelength component (Fig. 4b). The results indicate that the different spatial pattern of component II compared with the others (the middle-wavelength component and component II’) is due to the cross talk of the long-wavelength component. Further, it is shown that cross talk can be partially avoided by appropriately limiting the data used for PARAFAC.
Possible Assignments of Fluorescence Components
We observed the short-, middle-, and long-wavelength components in the elicitor-treated cells and the short- and long-wavelength components in the water-treated cells. First, we focused our attention on the long-wavelength components, which were observed in both samples and found to have the same fluorescence properties. From the fluorescence properties, this component is naturally assigned to chlorophyll.
Second, we moved on to the middle-wavelength component, which was only observed in the elicitor-treated cells. This suggests the possibility that the component is associated with avenanthramides. It was reported that avenanthramide A is a major component of induced avenanthramides and reaches a maximum at 36–48 h after treatment with the elicitor [39]. We measured time-resolved fluorescence spectra of avenanthramide A in an aqueous solution (pH 7.0) in vitro. The time-integrated fluorescence spectrum is broad and centered at 510 nm (Fig. 3a). The spectral shape is, in part, similar to that of P2 and very similar to that of component II. The comparison of the time-integrated fluorescence spectra supports the suggestion that the middle-wavelength component (or component II) originates from avenanthramide A. However, the time profiles of the fluorescence intensities are very different from each other (Fig. 3b and Table 1). Both time profiles have faster decay components than the time resolution (0.2 ns). The fast decay component of avenanthramide A is much faster than that of component II. This presumably indicates the different environments of avenanthramide A in vivo and in vitro because the fluorescence decay (or fluorescence quantum yield) is generally very sensitive to the surrounding environment of the molecules. Viscosity is known to be one of the parameters which affects fluorescence quantum yield. The relationship between fluorescence quantum yield and solvent viscosity has been studied for various dye molecules with a flexible structure, such as diphenyl-methane and polymethine-dyes, and discussed in terms of conformational changes induced by internal rotation [40, 41]. We measured the viscosity dependence of fluorescence for avenanthramide A in an aqueous solution of saccharides and observed an increase of fluorescence quantum efficiency with increasing solvent viscosity. Although this observation is preliminary, it seems that the discrepancy between the fluorescence decay profiles of the middle-wavelength component (or component II) and avenanthramide A in aqueous solution is due to the different environments of avenanthramide A in vivo and in vitro. To identify the middle-wavelength component (or component II) observed in this study more reliably, a combination of other analytical methods is essential and now in progress.
Finally, we discuss the short-wavelength components observed both in the elicitor- and in water-treated samples. These two components are similar in their fluorescence properties, implying that they have a common origin. Although a reasonable assignment for this component is difficult at the present stage, it is considered that nicotinic coenzyme, NAD(P)H, is a possible candidate for the short-wavelength component because the fluorescence spectra are similar to each other [15–19, 22]. It is noticed that the relative intensity of the short-wavelength component to that of the long-wavelength component is higher in the elicitor-treated cells than that in water-treated cells (cf. Fig. 4a, d). This indicates the increase of the short-wavelength component and/or the appearance of an additional fluorescence component induced by the elicitor treatment. Addition of further fluorescence parameters such as an excitation wavelength and anisotropy would help to identify the short-wavelength component and also to separate such an additional fluorescence component induced by the elicitor treatment.
Conclusions
For a study on spatiotemporal dynamics of autofluorescence molecules in living cells, we developed a method of fluorescence microscopy based on efficient acquisition and analysis of a multidimensional fluorescence dataset. The line-scanning method demonstrated the rapid acquisition of time-gated and spectrally resolved fluorescence images. A multidimensional dataset obtained was analyzed and decomposed by the PARAFAC modeling without any prior knowledge about spectroscopic properties of autofluorescent molecules in the cells.
We applied this method to the analysis of a plant defense response, accumulation of phytoalexin in oat leaves, induced by the elicitor treatment. In addition to the strong fluorescence from chlorophyll molecules, weak fluorescence components, one of which possibly originated from avenanthramide A as a phytoalexin was observed in oat leaves treated with an elicitor.
In this article, we presented the application of this method for the detection and analysis of autofluorescent molecules in living cells. In addition to autofluorescent molecules, fluorescence indicators for Ca2 + , pH, and so on [2, 3] may be unique targets for this method. In general, the fluorescence quantum efficiency of autofluorescence is much lower than that of the fluorescence of the indicators. Therefore, a technique for separating unknown fluorescence components with very different quantum efficiencies is essential. Simultaneous analysis of the spatiotemporal dynamics of autofluorescent molecules and fluorescence indicators would be a powerful approach for revealing complicated responses in living cells.
Acknowledgements
We thank Dr. A. Ishihara (Kyoto University, Kyoto) for helpful discussion and for providing avenanthramides. This work was supported by a grant from CREST of the Japan Science and Technology Agency and by the Grant-in-Aid for Scientific Research in Priority Area (432) “Molecular Nano Dynamics” from the Ministry of Education, Culture, Sports, Science, and Technology (No. 17034033).
References
- 1.Evanko, D. (ed.): Focus on fluorescence imaging—principles and practice of microscope techniques. Nat. Methods 2, 901–950 (2005) [DOI]
- 2.Invitrogen: The handbook—a guide to fluorescent probes and labeling technologies. Available via DIALOG. http://probes.invitrogen.com/handbook/ (2008)
- 3.Chudakov, D.M., Lukyanov, S., Lukyanov, K.A.: Fluorescent proteins as a toolkit for in vivo imaging. Trends Biotechnol. 23, 605–613 (2005) [DOI] [PubMed]
- 4.Richards-Kortum, R., Sevick-Muraca, E.: Quantitative optical spectroscopy for tissue diagnosis. Annu. Rev. Phys. Chem. 47, 555–606 (1996) [DOI] [PubMed]
- 5.Andersson-Engels, S., af Klinteberg, C., Svanberg, K., Svanberg, S.: In vivo fluorescence imaging for tissue diagnostics. Phys. Med. Biol. 42, 815–824 (1997) [DOI] [PubMed]
- 6.Bennett, M., Gallagher, M., Fagg, J., Beswick, C., Paul, T., Beale, M., Mansfield, J.: The hypersensitive reaction, membrane damage and accumulation of autofluorescence phenolics in lettuce cells challenged by Bremia lactucae. Plant J. 9, 851–865 (1996) [DOI]
- 7.Andersson, H., Baechi, T., Hoechl, M., Richter, C.: Autofluorescence of living cells. J. Microsc. 191, 1–7 (1998) [DOI] [PubMed]
- 8.Betz, C.S., Mehlmann, M., Rick, K., Stepp, H., Grevers, G., Baumgartner, R., Leunig, A.: Autofluorescence imaging and spectroscopy of normal and malignant mucosa in patients with head and neck cancer. Lasers Surg. Med. 25, 323–334 (1999) [DOI] [PubMed]
- 9.Inaguma, M., Hashimoto, K.: Porphyrin-like fluorescence in oral cancer. Cancer 86, 2201–2211 (1999) [DOI] [PubMed]
- 10.Rigacci, L., Alterini, R., Bernabei, P.A., Ferrini, P.R., Agati, G., Fusi, F., Monici, M.: Multispectral imaging autofluorescence microscopy for the analysis of lymph-node tissues. Photochem. Photobiol. 71, 737–742 (2000) [DOI] [PubMed]
- 11.Huang, S., Heikal, A.A., Webb, W.W.: Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 82, 2811–2825 (2002) [DOI] [PMC free article] [PubMed]
- 12.Ashjian, P., Elbarbary, A., Zuk, P., DeUgrate, D.A., Benhaim, P., Marcu, L., Hedrick, M.H.: Noninvasive in situ evaluation of osteogenic differentiation by time-resolved laser-induced fluorescence spectroscopy. Tissue Eng. 10, 411–420 (2004) [DOI] [PMC free article] [PubMed]
- 13.Zangaro, R.A., Silveira, L., Manoharan, R., Zonios, G., Itzkan, I., Dasari, R.R., Van Dam, J., Feld, M.S.: Rapid multiexcitation fluorescence spectroscopy system for in vivo tissue diagnosis. Appl. Opt. 35, 5211–5219 (1996) [DOI] [PubMed]
- 14.Zuluaga, A.F., Utzinger, U., Durkin, A., Fuchs, H., Gillenwater, A., Jacob, R., Kemp, B., Fan, J., Richards-Kortum, R.: Fluorescence excitation emission matrices of human tissue: a system for in vivo measurement and method of data analysis. Appl. Spectrosc. 53, 302–311 (1999) [DOI]
- 15.Coghlan, L., Utzinger, U., Drezek, R., Heintzelman, D., Zuluaga, A., Brookner, C., Richards-Kortum, R., Gimenez-Conti, I., Follen, M.: Optimal fluorescence excitation wavelengths for detection of squamous intra-epithelial neoplasia: results from an animal model. Opt. Express 7, 436–446 (2000) [DOI] [PubMed]
- 16.Zellweger, M., Grosjean, P., Goujon, D., Monnier, P., van den Bergh, H., Wagnieres, G.: In vivo autofluorescence spectroscopy of human bronchial tissue to optimize the detection and imaging of early cancers. J. Biomed. Opt. 6, 41–51 (2001) [DOI] [PubMed]
- 17.Brancaleon, L., Durkin, A.J., Tu, J.H., Menaker, G., Fallon, J.D., Kollias, N.: In vivo fluorescence spectroscopy of nonmelanoma skin cancer. Photochem. Photobiol. 73, 178–183 (2001) [DOI] [PubMed]
- 18.Shirakawa, H., Miyazaki, S.: Blind spectral decomposition of single-cell fluorescence by parallel factor analysis. Biophys. J. 86, 1739–1752 (2004) [DOI] [PMC free article] [PubMed]
- 19.Malik, Z., Cabib, D., Buckwald, R.A., Talmi, A., Garini, Y., Lipson, S.G.: Fourier transform multipixel spectroscopy for quantitative cytology. J. Microsc. 182, 133–140 (1996) [DOI]
- 20.Hanley, Q.S., Arndt-Jovin, D.J., Jovin, T.M.: Spectrally resolved fluorescence lifetime imaging microscopy. Appl. Spectrosc. 56, 155–166 (2002) [DOI]
- 21.Siegel, J., Elson, D.S., Webb, S.E.D., Parsons-Karavassilis, D., Leveque-Font, S., Cole, M.J., Lever, M.J., French, P.M.W., Neil, M.A.A., Juskaitis, R., Sucharov, L.O., Wilson, T.: Whole-field five-dimensional fluorescence microscopy combining lifetime and spectral resolution with optical sectioning. Opt. Lett. 26, 1338–1340 (2001) [DOI] [PubMed]
- 22.Haralampus-Grynaviski, N.M., Stimson, M.J., Simon, J.D.: Design and application of rapid-scan spectrally resolved fluorescence microscopy. Appl. Spectrosc. 54, 1727–1733 (2000) [DOI]
- 23.Veirs, D.K., Ager, J.W., Loucks, E.T., Rosenblatt, G.M.: Mapping materials properties with Raman spectroscopy utilizing a 2-D detector. Appl. Opt. 29, 4969–4980 (1990) [DOI] [PubMed]
- 24.Brakenhoff, G.J., Visscher, K.: Confocal imaging with bilateral scanning and array detectors. J. Microsc. 165, 139–146 (1991)
- 25.Brakenhoff, G.J., Squier, J., Norris, T., Bliton, A.C., Wade, M.H., Athey, B.: Real-time two-photon confocal microscopy using a femtosecond, amplified Ti:sapphire system. J. Microsc. 181, 253–259 (1996) [DOI] [PubMed]
- 26.Stimson, M.J., Haralampus-Grynaviski, N., Simon, J.D.: A unique optical arrangement for obtaining spectrally resolved confocal images. Rev. Sci. Instrum. 70, 3351–3354 (1999) [DOI]
- 27.Harshman, R.A., Lundy, M.E.: PARAFAC: parallel factor analysis. Comput. Stat. Data Anal. 18, 39–72 (1994) [DOI]
- 28.Bro, R.: PARAFAC. Tutorial and applications. Chemom. Intell. Lab. Syst. 38, 149–171 (1997) [DOI]
- 29.Bro, R.: Review on multiway analysis in chemistry—2000–2005. Crit. Rev. Anal. Chem. 36, 279–293 (2006) [DOI]
- 30.Anderson, C.M., Bro, R.: Practical aspects of PARAFAC modeling of fluorescence excitation–emission data. J. Chemom. 17, 200–215 (2003) [DOI]
- 31.Mayama, S., Tani, T., Matsuura, Y., Ueno, T., Fukami, H.: The production of phytoalexins by oat in response to crown rust, Puccinia-coronata f. sp. avenae. Physiol. Plant Pathol. 19, 217 (1981)
- 32.Mayama, S., Matsuura, Y., Iida, H., Tani, T.: The role of avenalumin in the resistance of oat to crown rust, Puccinia-coronata f. sp. avenae. Physiol. Plant Pathol. 20, 189–199 (1982) [DOI]
- 33.Collins, F.W.: Oat phenolics: avenanthramides, novel substituted N-cinnamoylanthranilate alkaloids from oat groats and hulls. J. Agric. Food Chem. 37, 60–66 (1989) [DOI]
- 34.Bordin, A.P.A., Mayama, S., Tani, T.: Potential elicitors for avenalumin accumulation in oat leaves. Ann. Phytopathol. Soc. Jpn. 57, 688–695 (1991)
- 35.Miyagawa, H., Ishihara, A., Nishimoto, T., Ueno, T., Mayama, S.: Induction of avenanthramides in oat leaves inoculated with crown rust fungus, Puccinia-coronata f. sp. avenae. Biosci. Biotechnol. Biochem. 59, 2305–2306 (1995)
- 36.Miyagawa, H., Ishihara, A., Kuwahara, Y., Ueno, T., Mayama, S.: Comparative studies of elicitors that induce phytoalexin in oats. J. Pestic. Sci. 21, 203–207 (1996)
- 37.Andersson, C.A., Bro, R.: The N-way toolbox for MATLAB. Chemom. Intell. Lab. Syst. 52, 1–4 (2000) [DOI]
- 38.Ishihara, A., Miyagawa, H., Matsukawa, T., Ueno, T., Mayama, S., Iwamura, H.: Induction of hydroxyanthranilate hydroxycinnamoyl transferase activity by oligo-N-acetylchitooligosaccharides in oats. Phytochemistry 47, 969–974 (1998) [DOI]
- 39.Ishihara, A., Ohtsu, Y., Iwamura, H.: Induction of biosynthetic enzymes for avenanthramides in elicitor-treated oat leaves. Planta 208, 512–518 (1999) [DOI]
- 40.Oster, G., Nishijima, Y.: Fluorescence and internal rotation: their dependence on viscosity of the medium. J. Am. Chem. Soc. 78, 1581–1584 (1956) [DOI]
- 41.Sharafy, S., Muszkat, K.A.: Viscosity dependence of fluorescence quantum yields. J. Am. Chem. Soc. 93, 4119–4125 (1971) [DOI]