

Abstract
Cepharanthine (CEP), a biscoclaurine (bisbenzylisoquinoline) alkaloid isolated from Stephania cepharantha Hayata, is widely used in Japan to treat variety of diseases. Among a plethora of its biological activities CEP was reported to be able to scavenge radicals and prevent lipid peroxidation. We have recently described the phenomenon of constitutive ATM activation (CAA) and histone H2AX phosphorylation (CHP), the events that report DNA damage induced by endogenously generated radicals, the product of oxidative metabolism in otherwise healthy, untreated cells. The aim of the present study was to explore whether CEP can attenuate the level of CAA and CHP, which would indicate on its ability to protect DNA against endogenous oxidants. The data show that indeed the levels of CAA and CHP in human lymphoblastoid TK6 cells were distinctly lowered upon treatment with CEP. Thus, exposure of cells to 8.3 μM CEP for 4 h led to a reduction of the mean level of CAA and CHP by up to 60% and 50%, respectively. At 1.7 μM CEP the reduction of CAA and CHP after 4 h was 35% and 25%, respectively. Cells exposure to CEP led to a decrease in the level of ondogenous oxidants as measured by the ability to oxidate the fluorescent probe 5-(and-6)-carboxy-2′,7′-dichlorodihydro-fluorescein diacetate. No evidence of apoptosis was seen during the first 8 h of treatment with CEP but initiation of apoptosis (caspase-3 activation) was detected in relatively few (< 10%) cells after exposure to 8.3 μM CEP for 24 h. The data strongly suggest that the scavenging properties of CEP provide a protection of DNA from the radicals generated endogenously during oxidative metabolism.
Keywords: oxidative DNA damage, DNA repair, reactive oxygen species, ROS, oxygen radicals, radicals' scavenger, cell cycle, apoptosis, bisbenzylisoquinoline alkaloid
Introduction
Cepharanthine (CEP) is a biscoclaurine (bisbenzylisoquinoline) amphipathic alkaloid isolated from Stephania cepharantha Hayata. CEP or extracts from this plant are widely used, primarily in Japan, to treat variety of acute and chronic diseases (for review see [10]). Among the conditions being treated with CEP are alopecia areata [24], venomous snakebites [18], radiotherapy-caused leukopenia [26], malaria [5] and septic shock [12, 34]. Other pharmacological activities reported to be mediated by CEP are: inhibition of plasma membrane lipid peroxidation that leads to membrane stabilization [40], inhibition of histamine release [25], immuno-modulation [22], anti-inflammatory effect [12], anti-allergic effect [21], anti-HIV effect [27], multidrug resistance-reversing effect [9], inhibition of platelet aggregation [35, 36] and antitumor activity [14, 51].
Such great multitude of diverse effects of CEP suggests that at least some of them may be mediated by a common mechanism. One mechanism that may be considered involves the free-radicals scavenging property of this alkaloid. Indeed there is strong evidence that CEP can effectively scavenge radicals such as superoxide anion, hydroxyl radical and nitric oxide [19, 20, 28, 49]. The potent radicals scavenging properties of CEP may mediate several of the effects listed above, such as inhibition of the lipid peroxidation [40], protection from carcinogens [51], anti-inflammatory properties [12] or protection from radiotherapy-induced leukopenia [26]. DNA in live cells is exposed to oxidants generated during metabolic activity and to external oxidants or oxidant-inducers. This leads to oxidative DNA damage which is considered the primary cause of cell aging, senescence and predisposition to cancer [4, 11, 50]. We have recently reported that DNA damage induced by endogenous oxidants manifests as constitutive activation of Ataxia telangiectasia mutated (ATM; CAA) protein kinase concurrent with constitutive phosphorylation of histone H2AX (CHP) [17, 41, 45–48, 52]. Both, activation of ATM through its phosphorylation on Ser-1981 [2, 3, 29] as well as phosphorylation of H2AX on Ser-139 [32, 38] is generally recognized markers of DNA damage. The damage that involves formation of DNA double-strand breaks (DSBs) triggers a characteristic response revealed by the concurrent activation of ATM and H2AX phosphorylation, which when detected immunocytochemically, manifest as characteristic distinct foci of immunofluorescence (IF) [3, 38].
Given the reported radicals-scavenging properties of CEP we were interested to explore whether this alkaloid can attenuate the extent of CAA and CHP. The attenuation of CAA and CHP by CEP would be an indication of CEP ability to protect DNA against endogenous oxidants. Our present data clearly demonstrate that indeed, the extent of CAA and CHP has been reduced in TK6 lymphoblastoid cells exposed to CEP. The reduction of CAA and CHP was paralleled by a decrease in the level of endogenous reactive oxygen species (ROS).
Materials and Methods
Cells and culture conditions
Human B cell lymphoblastoid TK6 cells were kindly provided by Dr. Howard Liber of Colorado State University, Fort Collins, CO [37]. The cells were grown in 25 ml FALCON flasks (Becton Dickinson Co., Franklin Lakes, NJ) in RPMI 1640 supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine (all from GIBCO/BRL Life Technologies, Inc., Grand Island, NY) at 37°C in an atmosphere of 5% CO2 in air. At the onset of the experiments, there were fewer than 5 × 105 cells per ml in culture such that the cells were at an exponential and asynchronous phase of growth.
Cell treatment
The cultures were treated with 1.7 μM or 8.3 μM CEP (AXXORA LLC, San Diego, CA) as described in the legends to figures. Control cultures were treated with the equivalent volumes of dimethylsulfoxide (Sigma Chemical Co., St. Louis MO) that was used to prepare stock solutions of the drug. In parallel experiments we also used preparations of CEP that are clinically used in Japan (Cepharanthin inj., Lot No KO22196). Following incubation with the drug the cells were then fixed in suspension in 1% methanol-free formaldehyde (Polysciences, Inc., Warrington, PA) in PBS for 15 min on ice, followed by suspension in 80% ethanol where they were stored at −20°C for up to 24 h.
Immunocytochemical detection of ATM activation, γH2AX and activated caspase-3
The fixed cells were washed twice in PBS and suspended in a 1% (w/v) solution of bovine serum albumin (BSA; Sigma) in PBS for 30 min to suppress non-specific antibody (Ab) binding. The cells were then incubated in 100 μl of 1% BSA containing anti-phospho-histone H2A.X (Ser-139) mAb (Upstate, Lake Placid, NY, 1:100) or anti-phospho-ATM (Ser-1981) mAb (Upstate, 1:100) or cleaved caspase-3 (Asp175) Ab (Cell Signaling, Danvers, MA, 1:100) and incubated for 2 h at room temperature. The cells were rinsed with 1% BSA in PBS (100 g, 5 min) and, after centrifugation, the cell pellets were resuspended in 100 μl of PBS containing 1% BSA and Alexa Fluor 488 goat anti-mouse or anti-rabbit IgG (H+L) (Molecular Probes/Invitrogen, Eugene, OR, 1:100) for 30 min at room temperature in the dark. The cells were then counterstained with 10 μg/ml PI (Molecular Probes) in the presence of 100 μg/ml of RNase A (Sigma) for 30 min at room temperature. Other details of immunocytochemical detection of activated ATM and γH2AX are given elsewhere [13, 15, 16].
Detection of intracellular ROS
The intracellular level of ROS was measured with the fluorescent probe 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA, Molecular Probes) [39]. Briefly, untreated or treated with 8.3 μM DEP cells suspended in culture medium were preincubated with 10 μM H2DCF-DA for 30 min at 37°C. A 1 ml aliquot of cell suspension was then taken from the flask and cell green fluorescence (FL1) was measured using a FACScan flow cytometer (Becton-Dickinson, San Jose, CA). Other details of the procedure are described elsewhere [42, 45, 46].
ATM-1981P and γH2AX IF measurements
Cellular green (FITC) and red (PI) fluorescence of cells in suspension was measured using a FASCcan flow cytometer (Becton-Dickinson). The red (PI) and green (FITC) fluorescence from each cell were separated and quantified using the standard optics and CELLQuest software (Becton-Dickinson). Gating analysis was carried out to obtain mean values of ATM-S1981P and γH2AX IF for G1 (DNA Index; DI = 0.9–1.1), S (DI = 1.2–1.8) and G2M (DI = 1.9–2.1) cells in each experiment. The mean values (± SEM) for cells in each of these cell cycle phases, after the subtraction of the mean values of the non-specific (background) fluorescence of the cells immunostained with the irrelevant IgG, are presented in Figure 2. Each experiment was run in duplicate or triplicate. All experiments were repeated at least twice. Other details are given in figure legends.
Fig. 2.

Effect of CEP on ATM-S1981P and γH2AX expression in TK6 cells in relation to their position in the cell cycle. The mean values of ATM-S1981P and γH2AX IF (± SEM) for cells in G1, S and G2M phases of the cell cycle were estimated by gating analysis of the bivariate distributions as shown in Figure 1, as described in Materials and Methods. Note a reduction of IF in the cells treated with CEP in relation to Ctrl, most pronounced in the case of ATM-S1981P IF of the S-phase cells
Results
Figure 1 illustrates the effect of CEP on CAA and CHP on TK6 cells. The respective bivariate distributions (scatterplots) show the level of expression of ATM-S1981P and γH2AX IF in individual cells in relation to their position in the cell cycle. It is quite evident that cell treatment with CEP reduced the level of expression of both ATM-S1981P and γH2AX. The reduction was CEP concentration dependent, more pronounced in cells treated with 8.3 μM than with 1.7 μM of CEP, and was apparent in cells regardless of their cell cycle phase. The inspection of DNA content frequency histograms (Fig. 1, insets) indicates a decrease in proportion of cells in S and G2M- and an increase in proportion of G1-phase cells in cultures treated with CEP, more pronounced at 8.3 μM concentration, compared to the untreated (Ctrl) cells.
Fig. 1.

Attenuation of CAA and CHP in TK6 cells by CEP.Exponentially growing TK6 cells were treated with 1.7 μM or 8.3 μM CEP for 4 h then fixed and their expression of ATM-S1981P and γH2AX, detected immunocytochemically, was measured by flow cytometry concurrent with measurement of cellular DNA content, which is expressed as DNA index (DI). Based on differences in DI one can identify cells in G1, S and G2M phases of the cell cycle as shown in the left (Ctrl) panel. The dashed skewed lines indicate the mean level of fluorescence of cells immunostained with irrelevant IgG used as negative control. DNA content frequency histograms from the respective cultures are shown in the three right panels
Using gating analysis the mean values of ATMS1981P and γH2AX IF were calculated for cells in G1, S and G2M phases of the cell cycle, respectively, of the untreated and CEP-treated cells (Fig. 2). The data show a reduction of in mean ATM-S1981P and γH2AX IF, more pronounced in the case of ATMS1981P than γH2AX IF, of the cells treated with CEP compared to control. The maximal effect was seen in reduction of ATM-S1981P IF of the S-phase cells, whose mean IF was decreased well over 50% after treatment with 8.3 μM CEP. The minimal extent of reduction was observed in expression of γH2AX IF in G1-phase cells.
Induction of apoptosis in cells treated with CEP has been monitored by immunocytochemical detection of activated (cleaved) caspase-3, as described before [31]. No evidence of caspase activation was observed during the initial 8 h of the treatment either with 1.7 or 8.3 μM CEP (not shown). However, the presence of cells characterized by activated caspase-3 was observed in cultures treated with 8.3 μM CEP for 24 h while treatment with 1.7 μM CEP for such length of time had no visible effect (Fig. 3). There was no distinct evidence of cell cycle phase specificity in terms of induction of apoptosis, although proportion of cells characterized by activated caspase-3 was somewhat higher among S- as compared to G1- or G2M-phase cells (Fig. 3).
Fig. 3.
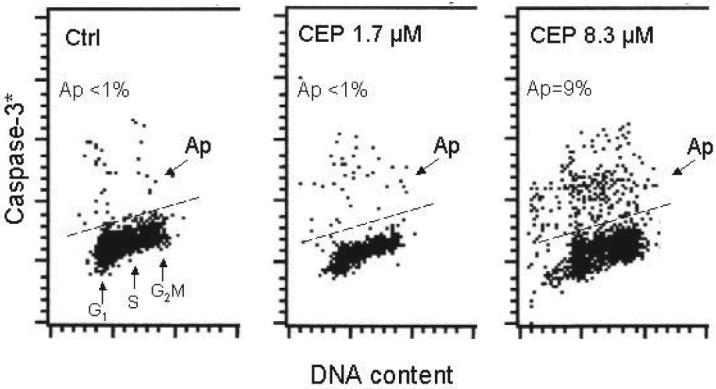
Induction of apoptosis in cells treated with CEP. The cells exposed in culture to 1.7 or 8.3 μM CEP for 24 h were harvested and the presence of activated caspase-3, the marker of induction of apoptosis, was detected immunocytochemically concurrently with measurement of DNA content. The skewed dashed line marks the upper level of immunofluorescence for cells showing no caspase-3 activation. While fewer cells than 1% showed activated caspase-3 (Ap) in control and in 1.5 μM CEP-treated cultures, 9% apoptotic cells were present in the culture containing 8.3 μM CEP
Figure 4 illustrates effect of treatment of TK6 cells on their ability to oxidize H2DCF-DA. This fluorochrome, which is widely used to probe the presence of reactive oxidants within the cell, penetrates plasma membrane and is not fluorescent unless oxidized. The raw data showing frequency histograms of intensity of H2DCF-DA fluorescence (left panel) clearly indicate that oxidation of H2DCF-DA was attenuated in cells treated with CEP (note that scale of H2DCF-DA fluorescence is exponential). When expressed in terms of mean values of fluorescence intensity (right panel) the data show that oxidation of this probe was reduced in CEP-treated cells in proportion to the time of their treatment, reaching nearly 50% reduction level after 4 h.
Fig. 4.

Effect of treatment of TK6 cells on with DEP on their ability to oxidize H2DCF-DA. The cells were left untreated (Ctrl) or were exposed in culture to 8.3 μM CEP for 2, 3 or 4 h, subsequently for 30 min to H2DCF-DA, and intensity of their green fluorescence was then measured by cytometry. The frequency distribution histograms shown in the left panel represent untreated cells and cells treated with CEP for 4 h (CEP), measured under identical conditions. The right panel shows mean values (± SD) of green fluorescence of cells untreated (zero time point), and exposed to CEP for 2, 3 and 4 h
Discussion
Exposure of cells to genotoxic agents such as radiation, radiomimetic antitumor drugs or certain carcinogens/mutagens was reported to trigger extensive ATM activation and H2AX phosphorylation (for reviews see [15, 43, 44]). Concurrent activation of ATM and phosphorylation of histone H2AX, thus, are being recognized as sensitive reporters of DNA damage, often reflecting formation of DSBs [2, 3, 38]. In contrast to the radiation-, drug-, or mutagen-induced activation of ATM and H2AX phosphorylation the CAA and CHP occur in normal- and in tumor-cells untreated with any genotoxic agents. Our recent findings provided strong evidence that a large portion of CAA and CHP occurs in response to DNA damage caused by metabolically generated ROS (for reviews see [41, 52]). Furthermore, cytometric analysis of the extent of CAA and CHP provided the means to measure the effectiveness of exogenous and endogenous factors, which either through lowering aerobic metabolism or neutralizing radicals protect DNA from such damage [52]. The present data demonstrate that CEP markedly reduced extent of CAA and CHP in TK6 cells as well as lowered the level of endogenous oxidants in these cells. The reduction of CAA and CHP was observed well prior to initiation of apoptosis, which was seen only at high CEP concentration and after 24 h (Fig. 3). Likewise there was no marked effect of CEP on cell cycle progression except of a minor prolongation of G1 phase as evidenced by the changes in DNA content histograms (Fig. 1). Neither apoptosis, thus, nor metabolic changes that otherwise accompany suppression of cell cycle progression [45], could be held accountable for the observed reduction of CAA and CHP in CEP-treated cells. Our findings thus indicate that CEP protects nuclear DNA from the damage induced by endogenous oxidants. Most likely the protection is mediated by the antioxidant properties of this biscoclaurine alkaloid.
As mentioned in the Introduction, there is indeed strong evidence that the scavenging of free radicals is one of many biological activities of CEP [19, 20, 28, 49]. In addition to direct scavenging of oxidants [19, 20] CEP can also reduce their generation by suppressing PKC and NADH oxidase activation [7, 23, 49]. Suppression of PKC may lead to a decrease in the overall metabolic activity, which as it was recently shown [45], markedly lowers constitutive oxidative DNA damage in conjunction with a decrease in quantity of ROS.
It should be noted that in addition to the deleterious effects such as induction of oxidative DNA damage or lipid peroxidation ROS function also as intracellular signaling molecules (for review see [6]). In this capacity, they can affect variety of metabolic and signaling pathways that are essential for cell survival or response to drugs. It is possible, therefore, that the observed antioxidant properties of CEP contribute to the multitude the diverse effects exerted by this biscoclaurine alkaloid, as reviewed in detail [10].
The lymphoblastoid TK6 cells express wt p53 [37]. It was observed that the level of CHP varies between the cells differing in expression of tumor suppressor p53, being the highest for cells with wt p53 and distinctly lower for cells with mt or null p53 status [48]. Among its many functions p53 induces transcription of genes that not only trigger the cell's response to DNA damage, but also modulate oxidative stress [1, 30, 33]. It was shown that cancer chemoprevention by some antioxidants is mediated by p53 [8]. It is possible, therefore, that the protective effect of CEP on oxidative DNA damage as observed in TK6 cells may vary in extent in the case of cells expressing dysfunctional p53.
Acknowledgement
Supported by NCI CA 28 704. The authors express gratitude to Kakenshoyaku Co, Ltd, in Osaka, Japan, for providing samples of cepharanthine used in this study.
Abbreviations
- ATM
Ataxia telangiectasia mediated protein kinase
- BSA
bovine serum albumin
- CAA
constitutive ATM activation
- CEP
cepharanthine
- CHP
constitutive histone H2AX phosphorylation
- DI
DNA index
- DSB
DNA double strand break
- H2DCF-DA-5-(and-6)-carboxy-2′,7′
dichlorodihydrofluorescein diacetate
- IF
immunofluorescence
- ROS
reactive oxygen species
References
- 1.Achanta G, Huang P. Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 2004;64:6233–6240. doi: 10.1158/0008-5472.CAN-04-0494. [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004;118:9–17. doi: 10.1016/j.cell.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 3.Bartkova J, Bakkenist CJ, Rajpert-De Meyts E, Skakkebaek NE, Sehested M, Lukas J, Kastan MB, Bartek J. ATM activation in normal human tissues and testicular cancer. Cell Cycle. 2005;4:838–845. doi: 10.4161/cc.4.6.1742. [DOI] [PubMed] [Google Scholar]
- 4.Beckman KB, Ames BN. Oxidative decay of DNA. J Biol Chem. 1997;272:13300–13305. doi: 10.1074/jbc.272.32.19633. [DOI] [PubMed] [Google Scholar]
- 5.Chea A, Hout S, Bun SS, Tabatadze N, Gasquet M, Azas N, Elias R, Balansard G. Antimalarial activity of alkaloids isolated from Stephania rotunda. J Ethnopharmacol. 2007;112:132–137. doi: 10.1016/j.jep.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 6.D'Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 7.Edashige K, Utsumi T, Utsumi K. Inhibition of 12-O-tetradecanoyl phorbol-13-acetate promoted tumor carcinogenesis by cepharanthine, a biscoclaurine alkaloid, in relation to the inhibitory effect on protein kinase C. Biochem Pharmacol. 1991;41:71–78. doi: 10.1016/0006-2952(91)90012-t. [DOI] [PubMed] [Google Scholar]
- 8.Erker L, Schubert R, Yakushiji H, Barlow C, Larson D, Mitchell JB, Wynshaw-Boris A. Cancer chemoprevention by the antioxidant tempol acts partially via the p53 tumor suppressor. Hum Mol Genet. 2005;14:1699–1709. doi: 10.1093/hmg/ddi181. [DOI] [PubMed] [Google Scholar]
- 9.Fujimura T, Shibata H, Maekawa I, Furusawa S, Kawauchi H, Sasaki K, Takayanagi Y. Reversal of resistance to doxorubicin with cepharanthine in murine P388 leukemia cells. Jpn J Pharmacol. 1990;54:464–467. doi: 10.1254/jjp.54.464. [DOI] [PubMed] [Google Scholar]
- 10.Furusawa S, Wu J. The effects of biscoclaurine alkaloid cepharanthine on mammalian cells: implications for cancer, shock, and inflammatory diseases. Life Sci. 2007;80:1073–1079. doi: 10.1016/j.lfs.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Gorbunova V, Seluanov A. Making ends meet in old age: DSB repair and aging. Mech Ageing Dev. 2005;126:621–628. doi: 10.1016/j.mad.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Goto M, Zeller WP, Hurley RM. Cepharanthine (biscoclaurine alkaloid) treatment in endotoxic shock of suckling rats. J Pharm Pharmacol. 1991;43:589–591. doi: 10.1111/j.2042-7158.1991.tb03542.x. [DOI] [PubMed] [Google Scholar]
- 13.Halicka HD, Huang X, Traganos F, King MA, Dai W, Darzynkiewicz Z. Histone H2AX phosphorylation after cell irradiation with UV-B: Relationship to cell cycle phase and induction of apoptosis. Cell Cycle. 2005;4:339–345. [PubMed] [Google Scholar]
- 14.Harada K, Supriatno, Yamamoto S, Kawaguchi S, Yoshida H, Sato M. Cepharanthine exerts antitumor activity on oral squamous cell carcinoma cell lines by induction of p27kip1. Anticancer Res. 2003;23:1441–1448. [PubMed] [Google Scholar]
- 15.Huang X, Halicka HD, Traganos F, Tanaka T, Kurose A, Darzynkiewicz Z. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005;38:223–243. doi: 10.1111/j.1365-2184.2005.00344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, Kurose A, Tanaka T, Traganos F, Dai W, Darzynkiewicz Z. Sequential phosphorylation of Ser-10 on histone H3 and Ser-139 on histone H2AX and ATM activation during premature chromosome condensation: relationship to cell-cycle and apoptosis. Cytometry. 2006;69A:222–229. doi: 10.1002/cyto.a.20257. [DOI] [PubMed] [Google Scholar]
- 17.Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol. 2006;29:495–501. [PubMed] [Google Scholar]
- 18.Kimoto T, Suemitsu K, Nakayama H, Komori E, Ohtani M, Ando S. Therapeutic experience of venomous snake-bites by the Japanese viper (Agkistrodon halys blomhoffii) with low dose of antivenin: report of 43 consecutive cases. Nippon Geka Hokan. 1997;66:71–77. [PubMed] [Google Scholar]
- 19.Kogure K, Goto S, Abe K, Ohiwa C, Akasu M, Terada H. Potent antiperoxidation activity of the bisbenzylisoquinoline alkaloid cepharanthine: the amine moiety is responsible for its pH-dependent radical scavenge activity. Biochim Biophys Acta. 1999;1426:133–142. doi: 10.1016/s0304-4165(98)00146-9. [DOI] [PubMed] [Google Scholar]
- 20.Kogure K, Tsuchiya K, Abe K, Akasu M, Tamaki T, Fukuzawa K, Terada H. Direct radical scavenging by the bisbenzylisoquinoline alkaloid cepharanthine. Biochim Biophys Acta. 2003;1622:1–5. doi: 10.1016/s0304-4165(03)00095-3. [DOI] [PubMed] [Google Scholar]
- 21.Kohno H, Inoue H, Seyama Y, Yamashita S, Akasu M. Mode of the anti-allergic action of cepharanthine on an experimental model of allergic rhinitis. Nippon Yakurigaku Zasshi. 1987;90:205–211. doi: 10.1254/fpj.90.205. [DOI] [PubMed] [Google Scholar]
- 22.Kondo Y, Imai Y, Hojo H, Hashimoto Y, Nozoe S. Selective inhibition of T-cell-dependent immune responses by bisbenzylisoquinoline alkaloids in vivo. Int J Immunopharmacol. 1992;14:1181–1186. doi: 10.1016/0192-0561(92)90053-n. [DOI] [PubMed] [Google Scholar]
- 23.Matsuno T, Orita K, Edashige K, Kobuchi H, Sato EF, Inouye B, Inoue M, Utsumi K. Inhibition of active oxygen generation in guinea-pig neuthrophils by biscoclaurine alkaloids. Biochem Pharmacol. 1990;39:1255–1259. doi: 10.1016/0006-2952(90)90271-l. [DOI] [PubMed] [Google Scholar]
- 24.Morita K, Nakamura M, Nagamachi M, Kishi T, Miyachi Y. Seventeen cases of alopecia areata: combination of SADBE topical immunotherapy with other therapies. J Dermatol. 2002;29:661–664. doi: 10.1111/j.1346-8138.2002.tb00199.x. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura K, Tsuchiya S, Sugimoto Y, Sugimura Y, Yamada Y. Histamine release inhibition activity of bisbenzylisoquinoline alkaloids. Planta Med. 1992;58:505–508. doi: 10.1055/s-2006-961536. [DOI] [PubMed] [Google Scholar]
- 26.Ohta T, Morita K. Effect of cepharanthin on radiotherapy induced leucopenia. Rinsho Hoshasen. 1990;35:471–474. [PubMed] [Google Scholar]
- 27.Okamoto M, Ono M, Baba M. Suppression of cytokine productionand neural cell death by the anti-inflammatory alkaloid cepharanthine: a potential agent against HIV-1 encephalopathy. Biochem Pharmacol. 2001;62:747–753. doi: 10.1016/s0006-2952(01)00692-x. [DOI] [PubMed] [Google Scholar]
- 28.Oyaizu H, Adachi Y, Yasumizu R, Ono M, Ikebukuru K, Fukuhara S, Ikahara S. Protection of T cells from radiation-induced apoptosis by Cepharantin. Int J Immunopharmacol. 2001;1:2091–2099. doi: 10.1016/s1567-5769(01)00127-8. [DOI] [PubMed] [Google Scholar]
- 29.Paull TT, Lee JH. The Mre11/Rad50/Nbs1 complex and its role as a DNA-double strand break sensor for ATM. Cell Cycle. 2005;4:737–740. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- 30.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–304. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 31.Pozarowski P, Huang X, Halicka DH, Lee B, Johnson G, Darzynkiewicz Z. Interactions of fluorochrome-labeled caspase inhibitors with apoptotic cells. A caution in data interpretation. Cytometry A. 2003;55A:50–60. doi: 10.1002/cyto.a.10074. [DOI] [PubMed] [Google Scholar]
- 32.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 33.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The anti-oxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1309. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakaguchi S, Furusawa S, Wu J, Nagata K. Preventive effects of a biscoclaurine alkaloid,cepharanthine on endotoxin or tumor necrosis factor-alpha-induced septic shock symptoms: involvement of from cell death in L929 cells and nitric oxide production in raw 264.7 cells. Int J Immunopharmacol. 2007;7:191–197. doi: 10.1016/j.intimp.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Sato T, Morita I, Fujita H, Ono M, Kimishima A, Tomiyama J, Murota S. Pharmacological characterization of cepharanthin in chronic idiopathic thrombocytopenic purpura. Platelets. 2001;12:156–162. doi: 10.1080/09537100120039334. [DOI] [PubMed] [Google Scholar]
- 36.Sato T, Ohnishi ST. In vitro anti-sickling effect on cepharanthine. Eur J Pharmacol. 1982;83:91–95. doi: 10.1016/0014-2999(82)90289-8. [DOI] [PubMed] [Google Scholar]
- 37.Schwartz JL, Jordan E, Evans HH, Lenarczyk M, Liber H. The TP53 dependence of radiation-induced chromosome instability in human lymphoblastoid lines. Radiat Res. 2003;159:730–738. doi: 10.1667/rr3005. [DOI] [PubMed] [Google Scholar]
- 38.Sedelnikova OA, Rogakou EP, Panuytin IG, Bonner W. Quantitative detection of 125IUdr-induced DNA double-strand breaks with γ-H2AX antibody. Radiat Res. 2002;158:486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 39.Sheng-Tanner X, Bump EA, Hedley DW. An oxidative stress-mediated death pathway in irradiated human leukemia cells mapped using multilaser flow cytometry. Radiat Res. 1998;150:636–641. [PubMed] [Google Scholar]
- 40.Shiraishi N, Arima T, Aono K, Inouye B, Morimoto Y, Utsumi K. Inhibition by biscoclaurine alkaloid of lipid peroxidation in biological membranes. Physiol Chem Physics. 1980;12:299–305. [PubMed] [Google Scholar]
- 41.Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006;5:1940–1945. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka T, Halicka HD, Traganos F, Darzynkiewicz Z. Phosphorylation of histone H2AX on Ser 139 and activation of ATM during oxidative burst in phorbol ester-treated human leukocytes. Cell Cycle. 2006;5:2671–2675. doi: 10.4161/cc.5.22.3472. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, Dai W, Darzynkiewicz Z. Cytometry of ATM activation and histone H2AXphosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A. 2007;71A:648–661. doi: 10.1002/cyto.a.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanaka T, Huang X, Jorgensen E, Gietl E, Traganos F, Darzynkiewicz Z, Albino AP. ATM activation accompanies histone H2AX phosphorylation in A549 cells upon exposure to tobacco smoke. BMC Cell Biology. 2007;8:26. doi: 10.1186/1471-2121-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka T, Kajstura M, Halicka HD, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation are strongly amplified during mitogenic stimulation of lymphocytes. Cell Prolif. 2007;40:1–13. doi: 10.1111/j.1365-2184.2007.00417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka T, Kurose A, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Nitrogen oxide-releasing aspirin induces histone H2AX phosphorylation, ATM activation, and apoptosis preferentially in S-phase cells; involvement of reactive oxygen species. Cell Cycle. 2006;5:1669–1674. doi: 10.4161/cc.5.15.3100. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka T, Kurose A, Halicka HD, Traganos F, Darzynkiewicz Z. 2-Deoxy-D-glucose reduces the level of constitutive activation of ATM and phosphorylation of histone H2AX. Cell Cycle. 2006;5:878–882. doi: 10.4161/cc.5.8.2681. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka T, Kurose A, Huang X, Traganos F, Dai W, Darzynkiewicz Z. Extent of constitutive histone H2AX phosphorylation on Ser-139 varies in cells with different TP53 status. Cell Prolif. 2006;39:313–323. doi: 10.1111/j.1365-2184.2006.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terasaka H, Machino M, Saito M, Fujisawa S, Satoh K, Jiang Y, Sakagami H. Re-evaluation of antitumor activity of Cepharanthin. Anticancer Res. 2002;22:165–170. [PubMed] [Google Scholar]
- 50.Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci USA. 2003;100:12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yasukawa K, Takido M, Takeuchi M, Akasu M, Nakagawa S. Cepharanthine inhibits two-stage tumor promotion by 12-O-tetradecanoylphorbol 13-acetate and mezerein on skin tumor formation in mice initiated with 7, 12-dimethylbenzá anthracene. J Cancer Res Clin Oncol. 1991;117:421–424. doi: 10.1007/BF01612761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao H, Tanaka T, Halicka HD, Traganos F, Zarebski M, Dobrucki J, Darzynkiewicz Z. Cytometric assessment of DNA damage by exogenous and endogenous oxidants reports the aging-related processes. Cytometry A. 2007;71A:905–914. doi: 10.1002/cyto.a.20469. [DOI] [PMC free article] [PubMed] [Google Scholar]