

Abstract
The role of hippocampal atrophy in the pathogenesis of major depression remains under investigation. Here, we show, within a neural network model, that the incorporation of atrophy reproduces the changes observed in cognitive impairment in depression and could also contribute to the maintenance of the depressed mood. Some other clinical observations, such as treatment resistance and frequent relapses of illness, could also be explained within the framework of the model. We also simulate the action of cognitive therapy and a combined treatment of cognitive therapy and antidepressant drugs. Our findings suggest that, in the presence of hippocampal atrophy, the incorporation of antidepressant drugs would be necessary for the reversal of symptomatology.
Keywords: Depression, Hippocampal atrophy, Cognitive impairment, Neural networks
Introduction
Depression in adulthood is a frequent disease that spans a wide range of severity, often incapacitating and occasionally lethal [1, 2]. Hippocampal atrophy (HA) has been constantly reported in depression, with levels of atrophy ranging from 8% to 19% [3–5]. However, the clinical correlates of this change and its connection with the duration of illness or treatment resistance remain under discussion [3]. Structurally, HA may arise due to neuronal loss, glial cell loss, inhibition of neurogenesis, dendritic atrophy, or reduction in neurotrophic factors [6–9]. Although the possible role of HA has not yet been elucidated, it is well known that antidepressant drugs may lead to neuronal sprouting and enhance neurogenesis in the hippocampus [6, 7]. Hence, it is thought that the antidepressant effect may be in part due to a capacity to oppose and/or reverse HA. Possibly linked to HA, depressive disorder is often accompanied by a substantial impairment of cognitive function, particularly mnemonic deficits and executive dysfunction [10].
Connectionist neural network models are useful tools for exploring the mechanisms underlying pathology because they link clinical presentation with underlying neural activity. To study the role of HA in depression, we need a model that can reproduce clinical aspects of depression and that includes a module representing the hippocampus. A model of these characteristics was developed by Siegle [11] to study certain features of depression. Siegle’s model is based on Le Doux’s [12] notion that emotional and nonemotional (conceptual or semantic) aspects of information are processed in parallel by brain systems, roughly represented by the amygdala system and the hippocampal system, respectively, with feedback occurring between these two systems.
The core symptom of depression that Siegle focused on is negativity. It is well known that depressed people pay excessive attention to negative information, preferentially remember negative events of their lives, and tend to interpret neutral or positive information as negative (see, for example, [5] and [10]). Based on Le Doux’s notion of parallel processing, Siegle designed an experiment to study what specific aspects of negative information depressive patients do principally attend to and to determine the stage of the process at which these attentional biases occur. This experiment is based on the measure of react- ion times of normal, dysphoric, and depressed people on two cognitive tasks that allow us to separately examine attention paid to the emotional and nonemotional aspects of infor- mation: a lexical (or semantic) decision task (in which participants are asked to identify whether a string of letters spells a word, which may be positive, negative, or neutral) that directs people’s attention to the nonemotional aspects of information, and a valence identification task (in which individuals are asked to name whether a word is positive, negative, or neutral) that directs participants’ attention to the emotional aspects of information.
Siegle’s neural network model was designed to simulate the processing of emotional information according to Le Doux’s concepts and to predict reaction times for the lexical and the valence tasks. When the network predictions are compared with the human data, it is observed that the model successfully reproduces the behavioral responses of normal and depressed individuals on reaction times for the two described cognitive tasks. The experience of a normal person was simulated in Siegle’s model by training the network on an equal number of positive, negative, and neutral stimuli. Depression was induced in the model by overtraining the network with a single memory trace representing a relevant negative experience. Consistently with human behavior, the network trained to simulate the experience of a depressed person was slower with positive than negative words on the valence task and slower with negative than positive words on the lexical task [11]. These tendencies are named within this framework as cognitive-processing biases.
The only assumptions of Siegle’s model are parallel processing of incoming information by the amygdala and hippocampal systems and the existence of feedback between these structures. The neurobiological foundations of these minimal assumptions appear to be consistent with the experimental evidence (see reviews and discussions in [13–15]). With these simple assumptions, the model reproduces the behavior of human beings in cognitive experiments.
In this article, we investigated the effect of HA on depression using this neural network model developed by Siegle. The architecture of the network is shown in Fig. 1. We validated our version of the model by reproducing the phenomenology of reaction times for the two described cognitive tasks (Fig. 2a and b). A detailed description of the model and the methods used throughout the article is found in the Appendix.
Fig. 1.

Architecture of the neural network model. a Modules of the network with their population of neurons. An input module (32 neurons) representing perceptual characteristics of a stimulus sends information to a semantic module (32 neurons) and to an affective module (16 neurons), which represent the semantic and the affective content of the stimulus, respectively. There are feedback connections between the semantic and the affective modules. Semantic and affective nodes send information to output semantic (32 neurons) and output affective (16 neurons) nodes, respectively. The names of the weight matrices are shown between brackets. b Connectivity between the nodes of the network. For visual help, only the connections of some representative neurons in each module are shown
Fig. 2.

Reaction times for positive, negative, neutral, and overtrained stimuli predicted by the normal and depressed network for the lexical (a) and valence tasks (b). The model reproduces temporal delays of reaction times consistent with human behavior in experiments reported by Siegle [11]. Points in the graphs represent averages over 1,000 random realizations. We follow the same graphical presentation used by Siegle to facilitate comparison. In particular, it is useful to remark that lines in the graphs are only for visual help
We studied the role of HA in depression by simulating it in the model and evaluating its effect on time delays on the semantic and valence tasks (Section 2). We also implemented in the depressed network (with and without HA) the effect of a cognitive therapy alone (Section 3) or combined with antidepressant drugs (Section 4). This allowed us to test the influence of HA over the effectiveness of these therapies to reverse depressive symptoms in the network. In Section 5, as a discussion of previous results, we show that HA in our model can explain additional clinical observations that usually appear in the evolution of illness. Finally, we state some concluding remarks in Section 6.
Incorporation of HA in the Model
We simulated the HA by randomly destroying synapses (from 5% to 20%) in memories attributed to the hippocampus. One of the notorious advantages of mesoscopic models that focus on the flux of information processing is precisely their underdetermination by the facts. That is, different possible structural alterations underlying HA (neuronal loss, inhibition of neurogenesis, dendritic atrophy, or reduction in neurotrophic factors) are all represented in the model by the same diffuse deletion of synapses (deleting neurons instead of synapses is known to be computationally equivalent in associative memory models [16] but was also tested in the model with equivalent results [17]). So, in contrast to detailed models, in which minimal changes can dramatically change their behavior, mesoscopic level neural network models are robust to changes in our knowledge of the true biological underlying substrate.
If a model reproduces the behavior of human experiments based on the interaction between a module attributable to the hippocampus and another module processing affective aspects of information, changes that model atrophy in the hippocampal module are expected to mimic the global effect on the information processing interacting system. In particular, if the behavior observed in the model reproduces or is in agreement with other clinical facts not specifically searched for, this assumption will be reinforced.
If HA is implemented on the normal network, reaction times on the lexical task increase uniformly for all types of stimuli as the level of atrophy grows (Fig. 3a), whereas, on the valence task, reaction times stay at levels similar to those of the normal network (Fig. 3b).
Fig. 3.

Simulation of HA in the model. Atrophy was implemented on the normal (a, b) and the depressed network (c, d). Four levels of atrophy (5%, 10%, 15%, and 20%) were simulated. For each curve, a new independent set of 1,000 realizations was randomly chosen
Adding HA to the depressed network, that is, once the net was overtrained with a relevant negative stimulus, reaction times increase for all types of stimulus on the lexical task as the level of atrophy grows. The biases in reaction times established by the previous overtraining (see Section 1) are maintained in the depressed network with HA (Fig. 3c). On the valence task, reaction times increase for positive stimuli because, in several cases, the network falls into confusion, identifying these stimuli as negative. When such errors happen, reaction times increase markedly (Fig. 3d).
Assuming that an increase in reaction times on the semantic task can be taken as a measure of cognitive impairment, our results suggest an active role for HA in the cognitive deficits observed in depression. This is consistent with imaging studies that associate HA with memory loss and executive dysfunction in depression [18, 19]. Furthermore, memory loss has been seen to correlate with the number of depressive episodes [20], which also correlates with increased HA [21].
If it is assumed that the negative bias on the valence task correlates with depressed mood, the effect of atrophy in the behavior of the depressed network is also in agreement with the suggestion that hippocampal damage not only affects cognitive functioning but also plays a role in the induction and maintenance of the depressed mood itself [22]. In the model, this is a consequence of the feedback between the semantic and affective modules.
It must be noted that the results remain unaltered if one reverses the order of application of atrophy and overtraining in the network (data not shown). Therefore, the model cannot help to clarify whether atrophy precedes or is a consequence of depression.
The Effect of Cognitive Therapies on the Depressed Network
Cognitive models of depression imply the possibility of cognitive – psychological – therapies by reinstructing a normal cognition. We simulated the effect of cognitive therapies in the network by progressively reducing the negative overtraining and reinstructing the memories with normal experience (see Appendix).
In our model, the simulation of cognitive therapy reverses the increment in response times on the lexical task in the depressed network without atrophy (Fig. 4a), but not in those cases in which HA has also been implemented (Fig. 5a). Whereas biases established by the overtraining disappear in both cases, in the depressed network with atrophy, reaction times in the lexical task do not return to normal levels, remaining at levels similar to those produced by the HA on the normal network shown in Fig. 3a (this behavior does not change if more relearning is added). These results suggest that, in depression accompanied with HA, cognitive therapy alone will not completely reverse cognitive impairment.
Fig. 4.

Simulation of cognitive therapy on the depressed network. The relearning curves represent the progress of the therapy. a Lexical task. The arrow indicates the direction of change in reaction times as relearning progresses. b Valence task. The arrow indicates the direction of change in reaction times for positive and neutral stimuli as relearning progresses. Reaction times for negative stimuli move in the opposite direction with the relearning
Fig. 5.

Simulation of cognitive therapy on the depressed network with atrophy. The relearning curves represent the progress of the therapy. Atrophy was applied at a level of 10%. a Lexical task. The arrow indicates the direction of change in reaction times for neutral and nonovertrained negative stimuli as relearning progresses. Reaction times for the overtrained negative stimulus move in the opposite direction with relearning. As relearning progresses, reaction times approach values similar to those obtained when a 10% level of atrophy is applied to the normal network. b Valence task. The arrow indicates the direction of change in reaction times as relearning progresses, for positive and neutral stimuli. Reaction times for negative stimuli move in the opposite direction with the relearning. As relearning progresses, reaction times approach normal levels
On the valence task, as relearning occurs, reaction times progressively return to normal levels, both for the depressed network (Fig. 4b) and for the depressed network with atrophy (Fig. 5b). Assuming that a return to normal reaction times on the valence task can be seen as the recovery of mood, our results suggest that a residual cognitive impairment (an elevation of reaction times on the lexical task) could persist in an euthymic patient (a patient with normal mood) if HA is present. This is in agreement with studies that report the persistence of cognitive impairment after clinical remission of the illness [10].
The Effect of Antidepressants and Combined Therapy on the Model
Antidepressant drugs are believed to stimulate neural growth factors and to reverse HA [6, 7, 23–25] since antidepressants
-
Increase the expression of brain-derived neurotrophic factor
Increase synaptic density in the CA1 and CA3 regions of the hippocampus
Prevent induction of HA by stressors
Stimulate neurogenesis in the hippocampus
To investigate whether the reversion of HA is in fact a necessary condition for recovery, we mimicked in our model the postulated effect of antidepressants on the hippocampus.
We simulated the effect of an antidepressant treatment provoking a reversion of the atrophy in our model by the reconnection of synaptic elements that were previously altered by atrophy (see Appendix). We tested that the effect of antidepressants did not distort the behavior of time delays in the model (data not shown). Reinstruction, simulating cognitive therapy applied after antidepressant treatment on the network with overtraining and atrophy, was found to revert depressive manifestations, normalizing reaction times on both lexical and valence tasks (Fig. 6). Obviously, with a combined treatment that only partially reverses HA, a residual level of cognitive impairment is expected to persist even in euthymia.
Fig. 6.

Simulation of a combined treatment of psychotherapy and antidepressant drugs on the depressed network with atrophy. The relearning curves represent the progress of the therapy. Atrophy was applied at a level of 10%. a Lexical task. The arrow indicates the direction of change in reaction times as relearning progresses. b Valence task. The arrow indicates the direction of change in reaction times for positive and neutral stimuli as relearning progresses. Reaction times for negative stimuli move in the opposite direction with the relearning. In both tasks, as relearning progresses, reaction times approach normal levels
Consequently, our results suggest that, in those cases where HA is suspected to be present, its reversion emerges as a condition for the effectiveness of cognitive therapies in the treatment of depression. In such cases, combined treatment (antidepressant drugs plus psychotherapy) would be more effective than psychotherapy alone in reversing the manifestations and impairments of depressive patients.
In a study using a three-layer feedforward perceptron model, Chambers et al. [26] simulated the effect of neuronal turnover (neuron loss and reposition in the hidden layer maintaining a constant number of neurons) to explore the cognitive significance of a possible functional association of apoptotic and neurogenic events in the dentate gyrus of the hippocampus. Their results show that elimination and replacement of neurons enhances the speed of learning new information but reduces accuracy in the recall of old information. This proposed mechanism of facilitating relearning and forgetting old information could be favoring our combined effect of restoring synapses by antidepressants and inducing relearning by cognitive therapies.
Additional Explanations of Clinical Observations
Taking into account the results for both the semantic and valence tasks, one could consider them as a picture of the clinical situation. Remembering that we proposed a correspondence between the elevation of reaction times in the semantic task with cognitive impairment, and the negative bias in the valence task as a feature of altered mood, we showed that the model could explain the observed persistence of cognitive impairment in euthymic patients (due either to a treatment based on psychotherapy alone or in combination with antidepressants that only partially reverses HA). This could be a consequence of HA, whose main manifestation within the limits of the model is an elevation of reaction times in the lexical task. These reaction time levels could represent a measure of the hidden biological alterations underlying depression, and we postulate that its exploration could be of clinical value to asses this impairment.
Unexpectedly, our model can also explain other clinical observations of depression that have been correlated with HA: the occurrence of frequent relapses of illness [21] and treatment resistance [27–30], reinforcing the validity of its assumptions. Even if the treatment was effective in improving mood (represented by a return to normal reaction times on the valence task), if cognitive impairment persists due to residual HA, an increased vulnerability to stressors is expected. An individual in real life is constantly subjected to stressful experiences. In the model, such experiences would manifest themselves as alterations in reaction times and response biases, which would become markedly accentuated if HA was present. Therefore, in the presence of HA, stressors that normally would not have clinical manifestations could provoke a relapse or prevent treatment from being effective. Hence, according to our model, the reversion of HA or its minimization is essential for obtaining a sustainable remission of the illness.
Consistent with the above, a combined treatment could have different levels of effectiveness, depending on the extent of HA, producing stable remission in one case, a remission with remaining HA (that implies vulnerability to stressors), or even treatment resistance (valence task reaction times do not normalize).
As already mentioned, varying the order of application of HA and overtraining is equivalent, so it does not help to clarify whether atrophy precedes or follows depression. However, if clinical studies were capable of showing a positive correlation between frequent relapses and treatment resistance with the progress of the illness, as these effects are explained in the model by HA, this would suggest a progression in atrophy with the evolution of depression. However, once more, this does not discount the possibility of a “priming” level of HA in young patients at the beginning of their illness, which could affect the natural history of its evolution.
As further evidence for the robustness of our results, another mechanism for the induction of depression (based not on the theory of overtraining of a single relevant stimulus but on a general reinforcement of all negative stimuli) was tested [17]. The results obtained with both types of overtraining were indistinguishable for the depressed network and for the networks with HA and treatments.
Final Remarks
In summary, our study offers an insight into some mechanisms that might underlie the pathogenesis of depression. In general, our results offer evidence of a mechanistic link between HA and the cognitive impairment observed in depression, and also suggest that HA could play a role in the maintenance of depressed mood. The model also gives an explanation for the persistent cognitive impairment seen after recovery from depression and highlights a need for reversion of HA to achieve a sustainable remission from illness.
Our results are also in general agreement with UK NICE recommendations [31] for psychosocial and psychological treatment of mild depression and a combination of antidepressants with psychotherapy in moderate to severe illness (where HA is more likely to be present).
Finally, our results suggest that cognitive impairment could be a sensitive marker of underlying neurobiological changes, and its monitoring could be useful in guiding pharmacological treatment. Therefore, further studies confirming this prediction and eventually developing standardized tests of the subclinical state of the illness could be of medical utility.
Acknowledgements
We wish to thank Miguel Arocena and Alfonso Pérez for their valuable technical help, and Dr. Douglas Steele for useful corrections.
Appendix
Methods
Architecture of the Network
The architecture of the network is inspired by the concept of parallel processing of emotional and nonemotional aspects of information (see Fig. 1). Activation of 32 input nodes represents perceptual characteristics of a stimulus. Input nodes feed activation forward to 32 semantic nodes representing the semantic content of the stimulus and to 16 affective (or valence) nodes representing the affective content of the stimulus. The semantic and the affective nodes are roughly associated with the hippocampal and the amygdala systems, respectively. Feedback occurs between the semantic and the affective nodes. Semantic and affective nodes transmit their activations to 32 output semantic and 16 output valence nodes, respectively, where the decisional process of choosing a possible response takes place. In Siegle’s model, the output module is roughly associated with the frontal lobe.
Representation of Input, Semantic and Valence Patterns
For every stimulus, the input and semantic patterns were represented with the same vector. For these representations, 32-dimensional orthogonal Walsh vectors were used after normalization; n-dimensional Walsh vectors were obtained as columns of n-dimensional Hadamard matrices (n = 2k; k ∈ N) of the form: H(2k) = H(2) ⊗ H(2k − 1), where H(2) = [1 1; 1 −1] and ⊗ is the Kronecker product. For valence representation, a near orthogonality between positivity and negativity was assumed. Valence patterns were constructed as linear combinations of two 16-dimensional Walsh vectors with the following linear coefficients: (cos10°, sin10°) for positivity, (cos80°, sin80°) for negativity, and (cos45°, sin 45°) for neutrality. Valence patterns were normalized.
Training of the Network
The network was trained on three positive, three negative, and three neutral stimuli. The training was implemented creating four weight matrices using the outer product rule [32]: the IS matrix (dimension 32 ×32, autoassociating a semantic pattern to the same input pattern), the IV matrix (dimension 16 ×32, associating a valence pattern to each in- put pattern), the VS matrix (dimension 32 ×16, associating a semantic pattern to a valence pattern), and the SV (dimension 16 ×32, associating a valence pattern with a semantic pattern).
Depression was induced overtraining the network on one negative stimulus from the training set, representing a relevant negative experience, and applying a forgetting rule to the old learning. During the overtraining, the feedback matrices (SV and VS) were adjusted according to the rule:
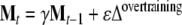 |
1 |
where γ (=0.89) is the forgetting coefficient, ε (=1.5) is the overtraining coefficient, and Mt is a feedback matrix (SV or VS) after t cycles of overtraining and Δovertraining is the outer product matrix for the overtrained stimulus. One overtraining cycle was applied to the matrix VS and seven overtraining cycles were applied to the matrix SV.
Network Activation
A unit in the network actualizes its activation as a function of its input and the activation already reached by the unit. We will represent this with s (v) as the activation of the semantic (valence) layer. During the presentation of the stimulus (lasting 10 iterations) bipolar and uniformly distributed noise with a magnitude of 0.005 also enters the net. Semantic and affective units activate according to the following rules:
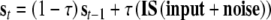 |
2 |
 |
3 |
where τ (=0.1) is the diffusion rate for inputs. After the stimulus presentation, only noise enters the network and feedback between semantic and valence nodes operates. Layers actualize activations according to the rules:
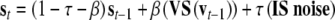 |
4 |
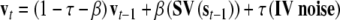 |
5 |
where β (=0.002) is the diffusion rate for feedback between semantic and valence layers. When the feedback is operating, semantic and valence layers normalize activations on every iteration.
Stimulus Identification at the Output Module
As in Siegle’s model, once a stimulus is presented to the network, evidence is progressively accumulated for each possible response until one of them reaches a threshold (Fig. 7). Following this idea, devices called counters, which accumulate evidence for every possible response of the network, were defined. Specifically, a counter was defined for each of the nine semantic and for each of the three valence stored patterns. At the beginning of a trial, counters are set to zero. In every iteration, counters change their value according to how well the output semantic or output valence node activation fits the semantic or valence patterns, respectively. The fit between the activation of output nodes and the stored patterns is evaluated as the cosine of the angle between the vectors. The amount that every counter i adds on each iteration is calculated according to the rule:
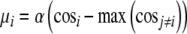 |
6 |
where α (=0.1) is the rate of evidence accumulation. If the semantic (valence) task is running, a reaction time is computed when one of the semantic (valence) counters reaches the threshold. The threshold values for the lexical and valence tasks were 4 and 1.5, respectively.
Fig. 7.

Computation of reaction times for the valence task when a positive stimulus enters the network. During the valence task, the network could identify a stimulus as positive, negative, or neutral. For each of these possible responses, counters are defined (C + : counter for positive valence; C − : negative valence; Cn: neutral valence). At the beginning of the trial, all counters are set to zero. In every iteration, each counter changes its value according to how well the output affective activation fits the valence pattern that the counter represents, evaluated as the cosine between the vectors (see Eq. 6 in the Appendix). When one of the counters reaches a threshold, the trial ends, and the number of iterations elapsed until that moment is computed as the reaction time of the network. In the figure, a positive stimulus enters the network and the counter that represents positivity is the first one in reaching the threshold (confusion may occur when a counter corresponding to a valence different from the stimulus reaches the threshold). For the lexical task, reaction times are computed in an analogous way, but in this case, nine counters are used
Implementation of HA in the Network
HA was simulated by randomly destroying synapses in memories attributed to the hippocampal system. Specifically, synaptic weights in matrices IS and VS were randomly selected and set to a zero value. According to data from anatomical studies, percentages of HA going from 5% to 20% were explored.
Effect of a Cognitive Therapy in the Network
The effect of a cognitive therapy was simulated by reinstructing the connection matrices with the whole set of stimuli and applying a forgetting rule to the old learning to withdraw previous overtraining on negative stimulus. During the relearning, the weight matrices were adjusted according to the rule:
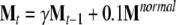 |
7 |
where γ (=0.89) is the forgetting coefficient, Mt is one of the weight matrices of the network, and Mnormal is the sum of the outer products for the whole set of stimuli. When the cognitive therapy is applied on the network with HA, the atrophied connections do not adjust their weights according to the rule (Eq. 7), staying with a zero value weight.
Effect of a Combined Treatment of Antidepressants and Cognitive Therapy
Antidepressant drugs might oppose or even reverse HA. In the model, the effect of antidepressants was simulated, reattributing basal synaptic weights to the previous atrophied connections (that were set to a value of zero), hence allowing them to adjust their weights with the cognitive therapy relearning mechanism. Basal synaptic weights were calculated as the average of the weights of the corresponding matrix (IS or VS) in the normal state, plus a random number normally distributed with mean zero and variance 1 ×10 − 4.
Statistical Analysis
Randomness was introduced in the model through the noise that enters the network, and also by the implementation of HA. The reaction times presented in the graphics are averages over 1,000 random realizations. For each curve in the graphs of the figures, a new independent set of 1,000 realizations was randomly chosen. A z test was used for comparing means. A z value of 2.33 was used to establish the rejection region. All results were significant with p < 0.001.
References
- 1.Kessler, R.C., Merikangas, K.R., Wang, P.S.: Prevalence, comorbidity, and service utilization for mood disorders in the United States at the beginning of the twenty-first century. Annu. Rev. Clin. Psychol. 3, 137–158 (2007). doi:10.1146/annurev.clinpsy.3.022806.091444 [DOI] [PubMed]
- 2.Ebmeier, K.P., Donaghey, C., Steele, J.D.: Recent developments and current controversies in depression. Lancet 367, 153–167 (2006). doi:10.1016/S0140-6736(06)67964-6 [DOI] [PubMed]
- 3.Videbech, P., Ravnkilde, B.: Hippocampal volume and depression: a meta-analysis of MRI studies. Am. J. Psychiatry 161, 1957–1966 (2004). doi:10.1176/appi.ajp.161.11.1957 [DOI] [PubMed]
- 4.Campbell, S., Marriott, M., Nahmias, C., MacQueen, G.M.: Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am. J. Psychiatry 161, 598–607 (2004). doi: 10.1176/appi.ajp.161.4.598 [DOI] [PubMed]
- 5.Davidson, R.J., Lewis, D.A., Alloy, L.B., Amaral, D.G., Bush, G., Cohen, J.D., Drevets, W.C., Farah, M.J., Kagan, J., McClelland, J.L., Nolen-Hoeksema, S., Peterson, B.S.: Neural and behavioral substrates of mood and mood regulation. Biol. Psychiatry 52, 478–502 (2002). doi:10.1016/S0006-3223(02)01458-0 [DOI] [PubMed]
- 6.Warner-Schmidt, J.L., Duman, R.S.: Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus 16, 239–249 (2006). doi:10.1002/hipo.20156 [DOI] [PubMed]
- 7.Duman, R.S., Monteggia, L.M.: A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59, 1116–1127 (2006). doi:10.1016/j.biopsych.2006.02.013 [DOI] [PubMed]
- 8.Sheline, Y.I.: 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol. Psychiatry 48, 791–800 (2000). doi:10.1016/S0006-3223(00)00994-X [DOI] [PubMed]
- 9.McEwen, B.S.: Stress and hippocampal plasticity. Annu. Rev. Neurosci. 22, 105–122 (1999). doi:10.1146/annurev.neuro.22.1.105 [DOI] [PubMed]
- 10.Austin, M.P., Mitchell, P., Goodwin, G.M.: Cognitive deficits in depression. Br. J. Psychiatry 178, 200–206 (2001). doi:10.1192/bjp.178.3.200 [DOI] [PubMed]
- 11.Siegle, G.J.: A neural network model of attention biases in depression. In: Reggia, J., Rupin, E. (eds.) Disorders of Brain, Behavior, and Cognition: The Neurocomputational Perspective, pp. 415–441. Elsevier, New York (1999) [DOI] [PubMed]
- 12.LeDoux, J.: The emotional brain. Simon & Schuster, New York (1996)
- 13.Packard, M.G., Cahill, L.: Affective modulation of multiple memory systems. Curr. Opin. Neurobiol. 11, 752–756 (2001). doi:10.1016/S0959-4388(01)00280-X [DOI] [PubMed]
- 14.Vuilleumier, P.: How brains beware: neural mechanisms of emotional attention. Trends Cogn. Sci. 9, 585–594 (2005). doi:10.1016/j.tics.2005.10.011 [DOI] [PubMed]
- 15.Phelps, E.A.: Emotion and cognition: insights from studies of the human Amygdala. Annu. Rev. Psychol. 57, 27–53 (2006). doi:10.1146/annurev.psych.56.091103.070234 [DOI] [PubMed]
- 16.Pomi-Brea, A., Mizraji, E.: Memories in context. Biosystems 50, 173–188 (1999). doi:10.1016/S0303-2647(99)00005-2 [DOI] [PubMed]
- 17.Gradin, V.B.: Aplicación de un modelo de red neuronal al estudio de los trastornos depresivos. MSc Thesis, PEDECIBA—Facultad de Ciencias, Universidad de la República, Montevideo (2007)
- 18.Hickie, I., Naismith, S., Ward, P.B., Turner, K., Scott, E., Mitchell, P., Wilhelm, K., Parker, G.: Reduced hippocampal volumes and memory loss in patients with early- and late-onset depression. Br. J. Psychiatry 186, 197–202 (2005). doi:10.1192/bjp.186.3.197 [DOI] [PubMed]
- 19.Frodl, T., Schaub, A., Banac, S., Charypar, M., Jäger, M., Kümmler, P., Bottlender, R., Zetzsche, T., Born, C., Leinsinger, G., Reiser, M., Möller, H.J., Meisenzahl, E.M.: Reduced hippocampal volume correlates with executive dysfunctioning in major depression. J. Psychiatry Neurosci. 31, 316–325 (2006) [PMC free article] [PubMed]
- 20.MacQueen, G.M., Galway, T.M., Hay, J., Young, L.T., Joffe, R.T.: Recollection memory deficits in patients with major depressive disorder predicted by past depressions but not current mood state or treatment status. Psychol. Med. 32, 251–258 (2002) [DOI] [PubMed]
- 21.MacQueen, G.M., Campbell, S., McEwen, B.S., Macdonald, K., Amano, S., Joffe, R.T., Nahmias, C., Young, L.T.: Course of illness, hippocampal function, and hippocampal volume in major depression. Proc. Natl. Acad. Sci. U. S. A. 100, 1387–1392 (2003). doi:10.1073/pnas.0337481100 [DOI] [PMC free article] [PubMed]
- 22.Reid, I.C., Stewart, C.A.: How antidepressants work. Br. J. Psychiatry 178, 299–303 (2001). doi:10.1192/bjp.178.4.299 [DOI] [PubMed]
- 23.Hajszan, T., MacLusky, N.J., Leranth, C.: Short-term treatment with the antidepressant fluoxetine triggers pyramidal dendritic spine synapse formation in rat hippocampus. Eur. J. Neurosci. 21, 1299–1303 (2005). doi:10.1111/j.1460-9568.2005.03968.x [DOI] [PubMed]
- 24.Czéh, B., Michaelis, T., Watanabe, T., Frahm, J., de Biurrun, G., van Kampen, M., Bartolomucci, A., Fuchs, E.: Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc. Natl. Acad. Sci. U. S. A. 98, 12796–12801 (2001). doi:10.1073/pnas.211427898 [DOI] [PMC free article] [PubMed]
- 25.Sheline, Y.I., Gado, M.H., Kraemer, H.C.: Untreated depression and hippocampal volume loss. Am. J. Psychiatry 160, 1516–1518 (2003). doi:10.1176/appi.ajp.160.8.1516 [DOI] [PubMed]
- 26.Chambers, R.A., Potenza, M.N., Hoffman, R.E., Miranker, W.: Simulated apoptosis/neurogenesis regulates learning and memory capabilities of adaptive neural networks. Neuropsychopharmacology 29, 747–758 (2004). doi:10.1038/sj.npp.1300358 [DOI] [PubMed]
- 27.Frodl, T., Meisenzahl, E.M., Zetzsche, T., Höhne, T., Banac, S., Schorr, C., Jäger, M., Leinsinger, G., Bottlender, R., Reiser, M., Möller, H.J.: Hippocampal and amygdale changes in patients with major depressive disorder and healthy controls during a 1-year follow-up. J. Clin. Psychiatry 65, 492–499 (2004) [DOI] [PubMed]
- 28.Shah, P.J., Glabus, M.F., Goodwin, G.M., Ebmeier, K.P.: Chronic, treatment-resistant depression and right fronto-striatal atrophy. Br. J. Psychiatry 180, 434–440 (2002). doi:10.1192/bjp.180.5.434 [DOI] [PubMed]
- 29.Vakili, K., Pillay, S.S., Lafer, B., Fava, M., Renshaw, P.F., Bonello-Cintron, C.M., Yurgelun-Todd, D.A.: Hippocampal volume in primary unipolar major depression: a magnetic resonance imaging study. Biol. Psychiatry 47, 1087–1090 (2000). doi:10.1016/S0006-3223(99)00296-6 [DOI] [PubMed]
- 30.Hsieh, M.H., McQuoid, D.R., Levy, R.M., Payne, M.E., MacFall, J.R., Steffens, D.C.: Hippocampal volume and antidepressant response in geriatric depression. Int. J. Geriatr. Psychiatry 17, 519–525 (2002). doi:10.1002/gps.611 [DOI] [PubMed]
- 31.National Institute for Clinical Excellence (NICE): Depression: Management of Depression in Primary and Secondary Care. Clinical Guideline 23. NICE, London (2004)
- 32.Anderson, J.A.: An Introduction to Neural Networks. MIT, Cambridge (1995)