

Abstract
The expression of 4 pluripotency genes (Oct4, Sox2, c-Myc and Klf4) in mouse embryonic fibroblasts can reprogramme them to a pluripotent state. We have investigated the expression of these pluripotency genes when human somatic 293T cells are permeabilised and incubated in extracts of mouse embryonic stem (ES) cells. Expression of all 4 genes was induced over 1 - 8 hours. Gene expression was associated with loss of repressive histone H3 modifications and increased recruitment of RNA polymerase II at the promoters. Lamin A/C, which is typically found only in differentiated cells, was also removed from the nuclei. When 293T cells were returned to culture after exposure to ES cell extract, the expression of the pluripotency genes continued to rise over the following 48 hours of culture, suggesting that long-term reprogramming of gene expression had been induced. This provides a methodology for studying the de-differentiation of somatic cells that can potentially lead to an efficient way of reprogramming somatic cells to a pluripotent state without genetically altering them.
Keywords: reprogramming, pluripotent, embryonic stem cells, gene expression, Oct-4 expression levels
INTRODUCTION
Reprogramming of differentiated somatic cells into pluripotent cell is a promising approach for regenerative medicine. The ability to reprogram somatic cells was first demonstrated in mammals by the generation of animals after nuclear transfer [1-3] and secondly by the derivation of embryonic stem cells (ES) from the inner cell mass (ICM) cells of SCNT embryos [4]. It has also been reported that embryonic stem (ES) cells, embryonal carcinoma (EC) cells and embryonic germ (EG) cells can induce nuclear reprogramming after hybridisation with somatic cells [5-8].
In a different approach Takahashi et al [9] showed that when over-expressed in mouse embryonic fibroblasts (MEFs), 4 genes (Oct4, Sox2, c-Myc and Klf4) could induce the morphology and growth properties of embryonic stem (ES) cells. The DNA methylation, gene expression and chromatin state of these induced pluripotent stem (iPS) cells were also similar to those of ES cells [9-12]. Further, iPS cells could form viable chimaeras, including contribution to the germ line [10-12]. Similar results have recently been reported for somatic cells [13-16]. These results suggest that to successfully re-programme somatic cells, expression of only these 4 genes is needed.
It would be advantageous to develop reprogramming methods that do not alter the genetic content of the reprogrammed cells. One approach used by Collas and co-workers is to transiently permeabilize the plasma membrane of cells with streptolysin-O (SLO), after which the membrane can spontaneously reseal so that cells can continue growth in culture [17]. When differentiated SLO-permeabilised cells were briefly incubated in ES cell extracts and then returned to cell culture for several weeks, colonies were obtained that had been stably reprogrammed to express a range of pluripotency genes [18, 19]. One drawback of these studies is that it is difficult to estimate the efficiency of reprogramming because the successive changes of medium leads to a selection for proliferating cells. Long incubations in culture also make it more time-consuming to analyse systematically factors involved in the reprogramming of gene expression.
In this paper, we have examined changes that occur directly after exposure of SLO-permeabilized somatic cells to ES cell extract. We demonstrate that the induction of the 4 key pluripotency genes (Oct4, Sox2, c-Myc and Klf4) can be detected within a few hours of ES cell treatment. This opens up a range of in vitro approaches to analysing biochemical mechanisms involved in the de-differentiation of somatic cells and that could ultimately lead to the development of new methods for efficiently reprogramming somatic cells.
MATERIALS AND METHODS
Cells
NCCIT and 293T (American Type Culture Cells, ATCC) were cultured in RPMI 1640 medium (Invitrogen) with 10% fetal calf serum (FCS), 2 mM L-glutamine, 1 mM sodium pyruvate, nonessential amino acids (complete RPMI 1640 medium) and 1% penicillin/streptomycin. NIH 3T3 (ATCC) were cultured in DMEM (Invitrogen) with 4.5 g/L L-glucose, 4 mM L-glutamine, 10% FCS, 1.5 g/L sodium bicarbonate and 1% penicillin/streptomycin. Mouse 46C and J1 ESCs (ATCC) were cultured in GMEM (Invitrogen) with 10% FCS, 2 mM sodium pyruvate, 0.1 mM β-mercaptoethanol, non essential amino acids, 1% penicillin/Streptomycin and 1000 U/ml (10 ng/ml) recombinant leukaemia inhibitory factor (LIF; Sigma) on gelatine-coated plates. MEFs (from CD1 foetuses, used at passage number 3 or 4) were cultured in DMEM with 10% FCS and 1% penicillin/streptomycin. Cells treated with extracts were seeded at 100,000 cells per well in a 24-well plate and cultured in 500 μl of complete RPMI 1640 medium with antibiotics.
Cell Extracts
To prepare ES or 3T3 cell extracts, cells were washed twice in PBS and once in cell lysis buffer (20 mM HEPES pH 8.2, 50 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.1% protease inhibitor cocktail (Sigma), sedimented at 400xg, resuspended in 0.8 vols cold cell lysis buffer and incubated 45 min on ice. Cells were sonicated on ice in 2 ml aliquots using a Soniprep 150 sonicator fitted with a 3-mm-diameter probe (MSE) until all cells and nuclei were lysed, as judged by microscopy. The lysate was cleared at 15,000xg for 15 min at 4°C and the supernatant frozen in beads with 2% glycerol and stored in liquid nitrogen. One bead of extract was used to determine protein concentration (∼30 mg/m) and pH (∼7.0).
Activated Xenopus egg extract was prepared as described [20]. Oocyte extract was prepared from ovaries rinsed in Barth buffer (88 mM NaCl, 2 mM KCl, 1 mM MgCl2, 15 mM Tris-HCl pH 7.6, 0.5 mM CaCl2) and digested 6 hours at room temperature in 5 volumes OR-2 buffer (82 mM NaCl, 2.5 mM KCl, 1 mM MgCl2, 5 mM HEPES pH 7.5) containing 0.2% collagenase type I (Sigma) with occasional agitation. Stage 4-6 oocytes were then selected on size, washed 4x in Barth buffer and 2x in extraction buffer (50 mM KCl, 50 mM HEPES, pH 7.4, 5 mM MgCl2, 2 mM β-mercaptoethanol) and prepared as for egg extracts. Xenopus extracts contained 40 - 60 mg/ml protein.
SLO-permeabilization and cell extract treatment
Cells were washed 2x in PBS, 1x in cold Acetate Buffer (115 mM KO Acetate, 25 mM HEPES pH 7.4, 2.5 mM KCl) and were incubated for 50 min in Acetate Buffer + 10 mM DTT + 1.5 μg/ml pre-activated SLO at 1000 cells/μl at 37°C with occasional agitation. Permeabilization was assessed by monitoring uptake of Trypan Blue (Invitrogen).
After permeabilization, cells were resuspended at 1000 cells/μl in 100 μl cell extracts containing 2 mM ATP, 10 mM creatine phosphate, 25 μg/ml creatine kinase; Sigma), 1.1 mM GTP, 1 mM of both CTP and UTP (Roche Diagnosis), 50 μg/ml ampicillin and 15 μg/ml fungin (optionally supplemented with 100 μM α-amanitin or 1 mg/ml cycloheximide) and incubated at 37°C (23°C for Xenopus extracts) in a H2O bath with occasional agitation. For membrane resealing, extract was diluted with 1 ml RPMI 1640 + 2 mM CaCl2, and incubated for 2 hr at 37°C in a H2O bath. Cells were pelleted through 100 μl sterile HBSS (Invitrogen) containing 10% sucrose at 400xg in a swinging bucket centrifuge for 5 min at 4°C. Pelleted cells was resuspended in 1 ml RPMI 1640 and seeded on 24-well plates. After culture, both floating and adherent cells were collected for analysis.
Real-time Reverse Transcription (RT)-PCR
Total RNA was isolated using a Qiagen RNeasy Mini kit with a DNaseI treatment during the purification and eluted in 35 μl DEPC-treated water. RT was performed with 8 μl total RNA using the Superscript First-Strand synthesis System (Invitrogen) and random hexamers. Quantitative RT-PCR reactions were performed in triplicates on an IQ5 real-time PCR detection system (BioRad). SYBR Green PCR conditions were 95°C for 5 min and 40 cycles of 95°C for 15 s, 60°C or 58°C for 10 s and 72°C for 20 s using Lamin B2 as standard. Primers and conditions are described in Supplementary Figure S1.
Chromatin Immunoprecipitation
Cells were fixed in 1 ml 1% formaldehyde, 50 mM Hepes, 50 mM KCl, 5 mM MgCl2, 2 mM DTT, protease inhibitors, pH 7.5 and pelleted through 10% sucrose and resuspended in 1% SDS, 10 mM EDTA, 50 mM Tris, pH 8, plus protease inhibitors. Following incubation on ice for 10 min, chromatin was sheared by sonication (Misonix microson XL2000) with 5×10 s bursts at 7-8W and with >1 min on ice between bursts. Insoluble material was pelleted at 13,000xg for 5 min and supernatant was diluted 10x with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 167 mM NaCl, 1.2 mM EDTA, 16.7 mM Tris, pH 8). Supernatant was precleared for 1 hour with salmon sperm DNA plus Protein A (Upstate 16-157C). Chromatin from 2×105 cells was incubated overnight at 4°C with antibodies, which were collected with 50 μl Protein A beads, and washed (low salt wash: 150 mM NaCl, 0.1% SDS, 1% Triton, 2 mM EDTA, 20 mM Tris, pH 8; high salt wash: as low salt wash except 500 mM NaCl; LiCl buffer: 0.25M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris, pH 8; TE: 1 mM EDTA, 10 mM Tris, pH 8) and eluted in 400 μl 1% SDS, 0.1 M sodium bicarbonate for 30 min at room temperature. Formaldehyde cross-links were reversed in 0.2 M NaCl, 4 hours at 65°C. Proteins were degraded with proteinase K (40 μg/ml plus 40 mM Tris, 10 mM EDTA pH 8, at 45°C, 1 hr) and DNA was recovered by phenol/chloroform extraction (using Eppendorf LPG tubes) and ethanol precipitation (0.1 M sodium acetate pH 5.8, 2 μg glycogen). Pellets were resuspended in 40 μl water. Usually, 1 μl was used for PCR reactions. The quantity of DNA used for the PCR was first titrated to find a range where the PCR signal is semi-quantitative, and then samples were normalised to levels of GAPDH.
Antibodies
ChIP antibodies: Histone H3-K4 trimethylated (Upstate, MC315 05-745), histone H3-K9 acetylated (Upstate, 07-352), histone H3-K9 dimethylated (Upstate, 07-441), histone H3-K9 trimethylated (Upstate, 07-442), histone H3-K27 dimethylated (Upstate, 07-452), histone H3-K27 trimethylated (Upstate, cat no. 07-449), RNA polymerase II (Santa Cruz, sc-899X; used at 10-20 μg/IP). Upstate antibodies were used at 2-5 μg/IP. Western Blot and Immunofluorescence antibodies: Lamin A/C antibody used at 1/1000 for immunofluorescence (Eurogentec, MUB1102) and 1/2500 for western blot (Santa Cruz); Lamin B1 antibody (Santa Cruz, sc-6217) used at 1/300 for immunofluorescence and 1/2500 for western blot; Oct4 antibody (Santa Cruz) at 1/1000; and Nanog antibody (Chemicon) at 1/10000.
Immunofluorescence
MEFs attached to 6 mm 10-well slides (VWR) were washed 2x with PBS, once with Acetate Buffer then incubated in SLO for 50 mins at 37°C as described above. Permeabilisation buffer was aspirated and cells were covered with 40 μl extract and incubated for 3 hours at 23°C for Xenopus egg extract or 37°C for ES cell extract. Cells were gently washed with PBS, fixed in 4% formaldehyde/PBS for 20 mins, permeabilised with 0.2% Triton for 10 mins and blocked with 2% BSA for 1 hr. Incubations of primary antibodies were carried out overnight at 4°C. Secondary antibodies were FITC-conjugated anti-mouse antibody for Lamin A and Cy-3-conjugated anti-goat antibody for Lamin B.
RESULTS
Removal of lamin A/C from somatic cells by ES cell extracts
Lamin A/C is a major component of the nuclear lamina that is expressed in differentiated but not in pluripotent cells (Figure 1A). Lamin A/C can be removed from permeabilised somatic cells by incubation in extracts of oocytes or eggs of the frog Xenopus laevis [21]. We investigated the ability of ES cell extracts to remove lamin A/C from permeabilised MEFs. Figure 1B and C show that extracts prepared from 46C mouse ES cell extract removed lamin A/C from nuclei in >55% of MEF cells. In contrast, the levels of lamin B, which is expressed in both ES and somatic cells, remained constant. The degree of lamin A/C removal was comparable to that seen with Xenopus egg extract. Similar to results with Xenopus extracts [21], when ATP was depleted from the ES cell extract by addition of apyrase, the percentage of cells undergoing lamin A/C removal was strongly reduced. This suggests that nuclear lamin remodelling is an energy-dependent process and can be promoted by extracts prepared from mouse ES cells.
Figure 1.

Removal of lamin A/C from cells incubated in ES cell or Xenopus egg extracts.
(A) Western blot of Nanog, Oct4, Lamin A/C and Lamin B in MEF and 46C ES cells. (B, C) MEF cells were permeabilized and incubated in 46C ES cell extract or Xenopus egg extract for 3 hours in plus or minus apyrase. Cells were then stained for lamin A/C (FITC, green), lamin B2 (Cy-3, red) or DNA (DAPI, blue). Quantification in (C) was performed by visual inspection. Scale bar, 5μm.
ES cell extracts promote pluripotency-associated gene expression
We next investigated whether mouse ES cell extracts could induce somatic cells to induce transcription of genes associated with pluripotency. Using real time PCR we showed that, as expected, the mRNA levels of the pluripotency-associated genes Oct4, Nanog, Sox2, c-Myc and Klf4 was higher in human undifferentiated embryonal carcinoma cells (NCCIT) than in differentiated human embryonic kidney 293T cells (Figure 2A). In contrast, the lamin A gene (LMNA) was down-regulated in NCCIT cells (Figure 2A). Lamin B2 (LMNB2), ubiquitously expressed in all cells, was used as normalization control. The primers we used were designed to be specific for the human genes, and were predicted not to amplify their mouse homologues. This specificity was confirmed by the absence of amplification of cDNAs from mouse ES or 3T3 cells (Figure 2A). Using a different set of primers capable of amplifying the mouse homologues of these genes we observed low expression levels of the pluripotency genes in somatic 3T3 cells (derived from embryonic fibroblasts) compared with high levels in 2 pluripotent mouse ES cell lines, 46C and J1 (Figure 2B).
Figure 2.

Activation of pluripotency genes in permeabilized 293T cells exposed to mouse ES cell extracts.
(A, B) Quantitative RT-PCR analysis of indicated genes in EC or ES cells relative to differentiated 293T or 3T3 cells. In A, human specific-primers were used, whilst in B, primers recognising both human and mouse genes were used. NA - no amplification. (C-F) Quantitative RT-PCR analysis using human-specific primers of gene expression in 293T cells treated for 0 to 8 hours with: (C, F) mouse J1 ES cell extract, (D) mouse 46C ES cell extract and (E) mouse 3T3 cell extract. In (F), unpermeabilised 293 cells were used.
We next optimised the SLO treatment of 293T cells to give ∼80% cell permeabilization (Supplementary Figure S2) and verified that this allowed the incorporation of both 10 kDa and 70 kDa dextrans (data not shown). Consistent with previous reports [17, 19], the majority of cells appeared viable after this treatment, and were capable of forming colonies. This therefore provided a mechanism for exposing differentiated cell nuclei to extracts with reprogramming potential, and then returning the cells to culture.
After SLO-permeabilization, 293T cells were incubated in extracts of mouse 46C or J1 ES cells. Using the human specific primers, we detected the induction of Oct4, Klf4, Nanog, Sox2 and c-Myc mRNA during the 8 hr of incubation (Figure 2C and D). In contrast, expression of LMNA, normally down-regulated in ES cells, remained stable. All PCR products were verified by restriction digestion to confirm that expression of the human genes was occurring (Supplemental Figure S3). As negative control we also showed that incubation of permeabilized 293T cells in somatic 3T3 cell extract caused no significant change in expression of the pluripotency genes (Figure 2E). Further, no significant change in gene expression was observed in non-permeabilised 293T cells after incubation in ES cell extract (Figure 2F).
Since Xenopus egg and oocyte extracts can remove lamin A/C from somatic nuclei (Figure 1 and [21]), we also investigated whether they could induce expression of pluripotency genes in permeabilised 293T cells. After permeabilization with SLO, 293T cells were exposed for 0 to 8 hours to either Xenopus egg (Figure 3A) or oocyte (Figure 3B) extracts. In contrast to ES cell extracts, no significant change of expression was observed for any of our pluripotency marker genes (Oct4, Nanog, Sox2, c-Myc, Klf4) or for LMNA.
Figure 3.

Absence of reprogramming activity in Xenopus egg or oocyte extracts.
Quantitative RT-PCR analysis of expression of indicated genes in 293T cells treated for 0 to 8 hours with: (A) Xenopus egg extract, (B) Xenopus oocyte extract or (C) mixes of ES cell and Xenopus oocyte extracts for 6 hours.
One possible explanation for the inactivity of Xenopus extracts is that one or more factors involved in the reprogramming process is unable to recognise a key target in human cells. To test this, mixtures with different ratios of mouse ES and Xenopus oocytes extracts were used to treat permeabilised 293T cells. Figure 3C shows that, consistent with our previous experiments, ES cell extract induced the expression of Nanog, Oct4, Sox2, c-Myc and Klf4. As the ES extract was replaced with Xenopus oocyte extract, the expression of all these genes declined. We therefore obtained no evidence that Xenopus oocyte extracts provide any stimulation of the activity provided by the mouse ES cell extract.
ES cell extracts induce RNA polymerase II binding at Nanog and Oct4 promoters
The binding of RNA polymerase II to a promoter is an essential step in the commitment to initiate transcription. We used chromatin immunoprecipitation (ChIP) to analyse the loading of RNA polymerase II onto promoters of pluripotency genes (Figure 4A and B). This revealed a modest increase in binding of RNA polymerase II at the Nanog and Oct4 promoters. Increased loading of RNA polymerase was also detected on the promoter of the Cripto gene, which is also specifically expressed in undifferentiated cells [22]. Induction of pluripotency genes was blocked by addition to the 46C ES cell extract of α-amanitin, an inhibitor of RNA polymerase II (Figure 4C). Taken together, these results strongly suggest that a range of pluripotency genes are induced when permeabilised 293T cells are of incubated in ES cell extract.
Figure 4.
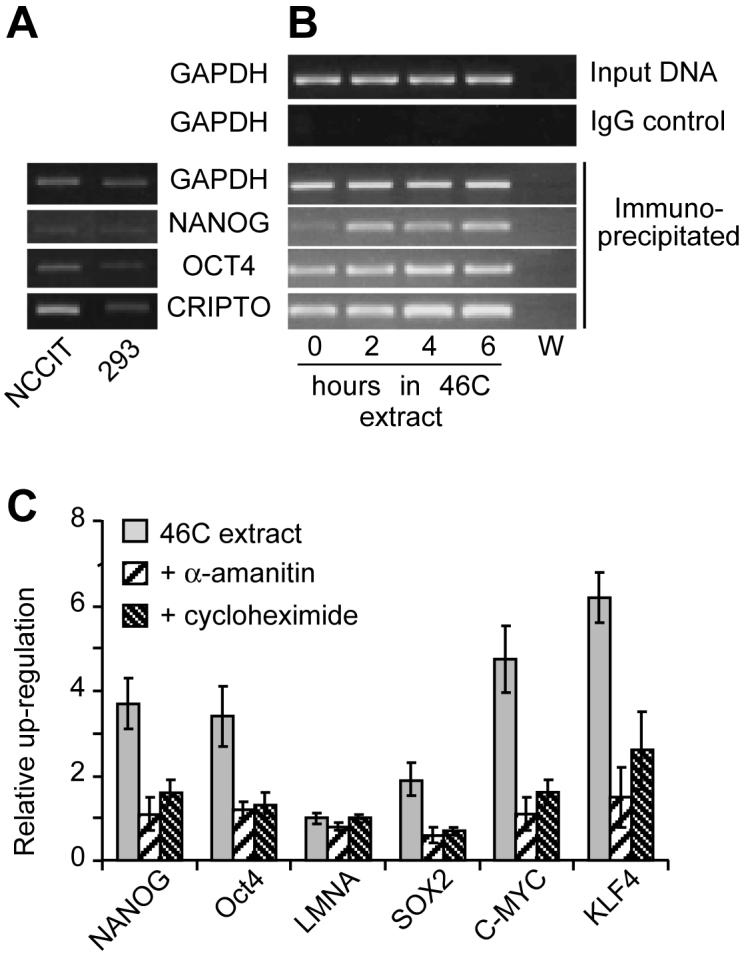
Efficient induction of pluripotency genes depends on transcription and translation.
(A, B) Chromatin immunoprecipitation, using an RNA polymerase II antibody, on either untreated NCCIT or 293 cells (A) or 293T cells incubated in 46C ES cell extract for 0 to 6 hours (B). Chromatin immunoprecipitates were subjected to PCR using primers specific for GAPDH, Nanog (promoter 1), Oct4 and CRIPTO. W, water only control for PCR reaction. The Nanog promoter (C) Quantitative RT-PCR analysis after incubation of 293T cells for 6 hours in mouse 46C ES extract. The ES extract was optionally supplemented with 100 μM α-amanitin or 1 mg/ml cycloheximide at the start of the incubation. Expression is expressed relative to untreated 293T cells.
Figure 4C also shows that addition of cycloheximide, an inhibitor of protein synthesis, to the ES cell extracts reduced the efficiency by which gene expression was induced. One possible explanation for this is that translation of key transcriptional regulators is rapidly induced during the incubation of somatic cells in ES cell extracts and is necessary for efficient activation of pluripotency genes.
Mouse ES cell extracts modify histone H3 on promoters of pluripotency genes
The post-translational modification of histone N termini, most notably on histone H3, plays a key role in regulating transcriptional activation or repression of genes [23, 24]. We used antibodies against acetylated and methylated lysines on histone H3 to perform ChIP on 293T cells before and after exposure to ES extracts. Figure 5A shows that the Nanog and Cripto promoters in pluripotent NCCIT cells had more acetylated H3K9 and trimethylated H3K4, typically associated with active promoters, than the same promoters in 293T cells. Conversely, dimethylated H3K9, which is typically associated with a repressed chromatin state, was more abundant on Nanog and Oct4 promoters in 293T than NCCIT cells. Trimethylated H3K27, also typically associated with a repressed chromatin state, was more abundant on the Cripto and Oct4 promoters in 293T cells than NCCIT cells. All these differences are consistent with higher activity of the Nanog, Oct4 and Cripto promoters in NCCIT cells [18]. In contrast, no significant differences were detected between NCCIT and 293T cells for any histone H3 modification on the GAPDH promoter.
Figure 5.

46C ES cell extract induces histone modifications at pluripotency gene promoters.
Chromatin immunoprecipitation was performed on (A) NCCIT and 293T cells or (B-D) on permeabilized 293T cells exposed to mouse ES cell extract for 0, 2, 4 or 6 hr. Chromatin was precipitated with antibodies to histone H3 modifications and DNA was amplified using primers against GAPDH, Nanog (promoter 2), Oct4 and CRIPTO. W, water only control for PCR reaction. In B-D, input DNA and chromatin precipitated with non-immune IgG
Treatment of 293T cells with 46C ES cell extract reduced the methylation of H3K9 (Figure 5C) and H3K27 (Figure 5D) on the Oct4 promoter and reduced methylation of H3K9 on the Nanog promoter (Figure 5C), consistent with them becoming de-repressed. There was also slight reduction in the methylation of H3K9 and H3K27 on the Cripto promoter. In contrast, there was little evidence for the appearance of the activating chromatin modifications, trimethylated H3K4 and acetylated H3K9 on these promoters (Figure 5B). Some transient changes were detected to the level of these modifications but were not maintained. These results suggest that treatment of somatic nuclei with ES cell extract lowers the level of certain repressive chromatin modifications, but does not strongly induce activating modifications. This is consistent with a partial chromatin remodelling that might be sufficient to allow the regulatory regions of some pluripotency genes to recruit RNA polymerase II.
The expression of pluripotent marker genes increases after treatment
To determine whether exposure to ES cell extract elicits a long-term induction of pluripotent genes, 293T cells were incubated in 46C ES cell extract for 6 hours, resealed and returned to cell culture. The level of marker gene expression was quantified by real time PCR after 16, 21, 41 or 48 hours in culture. Figure 6 shows the abundance of the different mRNA levels normalised to the abundance in untreated 293T cells. Over the whole 48 hours in culture, a continued up-regulation was observed for Nanog, Oct4, Sox2, c-Myc and Klf4 genes (note log scale). LMNA levels showed no significant changes over this period of time. The continued and increasing expression of pluripotency genes suggests that exposure of 293 nuclei to ES cell cytoplasm can induced a long-term induction of gene expression that drives the cells towards a more pluripotent state.
Figure 6.
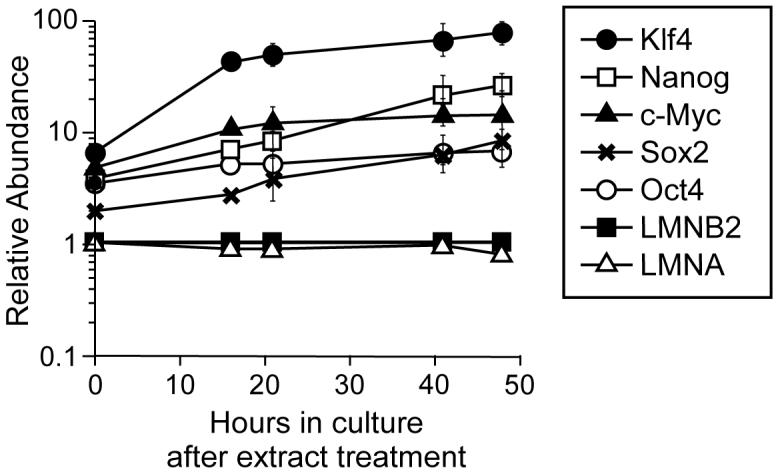
Reprogrammed gene expression increases after return to cell culture.
Human 293T cells were permeabilized with SLO and incubated for 6 hours in 46C ES cell extract. Cell membranes were then allowed to reseal for 2 hours, and the cells were returned to tissue culture medium. At the indicated times afterwards, quantitative RT-PCR was performed on selected genes. Expression was normalised to the level of lamin B2 expression and is expressed relative to levels in untreated 293T cells. Error bars show the standard deviation from 3 replicates.
DISCUSSION
This study provides insight into the molecular changes that take place during reprogramming of gene expression in mammalian somatic cells exposed to ES cell extracts and establishes a new approach for the identification of the active factors involved in this process. Exposure of transiently permeabilised human epithelial 293T cells to mouse ES cell extract induced loss of lamin A/C, activation of pluripotency gene expression and histone modification at the promoters of activated genes. These effects were maintained, and in fact increased, after treated cells were returned to culture, suggesting that long-term reprogramming of gene expression in the somatic cells had occurred.
In our experiments, 293T cells were permeabilized using SLO, after which cell membranes can reseal and cells can continue to grow in culture [17]. Consistent with work by Taranger et al [19] but in contrast to a separate report [25] we saw no induction of gene expression in the absence of membrane permeabilization. Genes that were up-regulated included Klf4, Oct4, Sox2 and c-Myc. It has recently been shown that forced expression of these genes in adult mouse fibroblasts is sufficient to turn them into pluripotent stem cells [9-12]. The changes that we have induced are therefore likely to have started to drive the epithelial 293T cells towards full pluripotency. We also observed up-regulation of Nanog, which along with Oct4 has been shown to function as a core transcription factor in maintaining pluripotency [26, 27]. Consistent with these changes being due to an induction of transcriptional activity, we detected increased RNA polymerase II binding to the promoters and showed that mRNA expression was inhibited by α-amanitin, an inhibitor of RNA polymerase II.
After exposure of 293T cells to mouse ES cell extract for 1 to 8 hours, a steady up-regulation of expression of pluripotency genes was observed. When the membranes were allowed to reseal and treated cells were returned to culture, the expression levels of pluripotency genes continued to rise over the following 48 hours, suggesting that long-term reprogramming of gene expression had occurred. The induction of Klf4 (∼70-fold) and c-Myc (∼12-fold) approached that of ES cells. However, induction of Sox2 (∼7-fold), Oct4 (∼6-fold) and Nanog (∼24-fold) was still significantly lower than the ∼200-fold, ∼250-fold and ∼1000-fold observed in the ES cells. This suggests that full reprogramming to a pluripotent state had not been achieved, but that exposure to ES cell extract had initiated a series of changes driving the somatic cells towards a more pluripotent state. Consistent with this interpretation, we observed chromatin changes over pluripotency gene promoters suggestive of a loss of transcriptional repression (demethylation of H3K9 and, to a lesser extent, of H3K27) but not of strong transcriptional activation (acetylation of H3K9 or methylation of H3K4). Similarly, although ES cell extracts reduced levels of lamin A/C protein in the permeabilized 293T cells, we did not observe a significant decrease in the expression of the LMNA gene as would be expected of pluripotent cells [19].
Microinjection of somatic cells into living Xenopus oocyte nuclei induces Oct4 gene expression and promoter demethylation after a period of 2 days [28-30]. We therefore investigated whether Xenopus egg and oocyte extracts could also induce pluripotency gene expression in permeabilised 293T cells. Unfortunately, we observed no induction of pluripotency genes relative to expression levels of housekeeping genes and in addition, no stimulation of gene expression was observed when Xenopus egg extract was supplemented with different quantities of ES cell extract. Our results are in contrast to the work of Hansis et al [25], who exposed digitonin-treated cells to Xenopus extract for 30 minutes and observed reprogramming of Oct4 expression after four days in media. We were unable to repeat this result (data not shown).
Our results build upon the work of Collas and colleagues who also used extracts of ES cells to reprogram SLO-permeabilized somatic cells [18, 19, 31]. In these studies, treated cells were grown out in culture for several weeks, after which cells could be identified that had been stably reprogrammed to express pluripotency genes. Freberg et al [18] also reported chromatin changes similar to the ones we report here. Because there is a long period of culture involved in this procedure, it is hard to determine the efficiency of reprogramming as an unknown proportion of the original somatic cells will be retained in culture. A second disadvantage of this protocol is that the long culture time means that experiments are relatively slow, making further biochemical analysis of the ES cell extracts hard to perform.
Our study demonstrates that the first stages of reprogramming do not depend on long-term culture and occur during short incubations. One limitation of the current study is that we have only investigated the response of one particular cell line (293T cells) to ES cell cytoplasm. Further work to validate this approach needs to determine the range of cell types that will respond in similar ways. This could open up a range of further in vitro approaches to study biochemical mechanisms involved in this process. We assayed changes in the total population of treated somatic cells and did not remove non-adherent cells when we returned the cells to culture. The extent to which we observe induction of pluripotency genes is therefore likely to represent a minimum value, since if only a proportion of permeabilized cells responded to the ES extract after treatment, the real level of expression of marker genes in these cells will have been under-estimated. Our approach provides a way of analysing features of the reprogramming process by, for example, fractionating the ES cell extracts to identify the factors involved activities. Use of such activities could eventually lead to an efficient way of reprogramming somatic cells to a pluripotent state without genetically altering them.
Supplementary Material
ACKNOWLEDGEMENTS
Thanks to Neil Perkins for comments on the manuscript. This work was supported by BBSRC grant G20421 (TB and CC) and CRUK grants C303/A3135 and C303/A7399 (JJB). The authors have no competing interests in the work presented here.
REFERENCES
- [1].Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–813. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- [2].Wakayama T, Perry AC, Zuccotti M, Johnson KR, Yanagimachi R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature. 1998;394:369–374. doi: 10.1038/28615. [DOI] [PubMed] [Google Scholar]
- [3].Gurdon JB, Byrne JA. The first half-century of nuclear transplantation. Proc Natl Acad Sci U S A. 2003;100:8048–8052. doi: 10.1073/pnas.1337135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wakayama T, Tabar V, Rodriguez I, Perry AC, Studer L, Mombaerts P. Differentiation of embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Science. 2001;292:740–743. doi: 10.1126/science.1059399. [DOI] [PubMed] [Google Scholar]
- [5].Tada M, Tada T, Lefebvre L, Barton SC, Surani MA. Embryonic germ cells induce epigenetic reprogramming of somatic nucleus in hybrid cells. Embo J. 1997;16:6510–6520. doi: 10.1093/emboj/16.21.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol. 2001;11:1553–1558. doi: 10.1016/s0960-9822(01)00459-6. [DOI] [PubMed] [Google Scholar]
- [7].Flasza M, Shering AF, Smith K, Andrews PW, Talley P, Johnson PA. Reprogramming in inter-species embryonal carcinoma-somatic cell hybrids induces expression of pluripotency and differentiation markers. Cloning Stem Cells. 2003;5:339–354. doi: 10.1089/153623003772032844. [DOI] [PubMed] [Google Scholar]
- [8].Do JT, Han DW, Scholer HR. Reprogramming somatic gene activity by fusion with pluripotent cells. Stem Cell Rev. 2006;2:257–264. doi: 10.1007/BF02698052. [DOI] [PubMed] [Google Scholar]
- [9].Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- [10].Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, Plath K, Hochedlinger K. Directly Reprogrammed Fibroblasts Show Global Epigenetic Remodeling and Widespread Tissue Contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- [11].Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- [12].Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- [13].Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- [14].Aoi T, Yae K, Nakagawa M, Ichisaka T, Okita K, Takahashi K, Chiba T, Yamanaka S. Generation of Pluripotent Stem Cells from Adult Mouse Liver and Stomach Cells. Science. 2008 doi: 10.1126/science.1154884. [DOI] [PubMed] [Google Scholar]
- [15].Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- [16].Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- [17].Walev I, Bhakdi SC, Hofmann F, Djonder N, Valeva A, Aktories K, Bhakdi S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci U S A. 2001;98:3185–3190. doi: 10.1073/pnas.051429498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Freberg CT, Dahl JA, Timoskainen S, Collas P. Epigenetic Reprogramming of OCT4 and NANOG Regulatory Regions by Embryonal Carcinoma Cell Extract. Mol Biol Cell. 2007;18:1543–1553. doi: 10.1091/mbc.E07-01-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Taranger CK, Noer A, Sorensen AL, Hakelien AM, Boquest AC, Collas P. Induction of dedifferentiation, genomewide transcriptional programming, and epigenetic reprogramming by extracts of carcinoma and embryonic stem cells. Mol Biol Cell. 2005;16:5719–5735. doi: 10.1091/mbc.E05-06-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chong JP, Thömmes P, Rowles A, Mahbubani HM, Blow JJ. Characterization of the Xenopus replication licensing system. Methods in Enzymology. 1997;283:549–564. doi: 10.1016/s0076-6879(97)83043-1. [DOI] [PubMed] [Google Scholar]
- [21].Alberio R, Johnson AD, Stick R, Campbell KH. Differential nuclear remodeling of mammalian somatic cells by Xenopus laevis oocyte and egg cytoplasm. Exp Cell Res. 2005;307:131–141. doi: 10.1016/j.yexcr.2005.02.028. [DOI] [PubMed] [Google Scholar]
- [22].Strizzi L, Bianco C, Normanno N, Salomon D. Cripto-1: a multifunctional modulator during embryogenesis and oncogenesis. Oncogene. 2005;24:5731–5741. doi: 10.1038/sj.onc.1208918. [DOI] [PubMed] [Google Scholar]
- [23].Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- [24].Lachner M, O’Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116:2117–2124. doi: 10.1242/jcs.00493. [DOI] [PubMed] [Google Scholar]
- [25].Hansis C, Barreto G, Maltry N, Niehrs C. Nuclear reprogramming of human somatic cells by Xenopus egg extract requires BRG1. Curr Biol. 2004;14:1475–1480. doi: 10.1016/j.cub.2004.08.031. [DOI] [PubMed] [Google Scholar]
- [26].Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- [28].Byrne JA, Simonsson S, Western PS, Gurdon JB. Nuclei of adult mammalian somatic cells are directly reprogrammed to oct-4 stem cell gene expression by amphibian oocytes. Curr Biol. 2003;13:1206–1213. doi: 10.1016/s0960-9822(03)00462-7. [DOI] [PubMed] [Google Scholar]
- [29].Koziol MJ, Garrett N, Gurdon JB. Tpt1 activates transcription of oct4 and nanog in transplanted somatic nuclei. Curr Biol. 2007;17:801–807. doi: 10.1016/j.cub.2007.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Simonsson S, Gurdon J. DNA demethylation is necessary for the epigenetic reprogramming of somatic cell nuclei. Nat Cell Biol. 2004;6:984–990. doi: 10.1038/ncb1176. [DOI] [PubMed] [Google Scholar]
- [31].Hakelien AM, Gaustad KG, Taranger CK, Skalhegg BS, Kuntziger T, Collas P. Long-term in vitro, cell-type-specific genome-wide reprogramming of gene expression. Exp Cell Res. 2005;309:32–47. doi: 10.1016/j.yexcr.2005.06.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.