

Abstract
Objective
Primary Pigmented Nodular Adrenocortical Disease (PPNAD) can occur as an isolated trait or part of Carney complex, a familial lentiginosis-multiple endocrine neoplasia syndrome frequently caused by mutations in PRKAR1A, which encodes the 1α regulatory subunit of protein kinase A (PKA). Because alterations in the insulin-like growth factor (IGF) axis, particularly IGF-II and IGF binding protein (IGFBP)-2 over- expression, have been implicated in sporadic adrenocortical tumors, we sought to examine the IGF axis in PPNAD.
Design
RNA samples and paraffin-embedded sections were procured from adrenalectomy specimens of patients with PPNAD. Changes in expression of IGF axis components were evaluated by real-time quantitative RT-PCR and immunohistochemistry. NCI-H295R cells were used to study PKA and IGF axis signaling in adrenocortical cells in vitro.
Results
IGFBP-2 mRNA level distinguished between the two genetic subtypes of this disease; increased IGFBP-2 expression in PRKAR1A mutation-positive PPNAD tissues was also confirmed by immunohistochemistry. Moreover, PKA inhibitors increased IGFBP-2 expression in NCI-H295R adrenocortical cells, and anti-IGFBP-2 antibody reduced their proliferation.
Conclusion
IGFBP-2 expression is increased in PPNAD caused by PRKAR1A mutations, and in adrenocortical cancer cells. This is the first evidence for PKA-dependent regulation of IGFBP-2 expression in adrenocortical cells.
Keywords: Insulin-like growth factor binding protein (IGFBP)-2, cAMP dependent protein kinase (PKA), PRKAR1A gene, adrenocortical cells, NCI-H295R cells, Primary Pigmented Nodular Adrenocortical Disease (PPNAD)
1. Introduction
Primary pigmented nodular adrenocortical disease (PPNAD) is a rare cause of ACTH-independent Cushing syndrome. Histologically, PPNAD is characterized by multiple brown-colored nodules, usually less than 4 mm in diameter, composed of large cells with granular, eosinophilic, pigment-containing cytoplasm, and surrounded by atrophic and disorganized extranodular cortex [1]. It can occur either as an isolated autosomal dominant trait or as part of Carney complex (MIM #160980), a familial lentiginosis-multiple endocrine neoplasia syndrome characterized by cardiac myxomas, spotty pigmentation of the skin and mucosae, and endocrine over-activity [1–3]. Carney complex is a dominantly inherited, genetically heterogeneous disease. Linkage analysis has attributed the genetic predisposition to chromosome loci 2p16 (CNC2) and 17q22–24 (CNC1). The responsible gene at 2p16 remains elusive [4], but the gene at 17q22–24 has been identified as PRKAR1A, which encodes the RIα regulatory subunit of cAMP dependent protein kinase (protein kinase A, PKA) [5].
PKA is a pivotal component of the cAMP signaling pathway. Its holoenzyme is made of two regulatory (R) and two catalytic (C) subunits. Regulatory subunits have 4 isoforms (RIα, RIβ, RIIα and RIIβ), which differ somewhat in their affinity for cAMP. When cAMP binds to the tandem cAMP-binding domains on R subunits, active C subunits are released and in turn phosphorylate specific substrates that include transcription factors. PRKAR1A inactivation leads to an increased ratio of type II to type I PKA activity, but the downstream mechanism of tumor growth in PPNAD is not yet clear and may involve the activation of other tumorigenic signaling pathways [6].
Alterations in the insulin-like growth factor (IGF) axis have been implicated in adrenocortical neoplasia [7]. Over-expression of IGF-II and IGF binding protein (IGFBP)-2 was found in adult sporadic adrenocortical tumors and associated with a malignant phenotype [8]. Plasma IGFBP-2 levels were significantly higher in adults with metastatic adrenocortical disease than normal controls and were inversely correlated with survival [9]. Because IGFBP-2 was not exclusively found in metastatic tumors, however, it was not specific enough to serve as a molecular marker for malignancy. IGF-II and IGFBP-2 over-expression was also demonstrated in NCI-H295R cells, a cell line derived from a human adrenocortical tumor, both in vitro and in xenografts in nude mice [10; 11].
We therefore hypothesized that the abnormal adrenocortical proliferation of PPNAD is associated with alterations in the IGF axis, most likely IGF-II and IGFBP-2. Quantitative real-time RT-PCR and immunohistochemistry revealed that IGFBP-2 expression distinguished PRKAR1A mutation-positive from mutation-negative PPNAD. Using NCI-H295R adrenocortical cells, we found the first evidence of IGFBP-2 regulation by PKA and demonstrated that IGFBP-2 inhibition reduced the proliferation of adrenocortical cells in vitro.
2. Materials and methods
2.1 Adrenal specimens
Nine human PPNAD adrenal tissues were obtained from prior adrenalectomies at the National Institute of Child Health and Human Development. The adrenalectomies had been performed for clinical indications, such as Cushing syndrome or adrenal mass. The frozen specimens had been snap-frozen in liquid nitrogen upon excision and stored at –80°C. Paraffin-embedded tissues were processed as in surgical pathology routines.
The use of human tissue specimens was approved by the Institutional Review Boards of both the Children’s Hospital of Philadelphia and the National Institute of Child Health and Human Development.
2.2 Adrenocortical Cell line
NCI-H295R cells, purchased from American Type Culture Collection (ATCC) (Manassas, VA), are NCI-H295 pluripotent adrenocortical carcinoma cells that had been adapted to grow in monolayer. These cells retain the ability to produce adrenal androgens, and are therefore generally used as a model for human adrenocortical cell biology [12]. NCI-H295R cells were maintained in DMEM/F12 (Meditech Corp., Herndon, VA) with addition of 2.5% Nu-Serum I (BD Biosciences, Mountain View, CA), 100 units/ml streptomycin/penicillin (Invitrogen, Carlsbad, CA) and ITS+ Premix (BD Biosciences), for final concentrations of 6.25 μg/ml insulin, 6.25 μg/ml transferrin, 6.25 ng/ml selenium, 1.25 mg/ml bovine serum albumin and 5.35 μg/ml linoleic acid, as recommended by ATCC. PRKAR1A mutation status was tested by sequencing, as previously reported [13].
2.3 RNA extraction and Real-Time RT-PCR
Total RNA was extracted from the frozen adrenocortical specimens using combined Trizol Reagent (Invitrogen) and RNeasy kit (Qiagen, Valencia, CA) procedure according to the manufacturer’s protocol. An in-column RNase-free DNase digestion (Qiagen) was also performed to exclude the possibility of amplification from genomic DNA. The integrity of each total RNA sample was checked by running 1% agarose RNA electrophoresis under denaturing conditions in 2.2M formaldehyde (Fisher Scientific, Fair Lawn, NJ) with the MOPS buffer system (Fisher Scientific). Commercially available normal human adrenocortical total RNA, which had been pooled from 61 individuals, (BD Clontech, Mountain View, CA) was used as the calibrator for the real time quantitative PCR.
Reverse transcription was performed with 20 μg of the total RNA from each specimen and SuperScript II reverse transcriptase according to the manufacturer’s protocol (Invitrogen). cDNA products were then diluted 1:10 and used for PCR templates. Real-time quantitative PCR was performed with SYBR green Master Mix (Applied Biosystems, Foster City, CA). Primer design was carried out with Primer Express software (Applied Biosystems) such that all primers spanned adjacent exons to further avoid the amplification of genomic DNA. The following primer sequences were used, at final concentrations of 167 nM each: IGF-I forward, 5’-CTG CTT CCG GAG CTG TGA TC-3’, IGF-I reverse, 5’-TCC CTC TAC TTG CGT TCT TCA AA-3’; IGF-II forward, 5’-ACC GTG CTT CCG GAC AAC-3’, IGF-II reverse, 5’-TGG ACT GCT TCC AGG TGT CA-3’; IGFBP-2 forward, 5’-ATG CGC CTT CC GGA TGA-3’, IGFBP-2 reverse, 5’-ACG CTG CCC GTT CAG AGA-3’; IGFBP-3 forward, 5’-GGA AGA CAC ACT GAA TCA CCT GAA-3’, IGFBP-3 reverse, 5’-CC TTT GGA AGG GCG ACA CT-3’; phosphoglycerate kinase (PGK) forward, 5’-GGG CTG CAT CAC CAT CAT AGG-3’, PGK reverse, 5‘-GAG AGC ATC CAC CCC AGG AAG-3’; Synaptophysin forward, 5’-GCC GTG TTT GCC TTC CTC TAC-3’, Synaptophysin reverse, 5’-CCC ATG CCG ATG AGC TAA CTA G-3’. The RT-PCR cycling conditions were 50°C for 2 min and 95°C for 10 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All real time PCR reactions were carried out on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) with the relative quantification method compared to the normal RNA sample as calibrator. Target gene mRNA levels were normalized to those of the housekeeping PGK gene in the replicated samples. Each sample was measured in quadruplicate for each experiment. Melting curves were checked to ensure specificity.
2.4 Immunohistochemistry
The 5 μm paraffin-embedded sections were de-paraffinized in xylene and rehydrated in serial decreasing concentrations of ethanol. Antigen retrieval was carried out by boiling slides in citrate buffer (pH 6.0) for 5 min twice. Endogenous peroxidase activity was blocked by incubation with 0. 3% H2O2 for 20 min. The primary antibodies used were: mouse anti-human IGF-I and IGF-II (Upstate, Charlottesville, VA) with a 1:100 dilution, rabbit anti-human IGFBP-2 (Cell Signaling Technology, Beverly, MA) at 1:50 and goat anti-human IGFBP-3 (Diagnostic Systems Laboratories, Webster, TX) at 1:100. Staining was performed by the standard Envision Plus (Dakocytomation, Carpinteria, CA) (for IGF-I, -II and IGFBP-2) or ABC Elite (Vector Lab., Burlingame, CA) (for IGFBP-3) procedure with 0.05% DAB (3, 3’-diaminobenzidine tetrahydrochloride) solution (Vector Lab) as coloring substrate. Sections were then counterstained with haematoxylin (Fisher Scientific).
2.5. Protein kinase A modulation
NCI-H295R cells were plated at 1x105 cells/well on 6-well plates and incubated for 24 hr in complete medium. PKA activator [5 μM Forskolin (Upstate)] or inhibitor [50 nM H89 (Upstate), or 10 μg/ml PKI (Biosource, Camarillo, CA)] was then added to duplicate wells for 6 more hr. Cell samples were harvested by directly adding 1 ml Trizol (Invitrogen) to each well and scraping with cell lifter, and total RNA was extracted by the standard Trizol method. RT-PCR was performed to quantify the IGF axis messages as described above.
2.6. Proliferation assay
NCI-H295R cells were plated at a density of 5x103 cells per 100μl complete medium per well on a 96-well plate and allowed to grow for 24 hr. Then, a final concentration of 1.2 μg/ml rabbit anti-human IGFBP-2 antibody (Cell Signaling Technology) or as negative control, rabbit anti-human CD8 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), was added. 72 hr later, 20 μl of PMS-MTS mixture was added to each well (CellTiter 96TM AqueousOne Proliferation Assay Kit, Promega, Madison, WI). Plates were incubated at 37°C in a humidified, 5% CO2 atmosphere for 3 hr before absorbance at 490 nm was read and recorded (Bio-Rad 3550 Plate Reader, Bio-Rad, Hercules, CA).
2.7. Statistical Analyses
For each IGF axis component tested, the Kruskal-Wallis nonparametric ANOVA was used to compare the IGF/synaptophysin ratios among normal (one), PRKAR1A mutation-positive and PRKAR1A mutation-negative PPNAD. ANOVA, followed by Tukey-Kramer Multiple Comparisons Test, was used to compare IGFBP-2 message levels in H295R cells among the different PKA modulators and the untreated condition. Changes in expression levels for IGF-II, IGFBP-2 and IGFBP-3 following H89 treatment were also compared by ANOVA, followed by Tukey-Kramer Multiple Comparisons Test, for each time point relative to baseline. Two-tailed, unpaired t test was used to compare H295R cell proliferation readings between untreated cells and either IGFBP-2- or CD-8-antibody treated cells. Statistical analyses were performed with InStat (GraphPad Software Inc., San Diego, CA). For all tests, significance was defined as P<0.05.
3. RESULTS
3.1. IGF axis expression in PPNAD
The clinical characteristics of the nine patients with PPNAD are summarized in Table 1. The mRNA expression of IGF-I, IGF-II, IGFBP-2 and IGFBP-3 in the PPNAD tissues was measured with real-time quantitative RT-PCR. To correct for the inhomogeneity of the adrenal tissues, synaptophysin, a neuroendocrine marker whose immunoreactivity was demonstrated in PPNAD nodules [14], was simultaneously tested and used to normalize for the amount of aberrant micronodular cells in each specimen.
Table 1. Clinical features of patients.
| Patient Code # | Sex | Age(yrs) | Diagnosis | Mutation | Presentation |
|---|---|---|---|---|---|
| Control Sample | F/M | 16–61 | Normal adrenal tissue | Unknown | Sudden death of unrelated reason |
| 4 | F | 32 | PPNAD (Carney) | IVS2+1G>A | Cushing |
| 6 | M | 21 | PPNAD (Carney) | IVS9+3A>G | Carney |
| 9 | F | 38 | PPNAD (Carney) | c.1A>G(Met1Val) | Carney |
| 21 | F | 19 | PPNAD (Carney) | c.101–105 del CTATT | Carney |
| 5 | F | 10 | PPNAD (isolated) | Negative | Cushing |
| 7 | F | 12 | PPNAD (isolated) | Negative | Cushing |
| 8 | F | 5 | PPNAD (isolated) | Negative | Cushing |
| 10 | F | 8 | PPNAD (isolated) | Negative | Cushing |
| 15 | M | 10 | PPNAD (isolated) | Negative | Cushing |
Figure 1 shows the relative amounts of the IGF axis messages, expressed as a ratio to the amount of synaptophysin message in each specimen. The expression of IGF-II, IGF-I, IGFBP-3 and IGFBP-2 were all significantly greater in PRKAR1A mutation-positive than in mutation-negative PPNAD specimens. Two of the PRKAR1A mutation-positive PPNAD specimens had IGF-II level at or below the normal level of 1. IGF-I and IGFBP-3 both had mutation-negative individuals with expression levels greater than the normal of 1. Only IGFBP-2 expression levels had no overlap between normal, PRKAR1A mutation-positive and mutation-negative PPNAD.
Fig. 1. IGF axis/synaptophysin mRNA ratios in PPNAD.
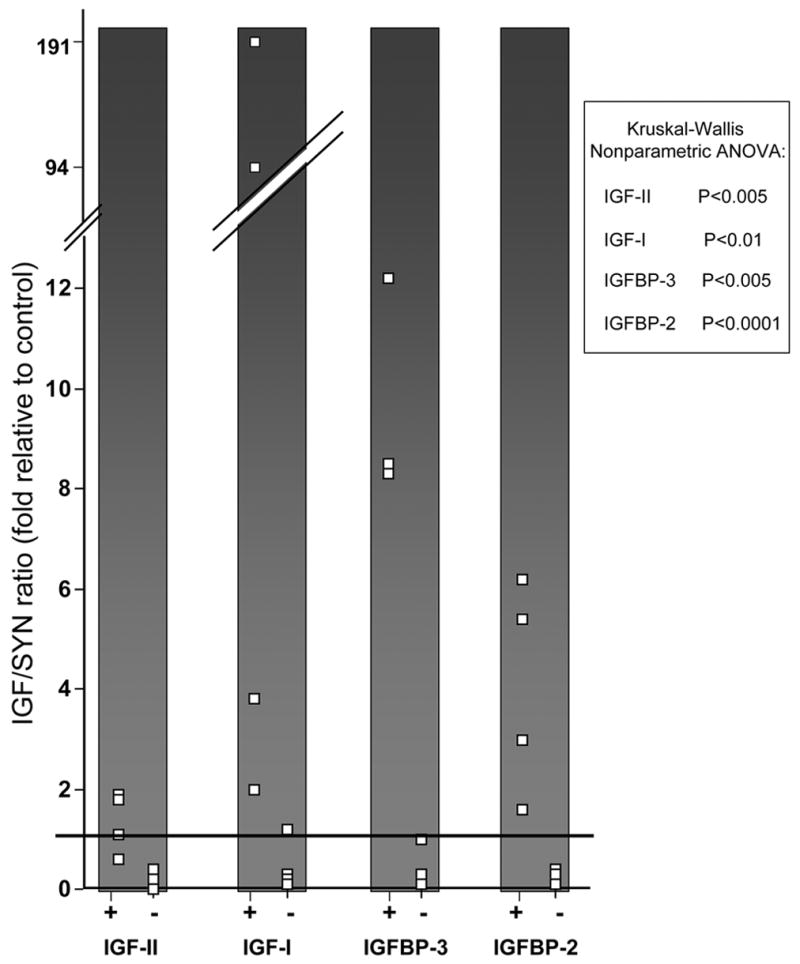
Expression of IGF-II, IGF-I, IGFBP-3 and IGFBP-2 were measured by Real-time quantitative RT-PCR. For each gene, the results are displayed according to PRKAR1A mutation status (positive,+; negative,−). Each white box depicts the message ratio of the IGF axis member to synaptophysin for an individual patient, expressed as fold difference from the ratio measured in the normal control RNA. Each box is the average of quadruplicate raw measures, each normalized to PGK. To facilitate visualization of any overlap between the patient groups, the normal control value of 1 is indicated by the black horizontal bar. Ratio values for each IGF axis member/synaptophysin significantly differed between patient groups, as shown.
Alterations in the in situ protein levels of IGF-I, IGF-II, IGFBP-3 and IGFBP-2, as detected by immunohistochemistry (IHC) in formalin-fixed paraffin blocks, paralleled the expression changes seen in Figure 1. Figure 2 shows representative immunohistochemistry results for IGFBP-2. Panel A, a 100X magnification of PRKAR1A mutation-positive PPNAD stained with rabbit anti-human IGFBP-2 antibody, shows high intensity staining of the nodules with much weaker staining of the extranodular cortical cells. Panel B is the same tissue stained with rabbit anti-human β amyloid precursor protein antibody as a negative control. Panels C and D depict the same IHC procedures applied to tissue from a patient with PRKAR1A mutation-negative PPNAD. The whole tissue stains positively for IGFBP-2, perhaps at a lesser intensity, reflecting the generalized hyperplasia seen in PRKAR1A mutation-negative PPNAD. Synaptophysin immunoreactivity has been similarly shown to sensitively and specifically localize to nodular cells in PRKAR1A mutation-positive PPNAD [14] but be more generalized in mutation-negative tissue [15].
Fig. 2. Representative IGFBP-2 immunohistochemistry in PPNAD.
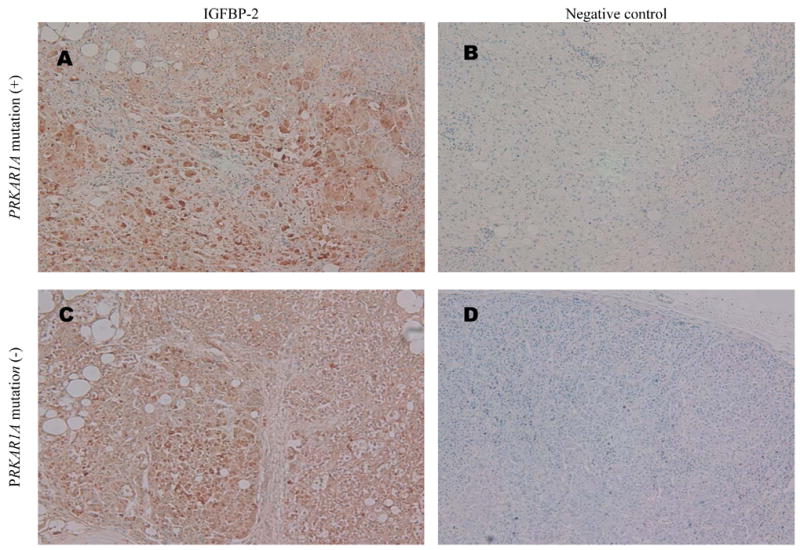
(A) PRKAR1A mutation-positive PPNAD section labeled with 1:50 rabbit anti-human IGFBP-2 antibody and visualized by the Envision Plus method using DAB color substrate and Harris’ haematoxylin counter-staining (original magnification X100).
(B) Adjacent section to that in Panel A, simultaneously processed the same way except using rabbit anti-human β amyloid precursor protein antibody as negative control primary antibody (original magnification X100).
(C) and (D) PRKAR1A mutation-negative PPNAD sections processed the same as (A) and (B), respectively (both original magnification X100).
3.2. IGFBP-2 induction by protein kinase A inhibitors in adrenocortical cells in vitro
The enrichment of IGFBP-2 in PRKAR1A mutation-positive PPNAD nodules relative to normal adrenal cortex and PRKAR1A mutation-negative PPNAD generated the hypothesis that IGFBP-2 may be regulated by the PKA pathway. We therefore tested NCI-H295R cells as an in vitro model of adrenocortical cell behavior to investigate the dynamic regulation of IGFBP-2 by PKA. We measured changes in IGFBP-2 expression when NCI-H295R cells were treated with known modulators of the PKA pathway: forskolin is a diterpene that ubiquitously activates eukaryotic adenylyl cyclase, thereby raising cAMP levels with subsequent PKA activation; H89 (N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide) is a synthetic, specific inhibitor of PKA; and protein kinase inhibitor (PKI) is an endogenous, specific and potent inhibitor of the catalytic subunit of PKA. Forskolin did not significantly alter IGFBP-2 message levels, but both H-89 and PKI increased IGFBP-2 message about 6-fold (P<0.001) (Figure 4). A time course experiment of H-89 effects revealed a rapid and progressive induction of IGFBP-2 message, with levels almost doubling at 3 hr, more than quadrupling by 6 hr and continuing to increase at 24 hr (P<0.0001 for trend)(Figure 5). H-89 induction was specific to IGFBP-2, as IGFBP-3 message levels did not change, and IGF-II message had only a late doubling at 24 hr (P<0.001).
Fig. 4. Time course of mRNA changes in NCI-H295R cells treated with 50 nM H89.
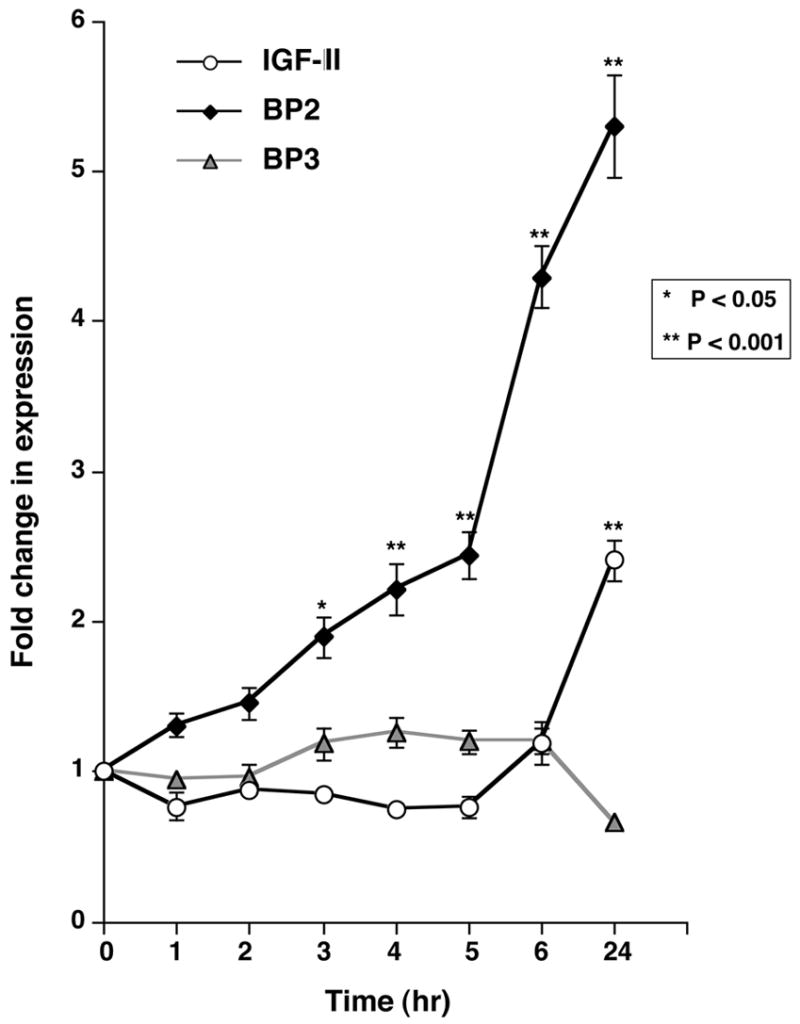
Real-time quantitative RT-PCR of IGFBP-2 (black diamonds), IGFBP-3 (gray triangles) and IGF-II (white circles) message levels was performed, normalized to PGK and expressed as fold change relative to control, untreated NCI-H295R cells plated at equal densities. Each point represents the mean ± SEM of 3 replications in each of two independent experiments.
Fig. 5. IGFBP-2 inhibition decreases NCI-H295R cell proliferation.
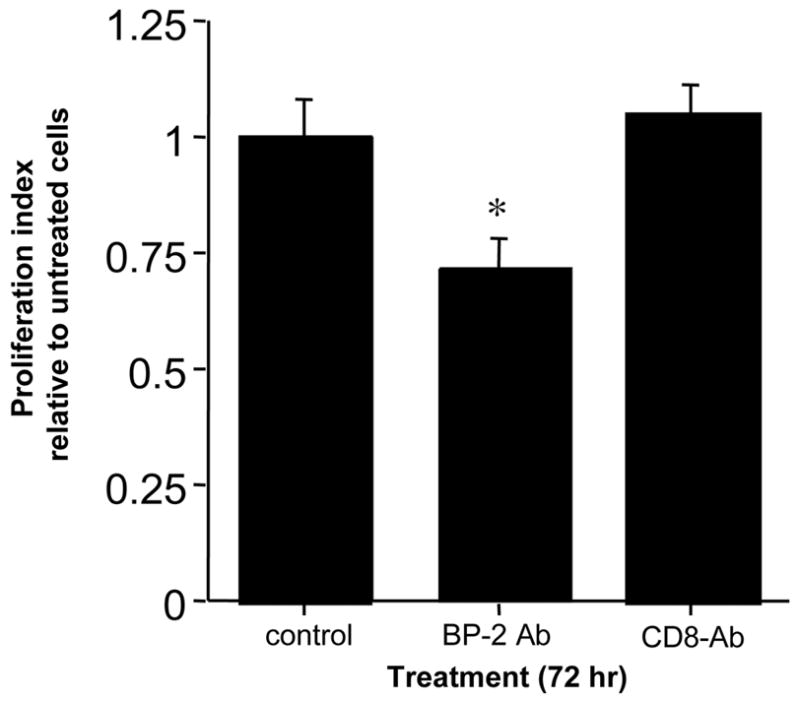
NCI-H295R cells in culture were treated with anti-human IGFBP-2 antibody or anti-human CD8 antibody at1.2 μg/ml. After 72 hr, cellular proliferation was measured by colorimetric assay and compared to untreated NCI-H295R cells. Each bar represents the mean of triplicate wells.
3.3. IGFBP-2 inhibition reduces adrenocortical cell proliferation in vitro
Evidence supporting IGFBP-2 action as pro- or anti-neoplastic is conflicting. To test the functional significance of IGFBP-2 induction in adrenocortical cells, we treated NCI-H295R cells in culture with anti-IGFBP-2 antibody or anti-CD8 antibody as negative control. After 72 hr, NCI-H295R cell proliferation was decreased by almost 30% by the IGFBP-2 antibody (P=0.01) and unchanged by the control antibody (P=0.71) (Figure 5).
4. Discussion
We discovered that the two genetic subtypes of PPNAD differentially involved the IGF axis; expression of IGFs I and II and IGFBP-2 and –3, when normalized to synaptophysin, were greater in PRKAR1A mutation-positive than mutation-negative PPNAD. Only IGFBP-2 expression level had no overlap among normal and the two types of PPNAD. Increased IGFBP-2 expression in PRKAR1A mutation-positive PPNAD nodules was also seen at the protein level. We further found specific induction of IGFBP-2 expression by PKA inhibitors in PRKAR1A+/+ H295R adrenocortical cells.
Thus, our findings suggested a role of PKA in IGFBP-2 regulation, and comparisons among the two genetic subtypes of PPNAD and the PRKAR1A+/+ H295R cells may reflect differential activation of the two types of PKA activity. Induction of IGFBP-2 in PRKAR1A mutation-positive nodules and other hyperplasias with complete loss of PRKAR1A may be associated with the PKA-II activation that has been reported in tissues with PRKAR1A losses [6]. PKI primarily inhibits PKA-I, whereas H89 inhibits both types of PKA activity equally. The fact that both H89 and PKI caused the same degree of IGFBP-2 induction means that IGFBP-2 induction is specific to PKA-I inhibition, akin to the absence of PRKAR1A in the mutation-positive PPNAD nodules. IGFBP-2 expression therefore seems to depend on the degree of PKA-I inhibition; expression is high when PRKAR1A is completely absent (nodules of mutation-positive PPNAD that have lost the normal allele via loss of heterozygosity and have a mutant allele that does not produce protein) and somewhat lower when PRKAR1A is present but there is deficient PKA-I (presumably the mutation-negative tissue, which is homogeneous without the loss of heterozygosity of the nodules).
In addition to IGFBP-2 induction, we found that IGFBP-2 inhibition reduced H295R cell proliferation. Two cAMP analogs that activate PKA induced differentiation and also reduced proliferation of H295R cells [16]. Altogether these findings support a model of PPNAD in which PRKAR1A mutation decreases PKA-I activity that in turn leads to IGFBP-2 induction and enhanced adrenocortical cell proliferation. This model may have implications for adrenocortical tumors as well; IGFBP-2 over-expression has been reported in sporadic adrenocortical tumors [8-11], and PRKAR1A-inactivating mutations, down-regulation and somatic allelic loss of the 17q22-24 region have also been found in sporadic adrenocortical tumors, especially those presenting with Cushing syndrome [17].
It is still unclear if IGFBP-2 regulation by PKA is a direct or indirect effect. For example, ERK1/2 inhibition by PKA has been implicated in the latter’s inhibition of cellular proliferation, and this inhibition was reversed in Carney complex cells [18]. c-myc was induced by PKA activation in adrenocortical cells and decreased their proliferation [16]. Finally, PRKAR1A may have PKA-independent functions [19]. An in silico search of the IGFBP-2 gene sequence for cAMP response elements (CREs) was essentially negative [20; 21], suggesting an indirect regulation of IGFBP-2 by PKA is more likely. A possible intermediary transcriptional repressor is NF-κB p50/p50 homodimer. DNA binding by NF-κB p50/p50 was found to require phosphorylation of serine-337 by the catalytic subunit of PKA in HeLa cells, COS-7 cells and adenovirally transformed murine kidney cells [22]. Four NF-κB binding sites were identified in the rat IGFBP-2 promoter and were involved in hyperoxia-induced NF-κB activity in rat lung alveolar epithelial cells [23]. The fore-mentioned phosphorylation of NF-κB p50/p50 by PKA’s catalytic subunit, and consequent transcriptional repression, were found to be constitutive and cAMP-independent in cells at rest [22]. Such a mechanism may explain why forskolin failed to further decrease IGFBP-2 levels in the H295-R cells.
PKA was found to affect other members of the IGF axis in non-adrenal tissues. cAMP/PKA stimulation induced IGF-I expression in cultured embryonic mouse mandibular mesenchymal cells [24] and through an identified CRE in promoter 1 in primary rat osteoblasts [25]. A CRE was similarly identified in the promoter of IGFBP-1 that mediated induction by cAMP/PKA in hepatocytes [26]. cAMP induced IGFBP-3, -4 and –5 in periosteal and osteoblast bone cell cultures, though CREs were not investigated [27]. PKA inhibitors blunted IGFBP-4 induction by platelet derived growth factor-BB in fetal rat lung fibroblasts [28], and in porcine ovarian granulosa cells, IGF-I and IGFBP-3 regulation by growth hormone [29] and IGFBP-3 induction by follicle stimulating hormone [30].
Our report provides the first evidence of IGFBP-2 regulation by PKA. It is particularly noteworthy that it occurs in adrenocortical cells, where PKA, as second messenger for ACTH, plays such a critical role in normal development [31]. The actions of IGFBP-2 in normal and pathologic adrenocortical cells remain unclear. IGFBP-2, like the five other high affinity IGFBPs, is generally understood to be growth inhibitory through competitive inhibition of IGF binding to the type 1 IGF receptor [32], and hence, reduction of IGF survival and mitogenic signaling [33]. IGF-independent actions of the IGFBPs are also coming to light [34]. Intracellular functions of IGFBP-2 are suggested by its cytosolic uptake by colon carcinoma cell lines [35], nuclear translocation in hyperoxia (95% O2) exposed lung adenocarcinoma cell line [36], and isolation in the peri/nuclear fractions of tissues from IGFBP-2 transgenic and non-transgenic mice [37].
There is conflicting evidence supporting both pro- and anti-neoplastic actions of IGFBP-2 [35]. IGFBP-2 expression has been shown to be elevated in a variety of tumors, to often correlate positively with tumor grade and/or stage, and to be increased in the serum of these patients [35]. Such associative data cannot distinguish if the IGFBP-2 elevations are a marker of disease or a pathogenic agent. In vitro IGFBP-2 over-expression has had conflicting results; IGFBP-2 inhibited proliferation of multiple cell lines in IGF-dependent cell culture systems [35], but increased proliferation of Y-1 murine adrenocortical tumor cells [38] and DU145 prostate cancer cells [39] and enhanced invasiveness of ovarian [40] and bladder [41] cancer cell lines. Proposed mechanisms include induction of metalloproteinase-2 in invasive bladder cancer [41] and glioblastoma cells [42], increased catalase activity in the malignant Y-1 and Caco-2 human colon tumor cells [43], and interactions with integrins in Ewing’s sarcoma and breast cancer cells [44; 45]. IGFBP-2’s heparin binding domain was implicated in its enhancement of neuroblastoma cell migration and invasiveness [46], while its RGD motif was found to bind invasion inhibitory protein 45 (IIp45) and play a role in the invasiveness of glioblastoma multiforme cells [47]. Much remains to be elucidated about IGFBP-2 effects on cell proliferation and neoplasia, and the field is likely complicated by tissue-specific mechanisms and modifiers. We herein report the first evidence of IGFBP-2 regulation by PKA and its pro-proliferative effect on adrenocortical cells.
Fig. 3. Protein kinase A inhibitors increase IGFBP-2 mRNA in NCI-H295R cells.
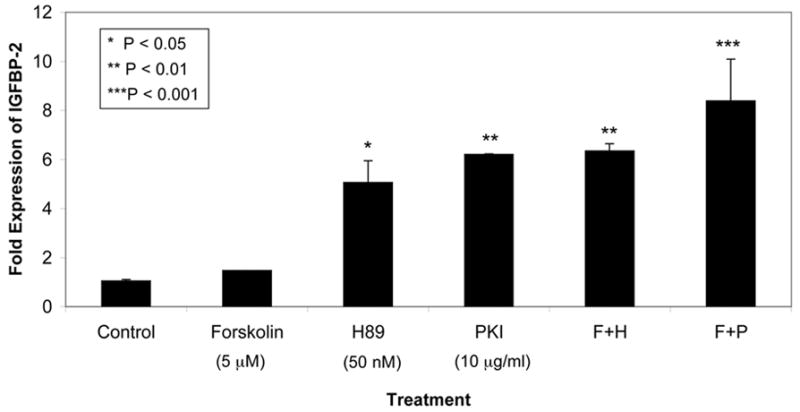
Real-time quantitative RT-PCR of IGFBP-2 message, normalized to PGK, in NCI-H295R adrenocortical cells treated with PKA modulators for 6 hr. Each bar depicts the mean ± SD of duplicate values expressed as fold-difference in IGFBP-2 mRNA level relative to untreated NCI-H295R cells plated at equal densities. The modulators were added singly or in combination at concentrations of 5 μM forskolin, 50 nM H89 and 10 μg/ml PKI. Difference among all treatments was highly significant (P<0.001, ANOVA).
Acknowledgments
This work was supported in part by NIH grants 5K08 DK64352 from the National Institute of Diabetes, Digestive and Kidney Disease (to A. Grimberg) and 1 Z01 HD000642-04 from the National Institute of Child Health and Human Development (to C.A. Stratakis). It was also supported by a Lester and Liesel Baker Foundation Award (to A. Grimberg) and T32 DK63688 (to M.J. Henwood).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shenoy BV, Carpenter PC, Carney JA. Bilateral primary pigmented nodular adrenocortical disease. Rare cause of the Cushing syndrome. Am J Surg Pathol. 1984;8:335–344. doi: 10.1097/00000478-198405000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Lacroix A, Bourdeau I. Bilateral adrenal Cushing's syndrome: macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin North Am. 2005;34:441–58. doi: 10.1016/j.ecl.2005.01.004. x. [DOI] [PubMed] [Google Scholar]
- 3.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86:4041–4046. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 4.Stratakis CA, Carney JA, Lin JP, et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest. 1996;97:699–705. doi: 10.1172/JCI118467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92. doi: 10.1038/79238. [DOI] [PubMed] [Google Scholar]
- 6.Groussin L, Cazabat L, Rene-Corail F, Jullian E, Bertherat J. Adrenal pathophysiology: lessons from the Carney complex. Horm Res. 2005;64:132–139. doi: 10.1159/000088586. [DOI] [PubMed] [Google Scholar]
- 7.Sutter JA, Grimberg A. Adrenocortical tumors and hyperplasias in childhood-etiology, genetics, clinical presentation and therapy. Pediatr Endocrinol Rev. 2006;4:366–373. [PMC free article] [PubMed] [Google Scholar]
- 8.Boulle N, Logie A, Gicquel C, Perin L, Le BY. Increased levels of insulin-like growth factor II (IGF-II) and IGF-binding protein-2 are associated with malignancy in sporadic adrenocortical tumors. J Clin Endocrinol Metab. 1998;83:1713–1720. doi: 10.1210/jcem.83.5.4816. [DOI] [PubMed] [Google Scholar]
- 9.Boulle N, Baudin E, Gicquel C, et al. Evaluation of plasma insulin-like growth factor binding protein-2 as a marker for adrenocortical tumors. Eur J Endocrinol. 2001;144:29–36. doi: 10.1530/eje.0.1440029. [DOI] [PubMed] [Google Scholar]
- 10.Logie A, Boulle N, Gaston V, et al. Autocrine role of IGF-II in proliferation of human adrenocortical carcinoma NCI H295R cell line. J Mol Endocrinol. 1999;23:23–32. doi: 10.1677/jme.0.0230023. [DOI] [PubMed] [Google Scholar]
- 11.Logie A, Boudou P, Boccon-Gibod L, et al. Establishment and characterization of a human adrenocortical carcinoma xenograft model. Endocrinology. 2000;141:3165–3171. doi: 10.1210/endo.141.9.7668. [DOI] [PubMed] [Google Scholar]
- 12.Rainey WE, Bird IM, Mason JI. The NCI-H295 cell line: a pluripotent model for human adrenocortical studies. Mol Cell Endocrinol. 1994;100:45–50. doi: 10.1016/0303-7207(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 13.Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney complex. Hum Mol Genet. 2000;9:3037–3046. doi: 10.1093/hmg/9.20.3037. [DOI] [PubMed] [Google Scholar]
- 14.Stratakis CA, Carney JA, Kirschner LS, et al. Synaptophysin immunoreactivity in primary pigmented nodular adrenocortical disease: neuroendocrine properties of tumors associated with Carney complex. J Clin Endocrinol Metab. 1999;84:1122–1128. doi: 10.1210/jcem.84.3.5549. [DOI] [PubMed] [Google Scholar]
- 15.Gunther DF, Bourdeau I, Matyakhina L, et al. Cyclical Cushing syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease or a new entity? J Clin Endocrinol Metab. 2004;89:3173–3182. doi: 10.1210/jc.2003-032247. [DOI] [PubMed] [Google Scholar]
- 16.Liu J, Li XD, Ora A, Heikkila P, Vaheri A, Voutilainen R. cAMP-dependent protein kinase activation inhibits proliferation and enhances apoptotic effect of tumor necrosis factor-alpha in NCI-H295R adrenocortical cells. J Mol Endocrinol. 2004;33:511–522. doi: 10.1677/jme.1.01535. [DOI] [PubMed] [Google Scholar]
- 17.Bertherat J, Groussin L, Sandrini F, et al. Molecular and functional analysis of PRKAR1A and its locus (17q22–24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res. 2003;63:5308–5319. [PubMed] [Google Scholar]
- 18.Robinson-White A, Hundley TR, Shiferaw M, Bertherat J, Sandrini F, Stratakis CA. Protein kinase-A activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum Mol Genet. 2003;12:1475–1484. doi: 10.1093/hmg/ddg160. [DOI] [PubMed] [Google Scholar]
- 19.Bossis I, Stratakis CA. Minireview: PRKAR1A: normal and abnormal functions. Endocrinology. 2004;145:5452–5458. doi: 10.1210/en.2004-0900. [DOI] [PubMed] [Google Scholar]
- 20. http://prospector.nci.nih.gov.
- 21.Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan H, Hou S, Ricciardi RP. DNA binding of repressor nuclear factor-kappaB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J Biol Chem. 2005;280:9957–9962. doi: 10.1074/jbc.M412180200. [DOI] [PubMed] [Google Scholar]
- 23.Cazals V, Nabeyrat E, Corroyer SKY, de Clement A. Role for NF-kappa B in mediating the effects of hyperoxia on IGF-binding protein 2 promoter activity in lung alveolar epithelial cells. Biochim Biophys Acta. 1999;1448:349–362. doi: 10.1016/s0167-4889(98)00095-0. [DOI] [PubMed] [Google Scholar]
- 24.Lambert HW, Weiss ER, Lauder JM. Activation of 5-HT receptors that stimulate the adenylyl cyclase pathway positively regulates IGF-I in cultured craniofacial mesenchymal cells. Dev Neurosci. 2001;23:70–77. doi: 10.1159/000048697. [DOI] [PubMed] [Google Scholar]
- 25.Thomas MJ, Umayahara Y, Shu H, Centrella M, Rotwein P, McCarthy TL. Identification of the cAMP response element that controls transcriptional activation of the insulin-like growth factor-I gene by prostaglandin E2 in osteoblasts. J Biol Chem. 1996;271:21835–21841. doi: 10.1074/jbc.271.36.21835. [DOI] [PubMed] [Google Scholar]
- 26.Suwanichkul A, DePaolis LA, Lee PD, Powell DR. Identification of a promoter element which participates in cAMP-stimulated expression of human insulin-like growth factor-binding protein-1. J Biol Chem. 1993;268:9730–9736. [PubMed] [Google Scholar]
- 27.Chen Y, Shu H, Ji C, et al. Insulin-like growth factor binding proteins localize to discrete cell culture compartments in periosteal and osteoblast cultures from fetal rat bone. J Cell Biochem. 1998;71:351–362. [PubMed] [Google Scholar]
- 28.Price WA. PDGF-BB regulates IGF-mediated IGFBP-4 proteolysis in fetal lung fibroblasts. Exp Lung Res. 2001;27:655–674. doi: 10.1080/019021401317138469. [DOI] [PubMed] [Google Scholar]
- 29.Sirotkin AV, Makarevich AV. Growth hormone can regulate functions of porcine ovarian granulosa cells through the cAMP/protein kinase A system. Anim Reprod Sci. 2002;70:111–126. doi: 10.1016/s0378-4320(01)00196-8. [DOI] [PubMed] [Google Scholar]
- 30.Ongeri EM, Verderame MF, Hammond JM. Follicle-stimulating hormone induction of ovarian insulin-like growth factor-binding protein-3 transcription requires a TATA box-binding protein and the protein kinase A and phosphatidylinositol–3 kinase pathways. Mol Endocrinol. 2005;19:1837–1848. doi: 10.1210/me.2004-0487. [DOI] [PubMed] [Google Scholar]
- 31.Hammer GD, Parker KL, Schimmer BP. Minireview: transcriptional regulation of adrenocortical development. Endocrinology. 2005;146:1018–1024. doi: 10.1210/en.2004-1385. [DOI] [PubMed] [Google Scholar]
- 32.Carrick FE, Hinds MG, McNeil KA, Wallace JC, Forbes BE, Norton RS. Interaction of insulin-like growth factor (IGF)-I and -II with IGF binding protein-2: mapping the binding surfaces by nuclear magnetic resonance. J Mol Endocrinol. 2005;34:685–698. doi: 10.1677/jme.1.01756. [DOI] [PubMed] [Google Scholar]
- 33.Grimberg A. Mechanisms by which IGF-I may promote cancer. Cancer Biol Ther. 2003;2:630–635. [PMC free article] [PubMed] [Google Scholar]
- 34.Grimberg A, Cohen P. Role of insulin-like growth factors and their binding proteins in growth control and carcinogenesis. J Cell Physiol. 2000;183:1–9. doi: 10.1002/(SICI)1097-4652(200004)183:1<1::AID-JCP1>3.0.CO;2-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoeflich A, Reisinger R, Lahm H, et al. Insulin-like growth factor-binding protein 2 in tumorigenesis: protector or promoter? Cancer Res. 2001;61:8601–8610. [PubMed] [Google Scholar]
- 36.Besnard V, Corroyer S, Trugnan G, et al. Distinct patterns of insulin-like growth factor binding protein (IGFBP)-2 and IGFBP-3 expression in oxidant exposed lung epithelial cells. Biochim Biophys Acta. 2001;1538:47–58. doi: 10.1016/s0167-4889(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 37.Hoeflich A, Reisinger R, Schuett BS, et al. Peri/nuclear localization of intact insulin-like growth factor binding protein-2 and a distinct carboxyl-terminal IGFBP-2 fragment in vivo. Biochem Biophys Res Commun. 2004;324:705–710. doi: 10.1016/j.bbrc.2004.09.111. [DOI] [PubMed] [Google Scholar]
- 38.Hoeflich A, Fettscher O, Lahm H, et al. Overexpression of insulin-like growth factor-binding protein-2 results in increased tumorigenic potential in Y-1 adrenocortical tumor cells. Cancer Res. 2000;60:834–838. [PubMed] [Google Scholar]
- 39.Chatterjee S, Park ES, Soloff MS. Proliferation of DU145 prostate cancer cells is inhibited by suppressing insulin-like growth factor binding protein-2. Int J Urol. 2004;11:876–884. doi: 10.1111/j.1442-2042.2004.00898.x. [DOI] [PubMed] [Google Scholar]
- 40.Lee EJ, Mircean C, Shmulevich I, et al. Insulin-like growth factor binding protein 2 promotes ovarian cancer cell invasion. Mol Cancer. 2005;4:7. doi: 10.1186/1476-4598-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miyake H, Hara I, Yamanaka K, Muramaki M, Gleave M, Eto H. Introduction of insulin-like growth factor binding protein-2 gene into human bladder cancer cells enhances their metastatic potential. Oncol Rep. 2005;13:341–345. [PubMed] [Google Scholar]
- 42.Wang H, Wang H, Shen W, et al. Insulin-like growth factor binding protein 2 enhances glioblastoma invasion by activating invasion-enhancing genes. Cancer Res. 2003;63:4315–4321. [PubMed] [Google Scholar]
- 43.Hoeflich A, Fettscher O, Preta G, et al. Increased activity of catalase in tumor cells overexpressing IGFBP-2. Horm Metab Res. 2003;35:816–821. doi: 10.1055/s-2004-814163. [DOI] [PubMed] [Google Scholar]
- 44.Pereira JJ, Meyer T, Docherty SE, et al. Bimolecular interaction of insulin-like growth factor (IGF) binding protein-2 with alphavbeta3 negatively modulates IGF-I-mediated migration and tumor growth. Cancer Res. 2004;64:977–984. doi: 10.1158/0008-5472.can-03-3056. [DOI] [PubMed] [Google Scholar]
- 45.Schutt BS, Langkamp M, Rauschnabel U, Ranke MB, Elmlinger MW. Integrin-mediated action of insulin-like growth factor binding protein-2 in tumor cells. J Mol Endocrinol. 2004;32:859–868. doi: 10.1677/jme.0.0320859. [DOI] [PubMed] [Google Scholar]
- 46.Russo VC, Schutt BS, Andaloro E, et al. Insulin-like growth factor binding protein-2 binding to extracellular matrix plays a critical role in neuroblastoma cell proliferation, migration, and invasion. Endocrinology. 2005;146:4445–4455. doi: 10.1210/en.2005-0467. [DOI] [PubMed] [Google Scholar]
- 47.Song SW, Fuller GN, Khan A, et al. IIp45, an insulin-like growth factor binding protein 2 (IGFBP-2) binding protein, antagonizes IGFBP-2 stimulation of glioma cell invasion. ProcNatlAcadSciUSA. 2003;100:13970–13975. doi: 10.1073/pnas.2332186100. [DOI] [PMC free article] [PubMed] [Google Scholar]