

Abstract
Due to their limited natural abundance and significant biochemical effects, we synthesized the alkaloids (±)-antofine (1a), (±)-deoxypergularinine (1b), and their dehydro congeners (2 and 3) starting from the corresponding phenanthrene-9-carboxaldehydes. We also evaluated their in vitro cytotoxic activity. Compounds 1a and 1b showed significant potency against various human tumor cell lines, including a drug-resistant variant, with EC50 values ranging from 0.16–16 ng/mL. Structure–activity correlations of these alkaloids and some of their synthetic intermediates were also ascertained. The non-planar structure between the two major moieties, phenanthrene and indolizidine, plays a crucial role in the cytotoxic activity of phenanthroindolizidines. Increasing the planarity and rigidity of the indolizidine moiety significantly reduced potency. A methoxy group at the 2-position (1a) was more favorable for cytotoxic activity than a hydrogen atom (1b).
Introduction
Phenanthroindolizidine alkaloids are pentacyclic natural products produced by various plants of the Asclepiadaceae family, including Cryptocarya and Ficus species. Biological activities of these compounds include antiamoebic, antibacterial, anti-inflammatory, and antifungal effects, as well as significant cytotoxic activity against various cancer cell lines.1,2 Prototype compounds, such as antofine (1a), not only exhibit IC50 values in the low nanomolar range, but are also equally effective against drug-sensitive and multidrug-resistant (MDR) human cancer cell lines.3,4 Alkaloid 1a exerts its cytotoxicity by inhibiting protein and nucleic acid synthesis. Recent mechanism studies by Lee et al. indicated that 1a exhibits inhibitory activity on cell proliferation by arresting the G2/M phase of the cell cycle.5 Compound 1a also interacts with the bulged regions of DNA and RNA.6,7
Due to low natural abundance of this compound type, various syntheses of 1a and other phenanthroindolizidine alkaloids, in both racemic and optically active forms, have been reported in an effort to further investigate the biochemical and pharmaceutical effects.8–14 The most commonly reported synthetic methodology toward the highly condensed natural alkaloids (±)-1a and its geometric isomer (±)-deoxypergularinine (1b) involves formation of the biaryl bond of the phenanthrene ring system in the final step, after coupling of the aromatic residues with the appropriate heterocycle. The other common sequence to these alkaloids uses differentially substituted phenanthrenes as starting materials, which are coupled with the appropriate heterocycle through a halomethyl moiety. Diverse strategies have been employed for attaching the pyrrolidine ring system. These methods include strategies based on the alkylation of pyrrole, alkylation of proline, use of a pyrrole-substituted Wittig-like reagent, and ortho metalation. Other strategies include a pyridine-based strategy and a cyclohexadienone photorearrangement approach. Enantioselective approaches based on alkene metathesis have recently been reported.
Chuang et al.15 previously published an efficient synthesis of the compound type, preparing both unsubstituted phenanthroindolizine and the racemic alkaloid tylophorine (2,3,6,7-tetramethoxyphenanthroindolizine). Herein, we describe the total synthesis of 1a and 1b, which are 2,3,6- and 3,6,7-trimethoxy-substituted phenanthroindolizidine isomers, respectively, using a similar synthetic protocol. Their dehydroiminium chlorides (2) and bromides (3) were also prepared. We evaluated in vitro cytotoxic activity and report structure–activity relationships (SAR) of these natural alkaloids, their salts, and certain synthetic intermediates.
Results and Discussion
Our retrosynthetic analysis of 1a and 1b was identical to that described and shown in reference 15. The starting materials for our synthesis were the known phenanthryl aldehydes 10a and 10b, which were synthesized from the commercially available acids 4a and 4b and aldehydes 5a and 5b via a conventional five-step sequence, according to a previously reported procedure, as shown in Scheme 1.15
Scheme 1.
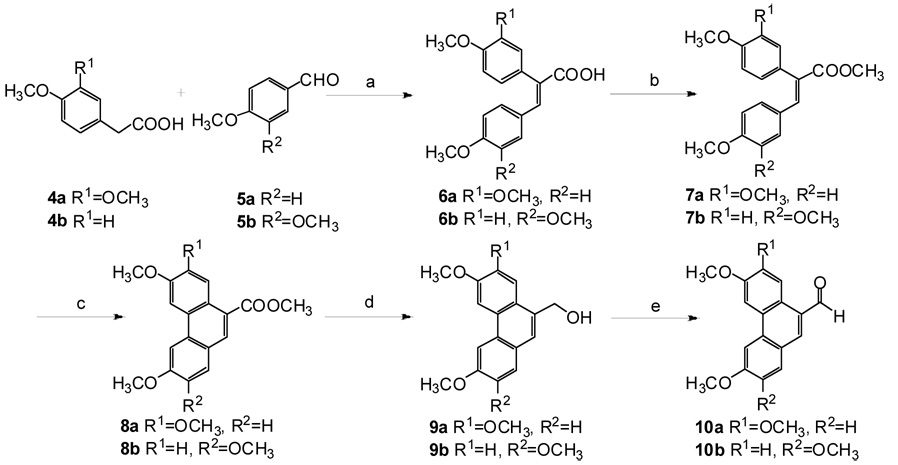
(a) (i)Ac2O, Et3N, reflux, 10 h; (ii) K2CO3(aq), 2 h; (b) H2SO4, CH3OH, reflux, 4 h; (c) VOF3, CH2Cl2, CF3COOH, (CF3CO)2, ice-salt bath, 1 h; (d) LiAlH4, THF, rt, 3 h; (e) PCC, CH2Cl2, rt, 4 h.
As shown in Scheme 2, aldehydes 10a and 10b were converted first to carboxylic acids 11a and 11b and then to acyl azides 12a and 12b through the reported sequence15 of Wittig reaction with (carboethoxymethylene)triphenyl phosphorane, hydrolysis to give the carboxylic acid, and treatment with oxalyl chloride and sodium azide to give the azide. These relatively unstable azides were used directly in the next reaction after purification by silica gel chromatography. A sequence involving a thermal Curtius rearrangement, an electrocyclic reaction, and hydrogen shift in boiling o-dichlorobenzene in the presence of I2, rather than Hg(OAc)2 as in reference 15, gave good yields of the isoquinolinones 13a and 13b from 12a and 12b, respectively. The pyrrolidine ring was added by the established methodology (N-alkylation with 1-bromo-3-chloropropane, conversion of bromide to iodide with excess sodium iodide in CH3CN, radical cyclization with n-Bu3SnH/AIBN in refluxing toluene)15 to afford phenanthroindolizidinones 14a and 14b. Finally, reduction of 14a and 14b with sodium bis(2-methoxyethoxy)aluminium hydride in refluxing dioxane gave racemic antofine 1a and deoxypergularinine 1b, respectively. Dehydroiminium chlorides 2a and 2b were prepared by exposing CHCl3 solutions of 1a and 1b, respectively to light and air,17 while the corresponding dehydroiminium bromides 3a and 3b were obtained from oxidation of 1a and 1b by N-bromosuccinimide in CHCl3.18
Scheme 2.
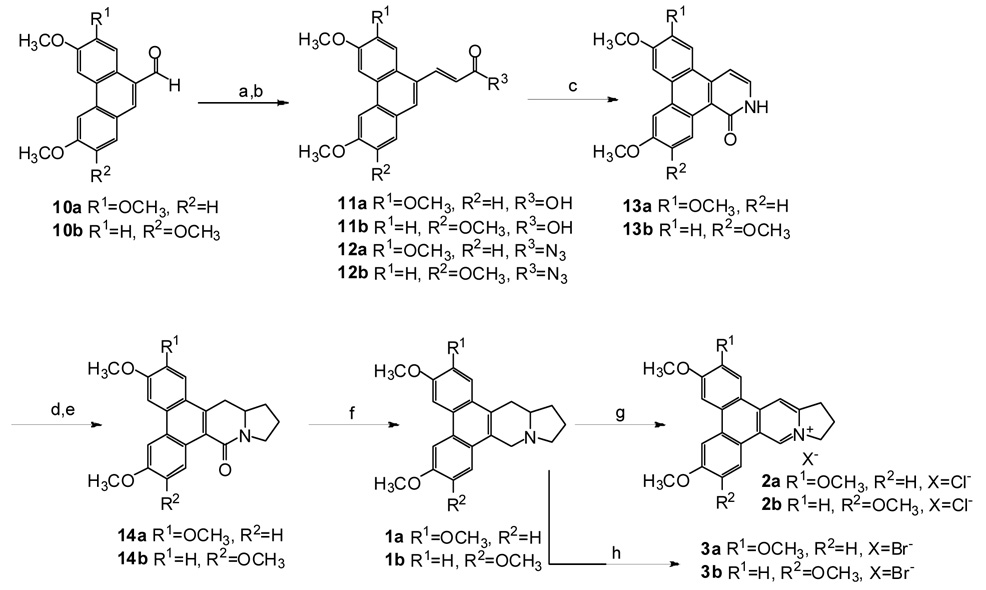
(a) (i) Ph3P = CHCO2Et, toluene, reflux, 4 h; (ii) KOH, EtOH-H2O, reflux, 3 h; (b) (i) (COCl)2, toluene, 80 °C, 5 h; (ii) NaN3, acetone, rt, 2 h; (c) I2, o-dichlorobenzene, reflux, 1 h; (d) (i) Br(CH2)3Cl, DMF, rt, overnight; (ii) NaI, CH3CN, reflux, overnight; (e) AIBN, n-Bu3SnH, toluene, reflux, 6 h; (f) NaAl(OCH2CH2OMe)2H2, dioxane, 2 h; (g) light, CHCl3; (h) NBS, CHCl3, rt, 1 h.
Biological Results
In order to explore their cytotoxic spectra and critical drug-resistance profiles, the target compounds and several synthetic intermediates were screened against four human tumor cell lines, A549 (lung), MCF-7 (breast), KB (nasopharyngeal) and KB-VIN (multidrug-resistant KB subline).19 The results are shown in Table 1. Compound 1a exhibited the best activity against all four cell lines, with EC50 values ranging from 0.16 to 4.5 ng/mL. Compound 1b also showed pronounced activity with EC50 values from 0.011 to 0.043 µg/mL; however, it was less potent (ca. 10- to 70-fold) than 1a. Both compounds showed no significant loss in potency against the drug-resistant KV-VIN cell line as compared with the parental KB cell line. Compounds 2a, 2b, 3a, and 3b, which have a dehydroiminium skeleton, showed reduced potency against all cell lines with EC50 values ranging from 0.2 to 8.26 µg/mL. Interestingly, 14b with an amide functionality also had moderate potency against all four cell lines, with EC50 values from 1.15 to 2.14 µg/mL. The remaining tested intermediates 11a–13a were inactive in this assay.
Table 1.
Effect of 1–3, 11a–13a, and 15b against Tumor Cell Line Replication
| EC50 (µg/mL) |
||||
|---|---|---|---|---|
| compound | A549 | MCF-7 | KB | KB-VIN |
| 1a | 0.00016 | 0.0045 | 0.00043 | 0.00083 |
| 1b | 0.011 | 0.043 | 0.023 | 0.016 |
| 2a | 0.73 | 1.72 | 0.93 | 0.33 |
| 2b | 1.06 | 1.49 | 0.95 | 5.66 |
| 3a | 0.49 | 1.29 | 5.37 | 5.14 |
| 3b | 0.32 | 1.18 | 0.29 | 8.26 |
| 11a | NA | NA | NA | 14.72 |
| 12a | NA | 8.29 | 13.52 | 9.36 |
| 13a | NA | NA | NA | 7.39 |
| 14b | 1.15 | 2.14 | 1.39 | 1.16 |
Cell line: A549 = lung; MCF-7 = breast; KB = nasopharynx; KB-VIN = nasopharynx MDR
Our data confirmed and extended SAR findings reported previously for this compound class.4,15,20 Introduction of double bonds adjacent to the phenanthrene nucleus reduced activity substantially. Compounds 1a/1b (non-planar) showed greater potency than their respective dehydro-counterparts, 2a/2b and 3a/3b (planar). The dehydro compounds have a charged pyridinium ring, which could decrease cellular permeability. In addition, compounds 1a/1b were much more potent than 14a/14b. The ketone at position-9 in the latter compounds changes the tertiary amine in the indolizidine nucleus to an amide, and increases the rigidity and planarity. The five-ring system is essential for cytotoxic activity; isoquinoline 13b (four-ring) and phenanthrenes 11a and 12a (three-ring) were inactive in all cell lines. Recently, several groups have reported that cytotoxic potency of phenanthroindolizidine is influenced by the substitution types and patterns on the phenanthrene ring.4,20 For example, Fu et al. found that changing the 2-methoxy of 1a to a bulkier isopropoxy or more polar hydroxy group decreased cytotoxic activity.4 In our study, we further found that the absence of the 2-methoxy as in 1b also decreases potency. Specifically, decreased cytotoxicity (10- to 70-fold) was observed for 1b relative to 1a, indicating that the C-2 methoxy group on the phenanthrene ring is important for optimal activity.
In summary, total syntheses of two natural phenanthroindolizidine alkaloids antofine (1a) and deoxypergularinine (1b) and their dehydroiminium chlorides (2a and 2b) and bromides (3a and 3b) were accomplished using a methodology based on thermal and radical cyclizations and a Curtius rearrangement. Alkaloids 1a and 1b showed profound cytotoxic activity against four cancer cell lines, with comparable potency in KB and KB-VIN cell lines. From activity results, we demonstrated that a non-planar five-ring structure and a methoxy group at the 2-position are important and favorable for potent activity of phenanthroindolizidine alkaloids.
Experimental Section
General Methods
Melting points were determined on a Yanaco MP-S3 melting point apparatus and are uncorrected. IR spectra were determined as KBr discs on a Shimadzu FT-IR Prestige 21. 1H and 13C-NMR spectra were recorded on Bruker Avance NMR 300 and AV 500 spectrometers, using tetramethylsilane (TMS) as an internal standard; all chemical shifts were reported in ppm (δ). All MS and HRMS spectra (EI) were obtained on a VG-70-250S mass spectrometer. FAB and HR-FAB mass spectra were obtained on a Joel JMS-700 spectrometer. Tetrahydrofuran was distilled over sodium, and dichloromethane was distilled over CaH2 unless otherwise stated. All other solvents and chemicals were used as purchased unless otherwise stated. Reactions were performed under Ar atmosphere in oven-dried flasks unless otherwise indicated.
Preparation of stilbene acids 6a and 6b
(E)-2-(3,4-Dimethoxyphenyl)-3-(4-methoxyphenyl)acrylic acid 6a
A solution of homoveratric acid (2.78 g, 20 mmol), p-anisaldehyde (3.96 g, 20 mmol), acetic anhydride (5.7 mL) and triethylamine (2.1 mL) was refluxed with stirring under Ar for 10 h. The resulting solution was cooled to room temperature, 10% K2CO3 solution (100 mL) was added, and then the resulting mixture was stirred for 2 h at 60 °C. The reaction mixture was cooled to room temperature and neutralized with concentrated HCl (pH = 4). The yellow stilbene acid obtained was collected by filtration and recrystallized from CH3OH to yield pure 6a (4.96 g, 79%): yellow needles, mp 217~218 °C, IR (KBr) cm−1: 3536, 2959, 1679, 1601; 1H-NMR (300 MHz, CDCl3) δ: 7.87 (1H, s, H-3″), 7.06 (2H, d, J = 8.4 Hz, H-2′ and H-6′), 6.91 (1H, d, J = 8.0 Hz, H-5), 6.82 (1H, d, J = 8.0 Hz, H-6), 6.76 (1H, s, H-2), 6.71 (2H, d, J = 8.4 Hz, H-3′ and H-5′), 3.93 (3H, s, OCH3-4), 3.81 (3H, s, OCH3-3), 3.93 (3H, s, OCH3-4′); 13C-NMR (75 MHz, CDCl3) δ: 172.5, 160.6, 149.2, 148.7, 142.1, 132.7, 128.6, 128.1, 127.0, 122.1, 113.8, 113.8, 112.8, 111.5, 55.9, 55.8, 55.2; EIMS m/z: 314 ([M+], 100), 178 (9), 137(17); anal. cacld for C18H18O5: C, 68.78; H, 5.77. Found: C, 68.68; H, 5.73.
(E)-3-(3,4-Dimethoxyphenyl)-2-(4-methoxyphenyl)acrylic acid 6b
Homoanisic acid 4b and 3,4-dimethoxybenzaldehyde 5b were used as starting materials in an analogous procedure as for the preparation of 6a to give 6b (4.75 g, 76%): yellow needles, mp 218~219 °C, IR (KBr) cm−1: 3536, 2983, 1663, 1595; 1H-NMR (300 MHz, CDCl3) δ: 7.86 (1H, s, H-3″), 7.20 (2H, d, J = 8.5 Hz, H-2 and H-6), 6.95 (2H, d, J = 8.0 Hz, H-3 and H-5), 6.81 (1H, dd, J = 8.3, 1.5 Hz, H-6′), 6.73 (1H, d, J = 8.3 Hz, H-5′), 6.53 (1H, d, J = 1.5 Hz, H-2′), 3.84 (3H, s, OCH3-4′), 3.81 (3H, s, OCH3-3′), 3.46 (3H, s, OCH3-4); 13C-NMR (75 MHz, CDCl3) δ: 173.1, 159.3, 150.3, 148.2, 142.2, 131.2×2, 128.0, 127.3, 125.8, 114.3×2, 112.5, 110.5, 55.8, 55.3, 55.2; EIMS m/z: 314 ([M+], 100), 178 (9), 137(17); anal. cazcld for C18H18O5: C, 68.78; H, 5.77. Found: C, 68.58; H, 5.77.
Preparation of stilbene methyl esters 7a and 7b
(E)-2-(3,4-Dimethoxyphenyl)-3-(4-methoxyphenyl)acrylic acid methyl ester 7a
Stilbene acid 6a (6.28g, 20 mmol) was dissolved in 120 mL water containing 8 mL concentrated H2SO4, and the resulting solution was refluxed for 4 h. The solvent was evaporated under reduced pressure, and the crude residue was purified by silica gel column chromatography (n-hexane–CHCl3 = 1:2) to afford 7a (4.21 g, 63.1%): yellow crystals, mp 111~112 °C, IR (KBr) cm−1: 3000, 2951, 1708, 1602; 1H-NMR (300 MHz, CDCl3) δ: 7.77 (1H, s, H-3″), 7.02 (2H, d, J = 8.8 Hz, H-2′ and H-6′), 6.90 (1H, d, J = 8.1 Hz, H-5), 6.78 (1H, dd, J = 8.1, 1.2 Hz, H-6), 6.74 (1H, d, J = 1.2 Hz, H-2), 6.70 (2H, d, J = 8.8 Hz, H-3′ and H-5′), 3.92 (3H, s, OCH3-4), 3.80 (3H, s, OCH3-3), 3.79 (3H, s, OCH3-1″), 3.76 (3H, s, OCH3-4′); 13C-NMR (75 MHz, CDCl3) δ: 168.7, 160.3, 149.1, 148.5, 140.2, 132.4×2, 129.6, 128.6, 127.3, 122.1, 113.7×2, 112.8, 111.4, 55.8, 55.7, 55.2, 52.3; EIMS m/z: 328 ([M+], 100), 269 (10), 151(58); anal. cacld for C19H20O5: C, 69.50; H, 6.14. Found: C, 69.63; H, 6.11.
(E)-3-(3,4-Dimethoxyphenyl)-2-(4-methoxyphenyl)acrylic acid methyl ester 7b
Esterification of 6b according to the above procedure afforded 7b (4.21 g, 63.1%): yellow crystals, mp 72~73 °C, IR (KBr) cm−1: 3005, 2950, 2838,1700, 1604; 1H-NMR (300 MHz, CDCl3) δ: 7.76 (1H, s, H-3″), 7.16 (2H, d, J = 8.5 Hz, H-2 and H-6), 7.04 (2H, d, J = 8.5 Hz, H-3 and H-5), 6.84 (1H, dd, J = 8.4, 2.2 Hz, H-6′), 6.71 (1H, d, J = 8.4 Hz, H-5′), 6.47 (1H, s, H-2′), 3.82 (3H, s, OCH3-4′), 3.80 (3H, s, OCH3-3′), 3.76 (3H, s, OCH3-1″), 3.44 (3H, s, OCH3-4); 13C-NMR (75 MHz, CDCl3) δ: 168.5, 159.0, 149.8, 148.0, 140.2, 131.0×2, 129.5, 128.4, 127.4, 125.2, 114.2×2, 112.3, 110.4, 55.6, 55.1, 55.0, 52.1; EIMS m/z: 328 ([M+], 100), 121 (34); anal. cacld for C19H20O5: C, 69.50; H, 6.14. Found: C, 69.48; H, 6.05.
Preparation of phenanthrene esters 8a and 8b
3,6,7-Trimethoxyphenanthrene-9-carboxylic acid methyl ester 8a
A solution composed of stilbene ester 7a (6.56 g, 20 mmol) and 1 mL of trifluoroacetic acid anhydride in 60 mL of dry CH2Cl2 was added dropwise to a solution of vanadium oxytrifluoride (4.96 g, 40 mmol) in 60 mL CH2Cl2 and 30 mL EtOAc containing 4 mL of trifluoroacetic acid and 1 mL of trifluoroacetic anhydride. During the addition (60 min), the reaction mixture was cooled in an ice-salt bath. The dark brown mixture was stirred for an additional 0.5 h in an ice-salt bath, then poured onto crushed ice, and was extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane–CHCl3 = 1:4) to afford 8a (4.82 g, 73.9%): colorless crystals, mp 150~151 °C, IR (KBr) cm−1: 3116, 2943, 1713, 1613; 1H-NMR (300 MHz, CDCl3) δ: 8.64 (1H, s, H-8), 8.42 (1H, s, H-10), 7.84 (1H, d, J = 8.6 Hz, H-1), 7.83 (1H, s, H-5), 7.78 (1H, d, J = 2.4 Hz, H-4), 7.20 (1H, dd, J = 8.6, 2.4 Hz, H-2), 4.10 (3H, s, OCH3-6), 4.08 (3H, s, OCH3-7), 4.02 (3H, s, OCH3-3), 4.00 (3H, s, OCH3-11); 13C-NMR (75 MHz, CDCl3) δ: 168.2, 160.1, 149.8, 148.8, 133.3, 131.8, 131.2, 125.1, 124.9, 124.2, 121.6, 116.0, 106.9, 103.6, 103.1, 55.8×2, 55.5, 52.0; EIMS m/z: 326 ([M+], 100), 283 (14); anal. cacld for C19H18O5: C, 69.93; H, 5.56. Found: C, 69.70; H, 5.53.
2,3,6-Trimethoxyphenanthrene-9-carboxylic acid methyl ester 8b
Phenanthrene ester 8b (4.73 g, 72.5%) was prepared from 7b according to the above procedure: colorless crystals, mp 154~155 °C, IR (KBr) cm−1: 3001, 2950, 1708, 1617; 1H-NMR (300 MHz, CDCl3) δ: 8.91 (1H, d, J = 9.2 Hz, H-8), 8.23 (1H, s, H-10), 7.79 (1H, s, H-5), 7.74 (1H, s, H-4), 7.24 (1H, d, J = 9.2 Hz, H-7), 7.18 (1H, s, H-1), 4.08 (3H, s, OCH3-6), 4.00 (9H, s, OCH3-2,3, and 6); 13C-NMR (75 MHz, CDCl3) δ: 168.1, 157.9, 159.8, 149.5, 131.5, 129.4, 128.2, 126.6, 125.4, 123.6, 123.1, 115.6, 109.1, 104.2, 103.0, 55.9, 55.8, 55.4, 52.0; EIMS m/z: 326 ([M+], 100), 181 (20); anal. cacld for C19H18O5: C, 69.93; H, 5.56. Found: C, 69.61; H, 5.52.
Preparation of phenanthrene alcohols 9a and 9b
(3,6,7-Trimethoxyphenanthrene-9-yl)methanol 9a
To a suspension of LiAlH4 (2.4 g, 20 mmol) in 70 mL of dry THF was added a solution of 8a (6.52 g, 20 mmol) in 100 mL of dry THF. The reaction mixture was stirred under Ar for 3h and quenched by sequential addition of 4 mL of water, 4 mL of 15% NaOH, and 8 mL of water. The mixture was filtered, and the filtrate was concentrated under reduced pressure. The residue was dissolved in CHCl3, and the solution was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3) to afford 9a (4.96 g, 83.2%): colorless crystals, mp 155~156 °C, IR (KBr) cm−1: 3386, 2933, 1610, 1525; 1H-NMR (300 MHz, CDCl3) δ: 7.83 (1H, s, H-5), 7.77 (1H, d, J = 1.6 Hz, H-4), 7.73 (1H, d, J = 8.6 Hz, H-1), 7.55 (1H, s, H-10), 7.49 (1H, s, H-8), 7.17 (1H, dd, J = 8.6, 1.6 Hz, H-2), 5.05 (2H, d, J = 4.1 Hz, H-11), 4.08 (3H, s, OCH3-6), 4.03 (3H, s, OCH3-7), 4.00 (3H, s, OCH3-3), 1.87 (1H, br s, D2O-exchangeable, OH-11); 13C-NMR (75 MHz, CDCl3) δ: 158.3, 149.4, 148.8, 131.3, 131.2, 130.2, 125.7, 125.5, 124.8, 124.5, 115.4, 104.8, 103.9, 103.7, 64.7, 55.9×2, 55.5; EIMS m/z: 298 ([M+], 100), 152 (21); anal. cacld for C18H18O4: C, 72.47; H, 6.08. Found: C, 72.37; H, 6.06.
(2,3,6-Trimethoxyphenanthrene-9-yl)methanol 9b
Phenanthrene alcohol 9b (4.97 g, 83.5%) was obtained using 8b according to the above procedure: colorless crystals, mp 185~186 °C, IR (KBr) cm−1: 3217, 2951, 1611, 1512; 1H-NMR (300 MHz, CDCl3) δ: 8.10 (1H, d, J = 9.0 Hz, H-9), 7.88 (1H, d, J = 2.2 Hz, H-5), 7.85 (1H, s, H-4), 7.55 (1H, s, H-10), 7.25 (1H, dd, J = 9.0, 2.2 Hz, H-7), 7.19 (1H, s, H-1), 5.13 (2H, s, H-11), 4.10 (3H, s, OCH3-6), 4.03 (6H, s, OCH3-2 and 3), 4.00 (3H, s, OCH3-3); 13C-NMR (75 MHz, CDCl3) δ: 158.1, 149.6, 149.4, 132.8, 131.7, 127.0, 126.1, 124.4, 124.1, 123.2, 115.2, 108.4, 104.7, 103.4, 64.3, 55.1, 55.9, 55.5; EIMS m/z: 298 ([M+], 100), 283 (6); anal. cacld for C18H18O4: C, 72.47; H, 6.08. Found: C, 72.43; H, 6.06.
Preparation of phenanthrene aldehydes 10a and 10b
3,6,7-Trimethoxyphenanthren-9-carbaldehyde 10a
To a solution of 9a (5.96 g, 20 mmol) in 150 mL CH2Cl2 was added pyridinium chlorochromate (5.17 g, 20 mmol), and the reaction mixture was stirred under Ar at room temperature for 2.5 h. Aqueous 100 mL NaHCO3 was added, the mixture was filtered on Celite, and the filtrate was extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3) to afford 10a [4.98 g (84.1%)]: colorless crystals, mp 169~170 °C, IR (KBr) cm−1: 2997, 2936, 1672, 1615,1518; 1H-NMR (300 MHz, CDCl3) δ: 10.06 (1H, s, CHO-9), 8.77 (1H, s, H-8), 7.80 (1H, s, H-10), 7.73 (1H, d, J = 8.7 Hz, H-1), 7.55 (2H, s, H-4 and H-5), 7.13 (1H, dd, J = 8.7, 1.6 Hz, H-2), 4.03 (3H, s, OCH3-6), 4.00 (3H, s, OCH3-7), 3.97 (3H, s, OCH3-3); 13C-NMR (75 MHz, CDCl3) δ: 193.6, 160.9, 150.3, 148.9, 140.0, 133.9, 131.9, 127.2, 124.5, 124.0, 123.6, 115.9, 106.0, 103.7, 102.7, 55.8, 55.6, 55.4; EIMS m/z: 296 ([M+], 100), 253 (20); anal. cacld for C18H16O4: C, 72.96; H, 5.44. Found: C, 72.88; H, 5.38.
2,3,6,-Trimethoxyphenanthren-9-carbaldehyde 10b
Phenanthrene aldehyde 10b (4.89 g, 82.6%) was prepared according to the above procedure: colorless crystals, mp 183~184 °C, IR (KBr) cm−1: 3011, 2960, 2938, 1679, 1616,1515; 1H-NMR (300 MHz, CDCl3) δ: 10.25 (1H, s, CHO-9), 9.31 (1H, d, J = 9.2 Hz, H-8), 7.97 (1H, s, H-10), 7.83 (1H, d, J = 2.2 Hz, H-5), 7.80 (1H, s, H-4), 7.30 (1H, dd, J = 9.2, 2.2 Hz, H-7), 7.29 (1H, s, H-1), 4.12 (3H, s, OCH3-6), 4.05 (3H, s, OCH3-3), 4.02 (3H, s, OCH3-2); 13C-NMR (75 MHz, CDCl3) δ: 193.5, 158.6, 151.9, 149.8, 138.3, 131.7, 128.8, 127.7×2, 125.6, 122.2, 116.1, 109.5, 104.6, 103.3, 56.1×2, 55.5; EIMS m/z: 296 ([M+], 100), 281 (6); anal. cacld for C18H16O4: C, 72.96; H, 5.44. Found: C, 72.99; H, 5.42.
Preparation of phenanthrene acrylic acids 11a and 11b
(E)-3-(3,6,7-Trimethoxyphenanthren-9-yl)acrylic acid 11a
A mixture of 10a (2.96 g, 10 mmol) and (carboethoxymethylene)triphenylphosphorane (4.76 g, 13 mmol) in 80 mL toluene was refluxed under Ar for 5 h. After cooling, the resulting solution was directly purified by silica gel column chromatography (n-hexane–CHCl3 = 1:4) and afforded the ethyl ester (3.19 g, 87.2%). Then, a solution of 30 mL 1 N KOH was added to a solution of the ethyl ester (3.19g, 8.7 mmol) in 60 mL EtOH, and the reaction mixture was heated under reflux for 3 h. After cooling, the solvent was evaporated under reduced pressure. The residue was dissolved in 30 mL water and acidified with 10% HCl. The yellow solid was filtered and recrystallized from CH3OH to afford pure 11a (2.68 g, 91.2%): yellow crystals, mp 258~259 °C, IR (KBr) cm−1: 3530, 2946, 1679, 1605,1519; 1H-NMR (300 MHz, CDCl3) δ: 12.50 (1H, br s, D2O-exchangeable, OH), 8.34 (1H, d, J = 15.7 Hz, H-11), 8.12 (1H, s, H-8), 8.10 (1H, s, H-10), 8.06 (1H, s, H-4), 7.92 (1H, d, J = 8.8 Hz, H-1), 7.47 (1H, s, H-5), 7.24 (1H, d, J = 8.8 Hz, H-2), 6.61 (1H, d, J = 15.7 Hz, H-12), 4.04 (3H, s, OCH3-7), 4.00 (3H, s, OCH3-6), 3.97 (3H, s, OCH3-3); 13C-NMR (75 MHz, CDCl3) δ: 167.5, 158.9, 149.5, 149.0, 141.4, 131.6, 130.9, 126.7, 124.9×2, 124.5, 124.1, 120.9, 116.5, 104.7, 104.1×2, 55.9, 55.6, 55.4; EIMS m/z: 338 ([M+], 100), 295 (37), 277(62); anal. cacld for C20H18O5: C, 70.99; H, 5.36. Found: C, 70.82; H, 5.35.
(E)-3-(2,3,6-Trimethoxyphenanthren-9-yl)acrylic acid 11b
Phenanthrene acrylic acid 11b (2.75 g, 92.5%) was prepared according to the above procedure: yellow crystals, mp 205~206 °C, IR (KBr) cm−1: 3530, 2953, 2937, 1689, 1616, 1512; 1H-NMR (300 MHz, CDCl3) δ: 12.40 (1H, br s, D2O-exchangeable, OH), 8.34 (1H, d, J = 15.6 Hz, H-11), 8.12 (1H, d, J = 2.0 Hz, H-5), 8.10 (1H, d, J = 9.2 Hz, H-8), 8.09 (1H, s, H-10), 8.06 (1H, s, H-4), 7.49 (1H, s, H-4), 7.29 (1H, dd, J = 9.2, 2.0 Hz, H-7), 6.57 (1H, d, J = 15.6, H-12), 4.04 (3H, s, OCH3-6), 4.00 (3H, s, OCH3-3), 3.92 (3H, s, OCH3-2); 13C-NMR (75 MHz, CDCl3) δ: 169.5, 158.0, 150.0, 149.6, 141.2, 131.1, 127.5, 126.4, 125.3, 125.0, 123.4×2, 120.8, 116.1, 109.1, 104.9, 104.1, 56.0, 55.6, 55.5; EIMS m/z: 338 ([M+], 100), 293 (8); anal. cacld for C20H18O5: C, 70.99; H, 5.36. Found: C, 70.95; H, 5.34.
Preparation of phenanthrene acryloyl azides 12a and 12b
(E)-3-(3,6,7-Trimethoxyphenanthren-9-yl)acryloyl azide 12a
A mixture of 11a (1.69 g, 5 mmol) and oxalyl chloride (1.27 g, 10 mmol) in 70 mL toluene was heated for 5 h at 80 °C. After cooling, the resulting mixture was concentrated under reduced pressure to afford the acyl chloride quantitatively. The acyl chloride was added immediately to a suspension of NaN3 (0.98 g, 15 mmol) in 60 mL dry acetone in an ice bath. The reaction mixture was stirred for 2 h at room temperature and filtered. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel column chromatography (CHCl3) to afford 12a (1.67 g, 92.1%): yellow crystals, mp 121~122 °C, IR (KBr) cm−1: 2933, 2140, 1674, 1604, 1509; 1H-NMR (300 MHz, CDCl3) δ: 8.45 (1H, d, J = 15.5 Hz, H-11), 7.84 (2H, s, H-8 and H-10), 7.78 (1H, d, J = 8.6 Hz, H-1), 7.76 (1H, d, J = 1.6 Hz, H-4), 7.38 (1H, s, H-5), 7.20 (1H, dd, J = 8.6, 1.6 Hz, H-2), 6.54 (1H, d, J = 15.5, H-12), 4.10 (3H, s, OCH3-7), 4.07 (3H, s, OCH3-7), 3.92 (3H, s, OCH3-3); 13C-NMR (75 MHz, CDCl3) δ: 172.0, 159.4, 149.8, 149.2, 144.5, 132.4, 131.2, 126.6, 125.5, 125.4, 125.1, 124.5, 120.4, 116.1, 104.0, 103.8, 103.7, 56.0×2, 55.5; EIMS m/z: 335 ([M+-N2], 100); anal. cacld for C20H17N3O4: C, 66.11; H, 4.72; N, 11.56. Found: C, 66.82; H, 4.80; N, 11.07.
(E)-3-(2,3,6-Trimethoxyphenanthren-9-yl)acryloyl azide 12b
Phenanthrene acryloyl azide 12b (1.64 g, 90.3%) was obtained using the above procedure: yellow crystals, mp 130~131 °C, IR (KBr) cm−1: 3008, 2938, 2834, 2141, 1678, 1613, 1513; 1H-NMR (300 MHz, CDCl3) δ: 8.51 (1H, d, J = 15.5 Hz, H-11), 8.07 (1H, d, J = 9.0 Hz, H-8), 7.83 (1H, s, H-5), 7.75 (1H, s, H-10), 7.24 (1H, d, J = 9.0 Hz, H-7), 7.16 (1H, s, H-1), 6.53 (1H, d, J = 15.5 Hz, H-12), 4.10 (3H, s, OCH3-6), 4.03 (3H, s, OCH3-3), 4.02 (3H, s, OCH3-2); 13C-NMR (75 MHz, CDCl3) δ: 172.0, 158.3, 150.3, 149.7, 144.4, 131.3, 127.8, 126.5, 125.8, 125.5, 124.1, 123.8, 120.5, 115.6, 108.7, 104.6, 103.1, 56.0, 55.9, 55.5; EIMS m/z: 335 ([M+-N2], 100), 320 (11); anal. cacld for C20H17N3O4: C, 66.11; H, 4.72; N, 11.56. Found: C, 66.46; H, 4.82; N, 11.08.
Preparation of isoquinolinones 13a and 13b
6,7,10-Trimethoxy-2H-2-azatriphenylene-1-one 13a
A mixture of azide 12a (1.1 g, 3 mmol) and I2 (0.02 g, catalytic amount) in 30 mL o-dichlorobenzene was refluxed for 2 h. After cooling, compound 13a was isolated by filtration and washed with n-hexane. The crude product was recrystallized from a CH3OH–CHCl3 mixture to afford pure isoquinolinone 13a (839 mg, 83.5%): pale yellow crystals, mp 269~270 °C, IR (KBr) cm−1: 3272, 2939, 2890, 1630, 1614, 1555,1503; 1H-NMR (300 MHz, DMSO-d6) δ: 11.70 (1H, br s, D2O-exchangeable, NH), 10.30 (1H, d, J = 9.5 Hz, H-12), 8.10 (2H, s, H-8 and H-9), 7.93 (1H, s, H-5), 7.47 (2H, m, H-3 and H-4), 7.27 (1H, d, J = 9.5 Hz, H-11), 4.07 (3H, s, OCH3-7), 4.01 (3H, s, OCH3-6), 3.99 (3H, s, OCH3-10); 13C-NMR (75 MHz, DMSO-d6) δ: 162.6, 157.7, 150.9, 149.6, 136.3, 130.3, 130.1, 129.0, 126.2, 123.2, 122.1, 117.0, 115.0, 106.1, 105.0, 104.7, 101.0, 55.9, 55.8, 55.3; EIMS m/z: 335 ([M+], 100), 292 (17); anal. cacld for C20H17NO4: C, 71.63; H, 5.11; N, 4.18. Found: C, 71.22; H, 5.17; N, 4.03.
7,10,11-Trimethoxy-2H-2-azatriphenylene-1-one 13b
Isoquinolinone 13b (820 mg, 81.6%) was prepared according to the above procedure: pale yellow crystals, mp 274~275 °C, IR (KBr) cm−1: 3267, 3219, 2957, 1627, 1554, 1517; 1H-NMR (300 MHz, DMSO-d6) δ: 11.70 (1H, br s, D2O-exchangeable, NH), 10.04 (1H, s, H-12), 8.56 (1H, d, J = 9.2 Hz, H-5), 8.11 (1H, d, J = 2.0 Hz, H-8), 8.10 (1H, s, H-9), 7.44 (1H, m, H-4), 7.43 (1H, m, H-3), 7.29 (1H, dd, J = 9.2, 2.0 Hz, H-6), 4.04 (3H, s, OCH3-11), 4.03 (3H, s, OCH3-10), 3.92 (3H, s, OCH3-7); 13C-NMR (75 MHz, DMSO-d6) δ: 162.7, 160.1, 149.0, 148.4, 137.3, 133.2, 130.4, 127.3, 125.0, 123.3, 120.6, 116.2, 115.7, 108.7, 105.1, 104.5, 100.6, 55.7, 55.6, 55.2; EIMS m/z: 335 ([M+], 100), 320 (12), 293 (17); anal. cacld for C20H17NO4: C, 71.63; H, 5.11; N, 4.18. Found: C, 71.78; H, 5.13; N, 4.22.
Preparation of phenanthroindolizidiones 14a and 14b
2-(3-Chloropropyl)-6,7,10-trimethoxy-2H-2-azatriphenylen-1-one
To a suspension of NaH (60% dispersion in oil, 80 mg, 2 mmol) in DMF (5 mL) cooled in an ice bath, a solution of 13a (335 mg, 1 mmol) in DMF (15 mL) was added with stirring. After the addition was completed, the mixture was added dropwise to a solution of 1-bromo-3-chloropropane (628 mg, 4 mmol) in 5 mL DMF. The mixture was stirred at room temperature overnight. The solvent was evaporated in vacuo, and water was then added. The mixture was extracted with CHCl3, washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (n-hexane–CHCl3 = 1:4) to afford the chloropropyl isoquinolinone of 13a (292 mg, 71.2%): colorless crystals, mp 183~184 °C, IR (KBr) cm−1: 2988, 2934, 1644, 1598, 1528; 1H-NMR (300 MHz, CDCl3) δ: 10.31 (1H, d, J = 9.5 Hz, H-12), 7.93 (2H, s, H-8 and H-9), 7.73 (1H, s, H-5), 7.48 (1H, d, J = 7.3 Hz, H-3), 7.32 (1H, d, J = 9.5 Hz, H-11), 7.21 (1H, d, J = 7.3 Hz, H-4), 4.32 (2H, t, J = 6.4 Hz, H-1′), 4.14 (3H, s, OCH3-7), 4.11 (3H, s, OCH3-6), 4.03 (3H, s, OCH3-10), 3.64 (2H, t, J = 5.9 Hz, H-3′), 2.40 (2H, tt, J = 6.4, 5.9 Hz, H-2′); 13C-NMR (75 MHz, CDCl3) δ: 162.1, 158.2, 151.0, 149.6, 135.6, 133.0, 131.1, 130.0, 127.0, 123.8, 122.3, 118.3, 114.5, 105.4, 105.2, 104.0, 101.4, 56.0×2, 55.5, 47.9, 42.2, 31.1; EIMS m/z: 413 ([M+2], 42), 411 ([M+], 80), 376 (46); anal. cacld for C23H22ClNO4: C, 67.07; H, 5.38; N, 3.40. Found: C, 67.79; H, 5.40; N, 3.39.
2,3,6-Trimethoxy-11,12,12a,13-tetrahydro-10H-9α-azacyclopenta[β]triphenylen-9-one 14a
A solution of chloropropyl isoquinolinone (251 mg, 0.5 mmol) and NaI (375 mg, 2.5 mmol) in dry CH3CN (15 mL) was placed in a sealed tube and then heated at 125 °C for 12 h. After cooling, the reaction mixture was filtered, and concentrated under reduced pressure to yield the iodopropyl isoquinolinone quantitatively. Subsequently, a solution of AIBN (33 mg, 0.2 mmol) in 2.4 mL toluene was added dropwise (syringe pump) to a degassed solution of iodopropyl isoquinolinone (251 mg, 0.5 mmol) and n-Bu3SnH (175 mg, 0.6 mmol) in 25 mL of toluene at refluxing temperature for 6 h. The reaction mixture was then cooled and the solvent was removed under reduced pressure. The residue was washed with n-hexane, purified by silica gel column chromatography (CHCl3) to give phenanthroindolizidione 14a (140 mg, 74.6%): colorless crystals, mp 252~253 °C, IR (KBr) cm−1: 2947, 1631, 1614, 1518; 1H-NMR (300 MHz, CDCl3) δ: 9.27 (1H, d, J = 9.3 Hz, H-8), 7.82 (2H, s, H-4 and H-5), 7.81 (1H, d, J = 9.3 Hz, H-7), 7.22 (1H, s, H-1), 4.09 (3H, s, OCH3-3), 4.01 (3H, s, OCH3-2), 3.99 (3H, s, OCH3-6), 3.86 (1H, m, H-11), 3.83 (1H, m, H-13), 3.80 (1H, m, H-11), 3.46 (1H, dd, J = 15.6, 3.9 Hz, H-14), 2.80 (1H, dd, J = 15.6, 12.0 Hz, H-14), 2.39 (1H, m, H-13), 2.12 (1H, m, H-12), 1.89 (1H, m, H-12), 1.82 (1H, m, H-13); 13C-NMR (75 MHz, CDCl3) δ: 164.3, 157.6, 149.9, 149.4, 132.5, 130.8, 129.5×2, 126.3, 124.2, 123.4, 115.1, 104.7, 104.0, 103.7, 55.9, 55.8, 55.4, 55.1, 45.2, 33.8, 32.5, 23.5; EIMS m/z: 377 ([M+], 100), 308 (57), 280 (45); anal. cacld for C23H23NO4: C, 73.19; H, 6.14; N, 3.71. Found: C, 73.09; H, 6.40; N, 3.59.
2-(3-Chloropropyl)-7,10,11-trimethoxy-2H-2-azatriphenylen-1-one
The chloropropyl isoquinolinone of 13b (297 mg, 72.2%) was prepared according to the above procedure: colorless crystals, mp 184~185 °C, IR (KBr) cm−1: 3119, 2957, 1644, 1614, 1599, 1519; 1H-NMR (300 MHz, CDCl3) δ: 10.05 (1H, s, H-12), 8.37 (1H, d, J = 9.2 Hz, H-5), 7.91 (1H, s, H-9), 7.90 (1H, d, J = 2.6 Hz, H-8), 7.47 (1H, d, J = 7.4 Hz, H-3), 7.29 (1H, d, J = 7.4 Hz, H-4), 7.24 (1H, dd, J = 9.2, 2.6 Hz, H-6), 4.31 (2H, t, J = 6.2 Hz, H-1′), 4.15 (3H, s, OCH3-11), 4.12 (3H, s, OCH3-10), 4.04 (3H, s, OCH3-7), 3.63 (2H, t, J = 6.2 Hz, H-3′), 2.40 (2H, p, J = 6.2 Hz, H-2′); 13C-NMR (75 MHz, CDCl3) δ: 162.6, 160.3, 149.6, 148.6, 136.8, 133.7, 133.2, 126.6, 125.6, 124.1, 120.9, 117.2, 115.1, 109.1, 104.9, 103.4, 101.5, 56.0,55.8, 55.5, 47.7, 42.1, 31.2; EIMS m/z: 413 ([M+2], 37), 411 ([M+], 100), 376 (28); anal. cacld for C23H22ClNO4: C, 67.07; H, 5.38; N, 3.40. Found: C, 67.07; H, 5.36; N, 3.37.
3,6,7-Trimethoxy-11,12,12a,13-tetrahydro-10H-9α-azacyclopenta[β]triphenylen-9-one 14b
Phenanthroindolizidinone 14b (143 mg, 75.8%) was obtained using the above procedure: colorless crystals, mp 200~201 °C, IR (KBr) cm−1: 2959, 2937, 1638, 1615, 1516; 1H-NMR (300 MHz, CDCl3) δ: 9.02 (1H, s, H-8), 8.00 (1H, d, J = 9.0 Hz, H-1), 7.88 (1H, d, J = 2.2 Hz, H-4), 7.86 (1H, s, H-5), 7.22 (1H, dd, J = 9.0, 2.2 Hz, H-2), 4.11 (3H, s, OCH3-7), 4.09 (3H, s, OCH3-6), 4.04 (3H, s, OCH3-3), 3.87 (1H, m, H-11), 3.83 (1H, m, H-13), 3.79 (1H, m, H-11), 3.66 (1H, dd, J = 15.6, 4.2 Hz, H-14), 2.93 (1H, dd, J = 15.6, 13.6 Hz, H-14), 2.42 (1H, m, H-13), 2.15 (1H, m, H-12), 1.93 (1H, m, H-12), 1.90 (1H, m, H-13); 13C-NMR (75 MHz, CDCl3) δ: 164.6, 159.3, 149.6, 148.6, 134.2, 133.1, 126.7, 125.3, 124.3, 123.0, 122.0, 115.5, 108.1, 104.4, 102.9, 55.9×2, 55.5, 55.3, 45.4, 33.9, 32.3, 23.5; EIMS m/z: 377 ([M+], 100), 308 (38), 280 (28); anal. cacld for C23H23NO4: C, 73.19; H, 6.14; N, 3.71. Found: C, 73.42; H, 6.17; N, 3.86.
Preparation of phenanthroindolizidines 1a and 1b
Antofine 1a
To a solution of 15a (39.7 mg, 1 mmol) in dry 5 mL dioxane was added a 3.5 M solution of sodium bis(2-methoxyethoxy)aluminium hydride in toluene (0.4 mL, 1.4 mmol) and the mixture was refluxed for 2 h in the dark. After evaporation of the solvents, the residue was diluted with 10 mL H2O and then basified with 10 mL 10% aqueous NaOH. The mixture was extracted with CHCl3, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–MeOH = 19:1) to yield 1a (32.8 mg, 90.3%): colorless crystals, mp 211~212 °C, IR (KBr) cm−1: 2914, 1615, 1529, 1512; 1H-NMR (300 MHz, CDCl3) δ: 7.90 (1H,s, H-4), 7.89 (1H, d, J = 2.1 Hz, H-5), 7.79 (1H, d, J = 9.0 Hz, H-8), 7.28 (1H, s, H-1), 7.19 (1H, dd, J = 9.0, 2.1 Hz, H-7), 4.66 (1H, d, J = 14.9 Hz, H-9), 4.09 (3H, s, OCH3-3), 4.05 (3H, s, OCH3-2), 4.00 (3H, s, OCH3-6), 3.66 (1H, d, J = 14.9 Hz, H-9), 3.45 (1H, td, J = 8.8, 2.0 Hz, H-11), 3.30 (1H, dd, J = 15.7, 2.3 Hz, H-14), 2.87 (1H, dd, J = 15.7, 10.5 Hz, H-14), 2.44 (1H, m, H-13a), 2.43 (1H, q, J = 8.8 Hz, H-11), 2.19 (1H, m, H-13), 1.99 (1H, m, H-12), 1.88 (1H, m, H-12), 1.72 (1H, m, H-13); 13C-NMR (100 MHz) δ: 157.4 (C-8), 149.3 (C-3), 148.3 (C-2), 130.1 (C-4b), 127.0 (C-14b), 126.6 (C-8b), 125.5(C-14a), 124.2 (C-8), 124.1 (C-8a), 123.5 (C-4a), 114.8 (C-7), 104.6 (C-5), 103.9 (C-1), 103.8 (C-4), 60.2 (C-13a), 56.0 (OCH3-3), 55.8 (OCH3-2), 55.5 (OCH3-6), 55.0 (C-11), 53.8 (C-9), 33.6 (C-14), 31.2 (C-13), 21.5 (C-12); EIMS m/z: 363 ([M+], 30), 294 (100); anal. cacld for C23H25NO3: C, 76.01; H, 6.93; N, 3.85. Found: C, 75.95; H, 6.80; N, 3.98.
Deoxypergularinine 1b
Deoxypergularinine 1b (33.4 mg, 92.0%) was prepared according to the above procedure: colorless crystals, 209~210 °C, IR (KBr) cm−1: 2959, 2912, 1612, 1512; 1H-NMR (300 MHz, CDCl3) δ: 7.91 (1H, d, J = 9.2Hz, H-1), 7.88 (1H, s, H-5), 7.86 (1H, d, J = 2.5Hz, H-4), 7.21 (1H, dd, J = 9.2, 2.5Hz, H-2), 7.09 (1H, s, H-8), 4.62 (1H, d, J = 14.5 Hz, H-9), 4.09 (3H, s, OCH3-7), 4.03 (3H, s, OCH3-6), 4.01 (3H, s, OCH3-3), 3.72 (1H, d, J = 14.5 Hz, H-9), 3.47 (1H, td, J = 8.1, 2.0 Hz, H-11), 3.40 (1H, dd, J = 16.5, 3.0 Hz, H-14), 3.00 (1H, dd, J = 16.5, 10.5 Hz, H-14), 2.59 (1H, m, H-13a), 2.58 (1H, dd, J = 8.1, 8.1 Hz, H-11), 2.25 (1H, m, H-13), 2.06 (1H, m, H-12), 1.96 (1H, m, H-12), 1.82 (1H, m, H-13); 13C-NMR (100 MHz) δ: 157.7 (C-3), 149.4 (C-7), 148.3 (C-6), 130.4 (C-4b), 126.6 (C-8b), 125.3 (C-14b), 125.2 (C-14a), 125.1 (C-1), 124.2 (C-8a), 123.4 (C-4a), 114.9 (C-2), 104.6 (C-4), 103.8 (C-5), 102.9 (C-8), 60.2 (C-13a), 56.0 (OCH3-7), 55.9 (OCH3-6), 55.5 (OCH3-3), 55.4 (C-11), 53.1 (C-9), 32.6 (C-14), 30.9 (C-13), 21.5 (C-12); EIMS m/z: 363 ([M+], 40), 294 (100); anal. cacld for C23H25NO3: C, 76.01; H, 6.93; N, 3.85. Found: C, 75.85; H, 6.81; N, 3.97.
Preparation of dehydrophenanthroindolizidine chlorides 2a and 2b
Dehydroantofine chloride 2a
A solution of antofine 1a (36.3 mg, 1 mmol) in 10 mL CHCl3 was exposed to light. After one day, a yellow product had formed, and was filtered and recrstallized from CH3OH to afford pure 2a (28.3 mg, 71.6%): yellow crystals, >290 °C, IR (KBr) cm−1: 2968, 1609, 1519; 1H-NMR (300 MHz, CD3OD) δ: 9.89 (1H, s, H-9), 8.74 (1H, s, H-14), 8.42 (1H, d, J = 8.9Hz, H-8), 7.77 (1H, s, H-1), 7.52 (1H, d, J = 2.2Hz, H-5), 7.50 (1H, s, H-1), 7.24 (1H, dd, J = 8.9, 2.2 Hz, H-7), 4.95 (2H, t, J = 7.4 Hz, H-11), 4.05 (6H, s, OCH3-2 and OCH3), 4.01 (3H, s, OCH3-6), 3.60 (2H, t, J = 7.4 Hz, H-13), 2.63 (2H, quin, J = 7.4 Hz, H-12); 13C-NMR (100 MHz) δ: 161.8, 154.3, 151.6, 151.3, 139.9, 137.9, 132.6, 128.9, 125.7×2, 120.9, 119.6, 117.3, 117.0, 107.0, 106.6, 105.4, 59.5, 56.6, 56.4, 56.0, 32.1, 22.8; EIMS m/z: 360 ([M+-Cl], 46), 154 (100); anal. cacld for C23H22ClNO3: C, 69.78; H, 5.60; N, 3.54. Found: C, 69.92; H, 5.45; N, 3.36.
Deoxypergularinine chloride 2b
Deoxypergularinine chloride 2b (27.8 mg, 70.3%) was prepared according to the above procedure: yellow crystals, >290 °C, IR (KBr) cm−1: 2964, 1608, 1517 ; 1H-NMR (300 MHz, CD3OD) δ: 9.51 (1H,s, H-9), 8.30 (1H, s, H-14), 8.09 (1H, d, J = 9.0 Hz, H-1), 7.13 (1H, s, H-8), 6.94 (1H, dd, J = 9.0, 1.5 Hz, H-2), 6.68 (1H, d, J = 1.5Hz, H-4), 6.61 (1H, s, H-5), 4.74(2H, t, J = 7.2 Hz, H-11), 3.89 (3H, s, OCH3-7), 3.86 (3H, s, OCH3-6), 3.80 (3H, s, OCH3-3), 3.41 (2H, t, J = 7.2 Hz, H-13), 2.54 (2H, p, J = 7.2 Hz, H-12); 13C-NMR (75 MHz) δ: 162.5, 151.1, 150.9, 150.8, 140.0, 137.7, 134.6, 128.0, 124.3, 124.1, 120.2, 118.5, 116.7, 116.0, 106.1, 104.1×2, 58.6, 57.3, 56.4, 56.3, 31.4, 22.2; EIMS m/z: 360 ([M+-Cl], 43), 154 (100); anal. cacld for C23H22ClNO3: C, 69.78; H, 5.60; N, 3.54. Found: C, 69.68; H, 5.55; N, 3.48.
Preparation of dehydrophenanthroindolizidine bromides 3a and 3b
Dehydroantofine bromide 3a
To a solution of antofine 1a (36.3 mg, 1 mmol) in 10 mL CHCl3 was added NBS (150 mg, 0.4 mmol) in small portions with stirring. After stirring for 1 h at room temperature, the orange solid formed was collected by filtration. The crude product was purified by aluminum oxide gel chromatography (CHCl3–MeOH = 9:1) to provide 3a (23.1 mg, 52.6%): yellow crystals, >290 °C, IR (KBr) cm−1: 2970, 1637, 1610, 1521; 1H-NMR (300 MHz, CD3OD) δ: 9.91 (1H, s, H-9), 8.76 (1H, s, H-14), 8.43 (1H, d, J = 9.0 Hz, H-8), 7.79 (1H, s, H-1), 7.53 (1H, d, J = 2.1 Hz, H-5), 7.52 (1H, s, H-1), 7.26 (1H, dd, J = 9.0, 2.1 Hz, H-7), 4.96 (2H, t, J = 7.4 Hz, H-11), 4.07 (6H, s, OCH3-2 and OCH3), 4.02 (3H, s, OCH3-6), 3.62 (2H, t, J = 7.4 Hz, H-13), 2.65 (2H, quin, J = 7.4 Hz, H-12); 13C-NMR (100 MHz) δ: 162.3, 154.8, 151.9, 151.8, 140.3, 138.3, 133.0, 129.3, 126.2×2, 121.2, 120.1, 117.7, 117.5, 107.4, 107.0, 105.9, 59.7, 57.0, 56.9, 56.3, 32.5, 23.1; EIMS m/z: 360 ([M+-Cl], 67), 154 (100); anal. cacld for C23H22BrNO3: C, 76.64; H, 6.15; N, 3.89. Found: C, 76.91; H, 6.25; N, 3.96.
Deoxypergularinine bromide 3b
Deoxypergularinine bromide 3b (24.2 mg, 55.1%) was prepared according to the above procedure: yellow crystals, >290 °C, IR (KBr) cm−1: 2965, 1639, 1608, 1516; 1H-NMR (400 MHz, CD3OD) δ: 9.51 (1H, s, H-9), 8.31 (1H, s, H-14), 8.10 (1H, d, J = 9.1 Hz, H-1), 7.13 (1H, s, H-8), 6.95 (1H, dd, J = 9.1, 1.6 Hz, H-2), 6.68 (1H, d, J = 1.6 Hz, H-4), 6.63 (1H, s, H-5), 4.75(2H, t, J = 7.1 Hz, H-11), 3.90 (3H, s, OCH3-7), 3.86 (3H, s, OCH3-6), 3.81 (3H, s, OCH3-3), 3.43 (2H, t, J = 7.1 Hz, H-13), 2.55 (2H, quin, J = 7.1 Hz, H-12); 13C-NMR (100 MHz) δ: 163.3, 151.4, 151.2, 151.0, 140.2, 138.0, 134.8, 128.4, 124.4, 124.1, 120.8, 118.8, 116.8, 116.4, 106.1, 104.9×2, 59.4, 57.4, 56.5, 56.3, 32.2, 23.0; EIMS m/z: 360 ([M+-Cl], 55), 154 (100); anal. cacld for C23H22BrNO3: C, 76.64; H, 6.15; N, 3.89. Found: C, 76.32; H, 6.00; N, 3.72.
In Vitro Cytotoxicity Assay
The sulforhodamine B assay was used according to the procedures developed and validated at NCI.18 Doxorubicin was used as the positive control antitumor drug. The in vitro anticancer activities are expressed as EC50 values, which is the test compound concentration (µg/mL) that reduced the cell number by 50% after 72 h of continuous treatment. The values were interpolated from dose-response data. Each test was performed in triplicate with variation less than 5%. The EC50 values determined in each of independent tests varied less than 10%. Compound stock solutions were prepared in DMSO with the final solvent concentration ≤1% DMSO (v/v), a concentration without effect on cell replication. The cells were cultured at 37 °C in RPMI-1640 supplemented with 25 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), 2% (w/v) sodium bicarbonate, 10% (v/v) fetal bovine serum, and 100 µg/mL kanamycin in a humidified atmosphere containing 5% CO2.
Acknowledgements
The authors are grateful for financial support from the National Science Council, Taiwan, Republic of China (NSC 95-2323-B-006-003) awarded to T. S.Wu. Thanks are also due to the support by NIH Grant No. CA-17625 from the National Cancer Institute awarded to K. H. Lee. We also thank Dr. T. H. Chuang (National Cheng Kung University) for his kind discussion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gellert E. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. New York: Academic Press; 1987. pp. 55–132. [Google Scholar]
- 2.Damu AG, Kuo PC, Shi LS, Li CY, Kuoh CS, Wu PL, Wu TS. Phenanthroindolizidine alkaloids from the stems of Ficus septica. J. Nat. Prod. 2005;68:1071–1075. doi: 10.1021/np050095o. [DOI] [PubMed] [Google Scholar]
- 3.Staerk D, Lykkeberg AK, Christensen J, Budnik BA, Abe F, Jaroszewski JW. In vitro cytotoxic activity of phenanthroindolizidine alkaloids from Cynanchum vincetoxicum and Tylophora tanakae against drug-sensitive and multidrug-resistant cancer cells. J. Nat. Prod. 2002;65:1299–1302. doi: 10.1021/np0106384. [DOI] [PubMed] [Google Scholar]
- 4.Fu Y, Lee SK, Min HY, Lee T, Lee J, Cheng M, Kima S. Synthesis and structure–activity studies of antofine analogues as potential anticancer agents. Bioorg. Med. Chem. Lett. 2007;17:97–100. doi: 10.1016/j.bmcl.2006.09.080. [DOI] [PubMed] [Google Scholar]
- 5.Lee SK, Nam KA, Heo YH. Cytotoxic activity and G2/M cell cycle arrest mediated by antofine, a phenanthroindolizidine alkaloid isolated from Cynanchum paniculatum. Planta Med. 2003;69:21–25. doi: 10.1055/s-2003-37021. [DOI] [PubMed] [Google Scholar]
- 6.Xi Z, Zhang R, Yu Z, Ouyang D, Huang R. Selective interaction between tylophorine B and bulged DNA. Bioorg. Med. Chem. Lett. 2005;15:2673–2677. doi: 10.1016/j.bmcl.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 7.Xi Z, Zhang R, Yu Z, Ouyang D. The interaction between tylophorine B and TMV RNA. Bioorg. Med. Chem. Lett. 2006;16:4300–4304. doi: 10.1016/j.bmcl.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 8.Leburn S, Couture A, Deniau E, Grandclaudon P. Total syntheses of (±)-cryptopleurine, (±)-antofine and (±)-deoxypergularinine. Tetrahedron. 1999;55:2659–2670. [Google Scholar]
- 9.Li Z, Jin Z, Huang R. Isolation, total synthesis and biological activity of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Synthesis. 2001:2365–2378. [Google Scholar]
- 10.Kim X, Lee J, Lee T, Park HG, Kim D. First asymmetric total synthesis of (−)-antofine by using an enantioselective catalytic phase transfer alkylation. Org. Lett. 2003;5:2703–2706. doi: 10.1021/ol0349007. [DOI] [PubMed] [Google Scholar]
- 11.Camacho-Davila A, Herndon JW. Total synthesis of antofine using the net [5+5]-cycloaddition of ,γ δ-unsaturated carbene complexes and 2-alkynylphenyl ketones as a key step. J. Org. Chem. 2006;71:6682–6685. doi: 10.1021/jo061053n. [DOI] [PubMed] [Google Scholar]
- 12.Furstner A, Kennedy JWJ. Total syntheses of the tylophora alkaloids cryptopleurine, (−)-antofine, (−)-tylophorine, and (−)-ficuseptine C. Chem. Eur. J. 2006;12:7398–7410. doi: 10.1002/chem.200600592. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Lee T, Lee E, Lee J, Fan GJ, Lee SK, Kim D. Asymmetric total syntheses of (−)-antofine and (−)-cryptopleurine using (R)-(E)-4-(tributylstannyl)but-3-en-2-ol. J. Org. Chem. 2004;69:3144–3149. doi: 10.1021/jo049820a. [DOI] [PubMed] [Google Scholar]
- 14.Ciufolini MA, Roschangar F. A unified strategy for the synthesis of phenanthroizidine alkaloids: Preparation of sterically congested pyridines. J. Am. Chem. Soc. 1996;118:12082–12089. [Google Scholar]
- 15.Chuang TH, Lee SJ, Yang CW, Wu PL. Expedient synthesis and structure-activity relationships of phenanthroindolizidine and phenanthroquinolizidine alkaloids. Org. Biomol. Chem. 2006;4:860–867. doi: 10.1039/b516152e. [DOI] [PubMed] [Google Scholar]
- 16.Bremmer ML, Khatri NA, Weinreb SM. Quinolizidine alkaloid synthesis via the intramolecular imino Diels-Alder reaction. epi-lupinine and cryptopleurine. J. Org. Chem. 1983;48:3661–3666. [Google Scholar]
- 17.Govindachari TR, Viswanathan N, Radhakrishan J. Quaternary alkaloids from Tylophora asthmatica. Indian J. Chem. 1973;11:1215–1216. [Google Scholar]
- 18.Rao KV, Kapicak LS. Action of N-bromosuccinimide on some indolizidine and quinolizidine systems. J. Heterocycl. Chem. 1976;13:1073–1077. [Google Scholar]
- 19.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks MR. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 20.Gao W, Bussom S, Grill SP, Gullen EA, Hu YC, Huang X, Zhong S, Kaczmarek C, Gutierrez J, Francis S, Baker DC, Yu S, Cheng YC. Structure-activity studies of phenanthroindolizidine alkaloids as potential antitumor agents. Bioorg. Med. Chem. Lett. 2007;17:4338–4342. doi: 10.1016/j.bmcl.2007.05.021. [DOI] [PubMed] [Google Scholar]