

Abstract
Cannabinoid receptor activation has been shown to modulate both neurotransmission (CB1) and neuroinflammatory (CB2) responses. There are conflicting reports in the literature describing the influence of cannabinoid receptor activation on ischemic/reperfusion injury. The goal of this study was to evaluate how changing the balance between CB1 and CB2 activation following cerebral ischemia influences outcome. CB1 and CB2 expression were tested at different times after transient middle cerebral artery occlusion (MCAO) in mice by real-time RT-PCR. Animals subjected to 1 hour MCAO were randomly assigned to receive different treatments: a CB1 antagonist, a CB2 antagonist, a CB2 agonist, a CB1 antagonist plus CB2 agonist, a CB2 antagonist plus CB2 agonist or an equal volume of vehicle as control. Cerebral blood flow was continuously monitored during ischemia; cerebral infarction and neurological deficit were tested 24 hours after MCAO. Cerebral CB1 and CB2 mRNA expression undertook dynamic changes during cerebral ischemia. The selective CB1 antagonist significantly decreased cerebral infarction by 47%; the selective CB2 antagonist increased infarction by 26% after 1 hour MCAO followed by 23 hours reperfusion in mice. The most striking changes were obtained by combing a CB1 antagonist with a CB2 agonist. This combination elevated the cerebral blood flow during ischemia and reduced infarction by 75%. In conclusion, during cerebral ischemia/reperfusion injury, inhibition of CB1 receptor activation is protective while inhibition of CB2 receptor activation is detrimental. The greatest degree of neuroprotection was obtained by combining an inhibitor of CB1 activation with an exogenous CB2 agonist.
Keywords: endogenous cannabinoids, cerebral ischemia/reperfusion injury, cerebral blood flow, inflammatory responses
INTRODCTION
The endocannabinoid system refers to two major types of cannabinoid receptors (termed CB1 and CB2), the endogenous ligands for those receptors and specific enzymes responsible for their degradation and inactivation (Rodriguez de Fonseca et al., 2005). The CB1 receptor is primarily expressed in the central nervous system (CNS), exhibiting a presynaptic location and playing a prominent role in synaptic neurotransmission (Pazos et al., 2005, Rodriguez de Fonseca et al., 2005). The CB2 receptor is expressed predominantly by cells of the immune system, such as lymphocytes and neutrophils, but is also expressed on resident inflammatory cells within the CNS. CB2 stimulation has been shown to have immunomodulatory properties, such as decreasing the activity of antigen presenting cells (APC) and down-regulating cytokine (IFN-γ and TNF-α) production during inflammatory responses (Berdyshev, 2000, Walter and Stella, 2004, Klein and Cabral, 2006, Lombard et al., 2007).
A number of investigations have shown that CB2 receptor activation has anti-inflammatory therapeutic potential in various CNS diseases, such as multiple sclerosis, traumatic brain injury and Alzheimer’s disease (Grundy et al., 2001, Molina-Holgado et al., 2002, Croxford, 2003, Ni et al., 2004, Ramirez et al., 2005). Because inflammatory responses have been shown to be important contributors to secondary injury following cerebral ischemia; the CB2 receptor has been investigated as a potential therapeutic target in stroke. It was demonstrated that selective activation of CB2 receptor attenuated cerebral ischemia/reperfusion injury in mice which was associated with decreased leukocyte/endothelial cell interactions (Zhang et al., 2007). The CB1 receptor has also been studied in cerebral ischemia/reperfusion injury (Nagayama et al., 1999, Jin et al., 2000, Parmentier-Batteur et al., 2002, Hayakawa et al., 2004). However, to date there are few studies focusing on the roles of CB1 and CB2 activation by endogenous cannabinoids in cerebral ischemic injury. In this investigation, we evaluated how ischemia/reperfusion injury influences cannabinoid receptor expression and how modification of the balance between CB1R and CB2R activation by endogenous and exogenous cannabinoids influences outcome after stroke.
EXPERIMENTAL PROCEDURES
Animals and surgical procedures
The cerebral ischemia/reperfusion studies were carried out in 8 week old male C57BL/6 mice (weighing 23 to 27g; Taconic, Hudson NY) and conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee at Temple University.
Middle Cerebral Artery Occlusion and Reperfusion (MCAO/R)
The animals were anesthetized by intraperitoneal injection of a mixture of Ketamine (100mg/ml) - Xylazine (20mg/kg) (1:1) at a dose of 1ml/kg. Body and cerebral temperature were maintained at 37±5°C by a heating lamp and heating pad. Middle cerebral artery occlusion was achieved by the intraluminal filament method (Hata et al., 1998). Briefly, a midline neck incision was made under an operating microscope. The right common carotid artery (CCA), external carotid artery (ECA) and internal carotid artery (ICA) were isolated. The ECA was ligated with 6-0 silk suture distal from the ICA-ECA branch and then cut distal from the ligated point. Another 6-0 silk suture was tied loosely around ECA close to the origin at the CCA. A blunted 5-0 monofilament nylon suture coated with poly-L-lysine (0.1% in deionized water, Sigma Inc, St Louis MO,USA) (Belayev et al., 1999) was introduced through a small incision in ECA and advanced into the circle of Willis, finally occluding the origin of the middle cerebral artery. The silk suture around the ECA stump was tied tightly to prevent bleeding and secured with a silk suture. The nylon suture was removed after 60 minutes occlusion and the ECA was permanently tied. Reperfusion was confirmed when pulsations were again observed in ICA.
Regional Cerebral Blood Flow (rCBF)
A laserPro Blood Perfusion Monitor (TSI Inc, Shoreview,MN,USA) was used to monitor and record regional cerebral blood flow (rCBF) prior to ischemia, during MCAO and reperfusion. A 1mm diameter microfiber laser-Doppler probe was attached to the skull 4mm lateral and 2mm posterior of Bregma. The MCAO was considered adequate if rCBF showed a sharp drop to 25% of baseline (pre-ischemia) level, otherwise, animals were excluded (Tsuchiya et al., 2003).
Injection of cannabinoid receptor agonist and antagonists in MCAO/R
The CB1 antagonist (SR141716) and CB2 antagonist (SR144528) were dissolved in a DMSO: cremophor: saline mixed solution (1:1:18). The antagonists (5mg/kg or 20mg/kg) or an equal volume of vehicle were administered 1 hour before MCAO i.p. The CB2 agonist (O-1966) was dissolved in a pure ethanol: emulphor: saline mixed solution at 1:1:18. The CB2 agonists (1mg/kg) or an equal volume of vehicle were administered as an intravenous injection into the jugular vein or intraperitoneally in a separate experimental group 1 hour before MCAO.
Real time PT-PCR
CB1 and CB2 expression were detected by the SYBR green-based real time RT-PCR technique. Animals were euthanized and transcardially perfused with cold PBS to remove the blood from vessels. Total RNA was isolated from brain specimens at 1, 3, 6 or 24 hours after MCAO by using Ultraspec reagent (Biotecx Laboratories, Huston TX, USA). Normal brain was used as sham. cDNA was prepared by reverse transcription. The 20μl (total volume) of the PCR mixture consists of 4μl diluted cDNA, 10μl SYBR green-containing PCR master mixture (2X) and 150μM of each primer. The CB1 and CB2 primers for real-time RT-PCR were designed by using the Primer Express software from Applied Biosystems (Fostercity, CA), and are as follows: CB1 sense: 5′-TGA AGT CGA TCT TAG ACG GCC-3′ and antisense: 5′GTG GTG ATG GTA CGG AAG GTA-3′; CB2 sense: 5′-TGA ATG AGC AGA CCG ACA GG-3′ and antisense: 5′-AGA GAT GTT TGC TGG GTG GC-3′; β-actin sense: 5′-TCC ACC ACC ACA GCT GAG AGG-3′, and antisense: 5′-CAG CTT CTC TTT GAT GTC ACG-3′ Real Time RT-PCR was performed using the Stratagene Mx3005P, and the cycling conditions used were 95°C for 15 sec, 60°C for 1 min, for 40 cycles, followed by a melting point determination or dissociation curves. The expression level of each gene is indicated by the cycle numbers needed for the cDNA to be amplified to reach a threshold. The amount of DNA is calculated from the cycle numbers by using standard curves and the results are normalized to the housekeeping gene β-actin from the same sample.
Infarct Volume Assessment
The animals were euthanized with an overdose of pentobarbital (200mg/kg i.p) 24 hours after MCAO and the brains were removed, and chilled on ice for 10 minutes to slightly harden the tissue. Five 2mm coronal sections were cut using a mouse brain matrix (Zivic lab, Pittsburg, PA, USA).The brain sections were placed in 2% triphenyltetrazolium chloride (TTC) (Sigma Inc, St Louis, MO, USA) dissolved in saline and stained for 20 minutes at 37°C in the dark. The brain sections were then fixed in 4% paraformaldehyde at 4°C for 24 hours and the anterior and caudal face of each section was scanned by a flatbed color scanner (Microtek Inc, Carson, CA, USA). The resulting images were captured as JPEG files and analyzed with NIH image software. The hemispheric infarct volumes were corrected for brain edema/swelling: the hemispheric infarct volume in each section was calculated by substracting the area of normal, TTC stained tissue in the hemisphere ipsilateral to the ligation from the contralateral nonischemic area to generate the infarct fraction (%), as described by Swanson et al. and Lin et al (Swanson et al., 1990, Lin et al., 1993).
Neurological Function Evaluation
The severity of neurological deficits was evaluated 24 hours after ischemic insult using a five-point deficit score (0=normal motor function; 1=flexion of torso and of contralateral forelimb upon lifting of the animal by tail; 2=circling to the contralateral side but normal posture at rest; 3=leaning to contralateral side at rest; and 4=no spontaneous motor activity) (Hata et al., 1998).
Statistical analysis
Bonferroni’s test after one way ANOVA was used for analyzing differences in infarct volume, neurological score and average of rCBF. The mRNA expression of CB1 and CB2 were analyzed by two way ANOVA (times, hemispheres) followed by Bonferroni’s test. Data were presented as means±SEM. A statistically significant difference was assumed at P<0.05.
RESULTS
CB1 and CB2 mRNA expression in brain during MCAO
There were no differences in CB1 mRNA expression in the non-ischemic hemisphere compared to normal control at 1, 3, 6 and 24 hours after MCAO. CB1 expression in the ischemic hemisphere increased at 1 hour after ischemia and was maximal at 6 hours. Similarly there were no significant differences in CB2 mRNA expression in the non-ischemic hemispheres compared to control. However, CB2 expression in the ischemic hemispheres decreased during first 3 hours following ischemia, followed by a gradual increase until the 24 hour measurement time (Figure 1).
Figure 1.
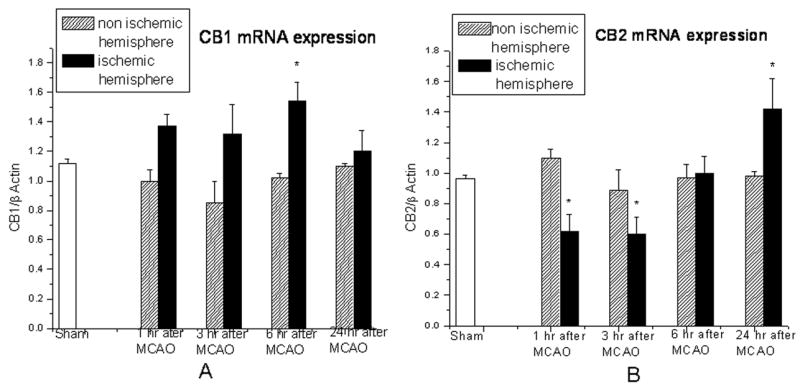
CB1 mRNA expression in ischemic hemisphere increased from 1 hour after ischemia and was highest at 6 hours after ischemia. However, CB2 mRNA expression in ischemic hemisphere decreased during first 3 hours following ischemia but then continued to increase until the 24 hour measurement time. There were no significant differences in both CB1 and CB2 mRNA expression in non-ischemic hemisphere compared to normal animals. (Samples were tested as triplicates, n=3 in each group, data were expressed as Mean±SEM, *p<0.05 vs sham)
Effects of CB antagonists and agonist on cerebral blood flow during Occlusion
During MCAO, rCBF decreased to approximately 25% of baseline value. Combined administration of the CB1 antagonist (SR141716, 20mg/kg, i.p.) and CB2 agonist (O-1966, 1mg/kg, i.v.) 1 hour prior to occlusion increased rCBF during the occlusion period when compared with the vehicle-treated group, whereas other treatments failed to alter rCBF during MCAO (Figure 2A). Lowering the dose of the CB1 and CB2 antagonist did not alter their influence on blood flow changes during occlusion; combination of CB2 agonist (i.p. injection) and low dose CB1 antagonist (5mg/kg) also increased rCBF (Figure 2B)
Figure 2.
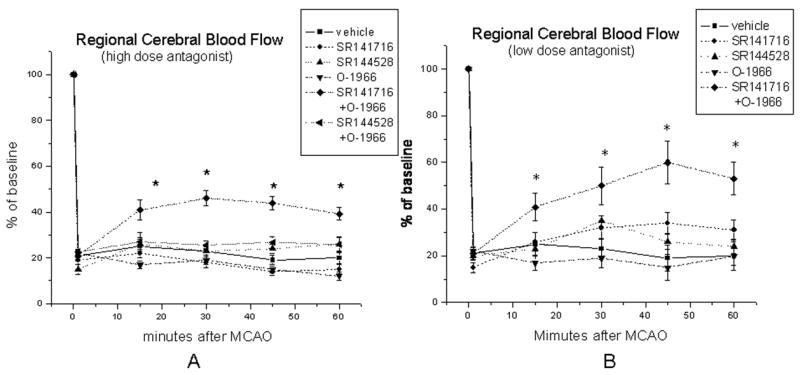
rCBF dropped to 25% of baseline level within the first 1 minute in all pretreatment groups. Administration of the high dose CB1 antagonist (SR141716, 20mg/kg, i.p.) and CB2 agonist (O-1966, 1mg/kg, i.v.) together 1 hour prior to occlusion increased rCBF during the 1 hour occlusion period when compared with the vehicle-treated group (A); similarly, administration of low dose CB1 antagonist (SR141716, 5mg/kg, i.p.) and CB2 agonist (O-1966, 1mg/kg) by i.p. injection also increased rCBF, whereas other treatments failed to alter rCBF during MCAO (B). (*p<0.05 vs vehicle treated control group, n=8 in each group)
Effects of CB antagonists and agonist on cerebral infarction
Administration of the CB1 antagonist (SR141716, 20mg/kg, i.p.) 1 hour before MCAO significantly decreased cerebral infarct fraction (12.3±0.86%) while administration of the CB2 antagonist (SR144528, 20mg/kg i.p.) alone increased cerebral infarct volume (31.1±2.45%) compared with vehicle-treated group (24.7±1.16%) (Figure 3A). Administration of a CB2 agonist (O-1966, 1mg/kg, i.v.) alone decreased cerebral infarction (15.6±1.67%) compared with vehicle treated group (22.4±1.08%), and the protection was totally reversed by co-administration of a CB2 antagonist (23.4±3.2%). Administration of the CB1 antagonist plus CB2 agonist 1 hour before MCAO further dramatically reduced infarct volume (6.1 ±2.33%) (Figure 4B). Reducing the dose of the antagonists to 5mg/kg and administration of the CB2 agonist at same dose by i.p. did not change the results obtained (Figure 3C).
Figure 3.
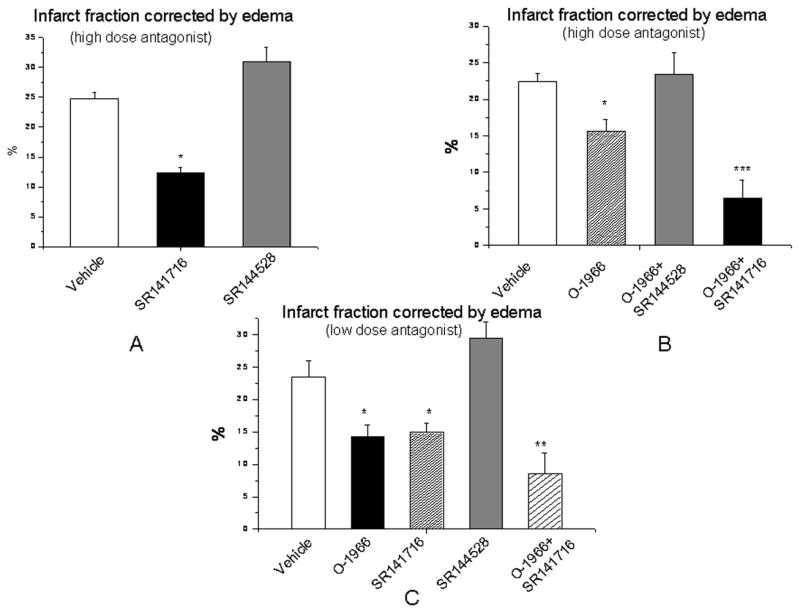
Effects of modulation of CB1 and CB2 receptor activation on cerebral infarction after MCAO. Administration of the CB1 antagonist (SR141716, 20mg/kg, i.p.) 1 hour before MCAO significantly decreased cerebral infarction while administration of CB2 antagonist (SR144528, 20mg/kg, i.p.) alone increased cerebral infarction compared with vehicle-treated group (A). CB2 agonist (O-1966, 1mg/kg, i.v.) alone decreased infarct volume which was completely reversed by CB2 antagonist; while administration of CB2 agonist with CB1 antagonist together dramatically further reduced cerebral infarction following MCAO (B). When the CB2 agonist (O-1966) was administered i.p. and antagonists were given at a lower dosage at 5mg/kg i.p.1 hour before MCAO, both the CB2 agonist and CB1 antagonist still significantly reduced cerebral infarction, while CB2 antagonist tended to increase the cerebral infarction although not achieving statistical significance. The combination of both CB2 agonist (1mg/kg, i.p.) and CB1 antagonist (5mg/kg, i.p.) dramatically reduced cerebral infarction compared to vehicle treated group (C). (n=5–8 in each group, data were expressed as Mean±SEM, *p<0.05 vs vehicle treated control group, **p<0.01 vs vehicle treated control group, ***p<0.001 vs vehicle treated control group)
Figure 4.
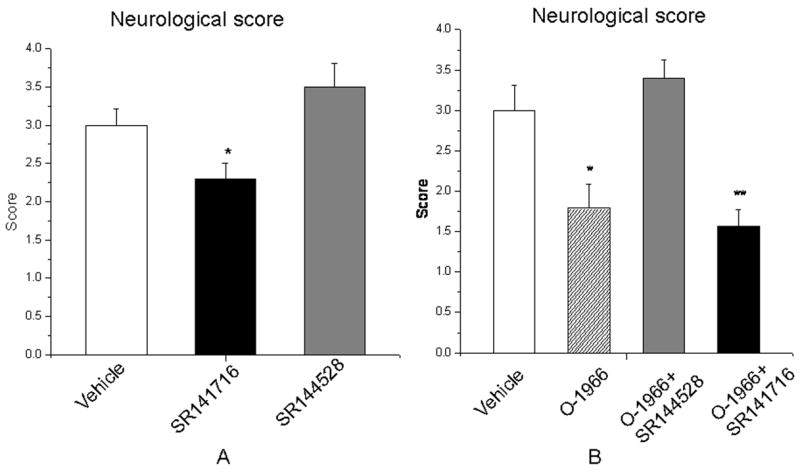
Effects of modulation of CB1 and CB2 receptor activation on neurological function after MCAO. In parallel with the changes in cerebral infarction, administration of a CB1 antagonist (20mg/kg, i.p.) improved motor function, while administrations of a CB2 antagonist tended to worsen motor function (A). Administration of CB2 agonist (1mg/kg, i.v.) alone improved motor function and this protection which was totally reversed by CB2 antagonist (20mg/kg, i.p.); while administration of CB2 agonist with CB1 antagonist together significantly attenuated neurological deficits after MCAO (B). (n=7–8 in each group, data were expressed as Mean±SEM, *p<0.05 vs vehicle treated control group, **p<0.01 vs vehicle treated control group)
Effects on neurological function
In parallel with the changes in infarct size, administration of a CB1 antagonist (20mg/kg i.p.) improved motor function significantly, while administrations of a CB2 antagonist (20mg/kg i.p.) tended to worsen motor function, although this value did not reach statistical significance (Figure 4A). Animals treated with either CB2 agonist (1mg/kg, i.v.) alone or the combined CB1 antagonist and CB2 agonist had significantly improved motor function compared to the vehicle treated animals (Figure 4B).
DISCUSSION
The primary goal of this study was to investigate whether modification of the endocannabinoid system could influence outcome following cerebral ischemia/reperfusion injury. The hypothesis that modification of the endocannabinoid system could influence outcome following ischemia was based upon prior reports that this system can have direct effects on neuronal function and can also modify inflammatory responses (Baker et al., 2001, van der Stelt et al., 2001, Muthian et al., 2004, McCollum et al., 2007). Both of these actions could have significant impact on cerebral ischemia/reperfusion injury.
Endogenous cannabinoids, are derivatives of arachidonic acid, and serve as natural ligands for the cannabinoid receptors with similar pharmacological properties to those of plant derived cannabinoids and synthetic analogs. 2-arachidonoyglycerol (2-AG) and anandamide are two major endogenous cannabinoids and are produced in relatively high concentrations in the CNS (Pazos et al., 2005, Rodriguez de Fonseca et al., 2005). Neuromodulatory properties of endogenous cannabiniods have been investigated in animal models of multiple sclerosis and brain trauma as well as in vitro studies (Baker et al., 2001, Panikashvili et al., 2001). It has previously been reported that the production of certain endogenous cannabinoids such as anandamide was increased in brain following ischemia (Muthian et al., 2004). There are also reports showing that the expression of the cannabinoid receptors were up-regulated in rat brain after cerebral ischemia (Jin et al., 2000, Ashton et al., 2007), but the changes of cerebral cannabinoid receptors expression in a mouse model of ischemic injury have not been previously examined. We found that CB1 receptor mRNA expression increased following ischemia and reached at peak 6 hours post ischemia in the ischemic hemisphere, while the non ischemic hemisphere CB1 mRNA content remained unchanged. Interestingly, cerebral CB2 mRNA content decreased over the first 3 hours after MCAO in the ischemic hemisphere, but increased after that. The difference in the time course of expression of these two receptors could, at least in part be due to changes in the cell types that express them. The delayed increase in CB2 receptor expression could reflect the time required for immune cell invasion of the CNS following ischemia (Heinel et al., 1994, Tomita and Fukuuchi, 1996). Microglia expression of CB2 receptors may also contribute (Nunez et al., 2004, Maresz et al., 2005).
Previous investigations performed in our laboratory have demonstrated the neuroprotective effects of administration of an exogenous CB2 ligand in a mouse model of cerebral ischemia/reperfusion injury (Zhang et al., 2007). Prior to this investigation, most studies have focused on the activation of the CB1 receptor rather than the activation of the CB2 receptor. WIN55212-2, which stimulates both the CB1 and CB2 receptor, with greater affinity for the CB2 receptor, has been shown to be neuroprotective in both global and focal models of ischemia. Based upon the use of WIN55212-2 in combination with a CB1 antagonist, these effects were interpreted to be the result of CB1 receptor activation (Nagayama et al., 1999). A later study indicated that the protective effect derived from stimulation of the CB1 receptor was the result of induced hypothermia, and was eliminated when temperature was maintained at the normal level (Hayakawa et al., 2004). Another study implicated a protective effect for the CB1 receptor by utilizing CB1 knockout mice which shown an increase in infarct size compared to wild type animals (Parmentier-Batteur et al., 2002). When interpreting these results it must be recognized that embryonic deletion of CB1 receptor may lead to abnormal CNS development, making these animals more susceptible to ischemia and may not reflect the acute contributions of the CB1 receptor in attenuating ischemic damage. In a separate investigation, and in agreement with the results presented in our study, other investigators have reported that the CB1 receptor antagonist SR141716 was found to reduce infarct volume in a rat MCAO model (Muthian et al., 2004).
Importantly, all of the investigations previously described have involved manipulation of either the CB1 or CB2 receptor alone. The current study examined the effects of changing the activity of both receptors simultaneously through either endogenous or exogenous cannabinoids. The results of this study showed that that the inhibition of CB1 receptor and activation of the CB2 receptor are both neuroprotective. Animals treated with either a CB1 antagonist alone or CB1 and CB2 antagonist together had smaller infarct volume compared to control animals, while CB2 antagonist treatment resulted in a larger infarct volume. This would indicate that the two major cannabinoid receptors, CB1 and CB2, play different roles in lesion formation during cerebral ischemia/reperfusion injury. These findings are consistent with results from our previous study showing that exogenous selective CB2 agonists reduced infarct volume and improved motor function in a mouse cerebral ischemia/reperfusion model (Zhang et al., 2007). Since the CB2 receptor is important in signaling immune cells as well as in the modulation of inflammatory responses, it is likely that CB2 activation exerts its protective mechanism at least in part through attenuation of inflammatory responses after stroke. Inflammation has been shown to be an important contributor to damage to the brain following ischemia/reperfusion injury (White et al., 2000, Danton and Dietrich, 2003). Within minutes of ischemia, cerebral vascular endothelium is activated and leukocytes begin to roll and adhere on inflamed endothelial cells, followed by transmigration into brain tissue (Kishimoto and Rothlein, 1994). Neutrophils are the first leukocytes to infiltrate at the site of inflammation and monocytes are subsequently recruited. Leukocytes activation and migration have been implicated as primary contributors to ischemia/reperfusion injury (Vasthare et al., 1990). In addition to their role in physical obstruction of capillaries, they participate in inflammatory responses and cause brain tissue damage by various mechanisms. For example, pro-inflammatory cytokines (TNF-α and IL-1β) secreted by leukocytes not only activate vascular endothelial cells and amplify inflammatory response but also directly induce neuronal injury (Wood, 2003). These studies highlight the involvement of immune cells and inflammatory cytokines in exacerbating ischemic injury, and numerous studies have shown that protection could be offered by interfering the inflammatory responses following ischemic/reperfusion injury (Kanemoto et al., 2002, Weaver et al., 2002, Sughrue et al., 2004).
CB2 is a Gi protein coupled-receptor and its activation triggers a series of signal transduction pathways which eventually leads to either up-or down-regulation of gene transcription. In most cases, the genes involved are coded for pro-inflammatory cytokines (Klein et al., 2001) and CB2 activation has been shown to inhibit some pro-inflammmatory cytokines such as TNF-α and IL-6 in in vivo and in vitro studies (Klein and Cabral, 2006). We have shown in our previous experiments that CB2 activation is protective in cerebral ischemic injury as well as CNS demyelinating diseases, and this protection is associated with attenuated leukocyte/endothelial interactions in brain (Ni et al., 2004, Zhang et al., 2007). Whether this attenuation is mediated through leukocytes, endothelial cells, or both is still under investigation.
Our most striking result was the finding that simultaneous administration of a CB1 antagonist and a CB2 agonist exerted the strongest effect in reducing the cerebral infarction following ischemic injury, which was associated with improved regional cerebral blood flow during ischemia. This might indicated a separate mechanism of action for the agonist and antagonist, which appear to have a synergistic neuroprotective effect. Although neither a CB1 antagonist alone nor a CB2 agonist alone had an effect on rCBF during occlusion, the combination significantly improved cerebral blood flow during ischemia. The underlying mechanisms contributing to this phenomenon are still under investigation. There are however, a number of potential targets that may be responsible. It has been reported that endogenous cannabinoids such as 2-AG and anandamide may induce vasodilation through a non-cannabinoid receptor (such as vanilloid receptor) present on endothelial cells (Golech et al., 2004, McCollum et al., 2007). It has been shown that cannabidiol, the nonpsychoactive constituent of cannabis, also reduced cerebral infarction due to increases in rCBF during ischemia, through activiation of the serotonergic 5-hydroxytryptamine1A receptor (Mishima et al., 2005). An additional mechanism could be through changes in the rheological contribution of leukocytes during the ischemic period. During periods of reduced cerebral perfusion pressure in stroke, activated leukocytes exaggerate microcirculatory dysfunction by direct occlusion of microvessels or releasing vasoactive factors such as thromboxane A2 causing platelets aggregation and increasing microvascular procoagulant activity (Ritter et al., 2000, Ishikawa et al., 2004), which further decrease perfusion in the ischemic brain. The increased blood flow in the ischemic region during occlusion in animals treated with the combination of a CB1 antagonist and a CB2 agonist could be the result of a reduction in vascular resistance due to an enhanced effect on the inhibition of leukocytes activation through greater CB2 receptor activation in the presence of the CB1 antagonist. It is also possible that the elevation in flow to the ischemic region during occlusion is the result of improved collateral blood flow. It has been reported that CB1 and CB2 receptors are found on different types of endothelial cells and their activation may participate in the control of vascular resistance (Zoratti et al., 2003, Golech et al., 2004).
In conclusion, the results of this investigation demonstrate dynamic changes in the expression of CB1 and CB2 receptors during cerebral ischemic/reperfusion injury in mice. The effects of stimulation of these receptors on damage ischemia/reperfusion injury differed dramatically. Stimulation of the CB2 receptor was found to be neuroprotective, while inhibition of the CB1 receptor was also protective,too. The combination of a CB2 agonist and a CB1 antagonist provided the greatest degree of protection and indicated a synergistic effect derived from combining these agents. Therefore, changing the balance of stimulation of these receptors by endogenous cannabinoids may provide an important therapeutic strategy during stroke.
Acknowledgments
This project is funded, in part, under a grant with the Pennsylvania Department of Health, a contract from BTG (London) and grants from DA P30 13429, DA 03672 and DA 05488 from the National Institute on Drug Abuse.
List of Abbreviations
- CB1R
cannabinoid CB1 receptor
- CB2
cannabinoid CB2 receptor
- MCAO
middle cerebral artery occlusion
- CNS
central nervous system
- APC
antigen presenting cell
- CCA
common carotid artery
- ECA
external carotid artery
- ICA
internal carotid artery
- rCBF
regional cerebral blood flow
- TTC
triphenyltetrazolium chloride
- 2-AG
2-arachidonoyglycerol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Ming Zhang, Center for Substance Abuse Research, Temple University School of Medicine, 208 MRB, 3420 N Broad Street, Philadelphia, PA 19140; Department of Physiology, Temple University School of Medicine, 208 MRB, 3420 N Broad Street, Philadelphia, PA 19140; Fax 2157074003; Phone 2157073398; Email mzhang@temple.edu.
Billy R Martin, Department of Pharmacology and Toxicology, Virginia Commonwealth University School of Medicine, 410 North 12th Street, Richmond, VA 23298; Fax 8048282117; Phone: 8048088407; Email martinb@vcu.edu.
Martin W. Adler, Center for Substance Abuse Research, Temple University School of Medicine, 305 OMS, 3400 N Broad Street, Philadelphia, PA 19140; Fax 2157071904; Phone 2157073243; Email baldeagl@temple.edu.
Raj K. Razdan, Organix Inc, 240 Salem Street, Woburn, MA 01801; Fax 7819336695; Phone 7819324142; Email: razdanrk@aol.com.
Doina Ganea, Center for Substance Abuse Research, Temple University School of Medicine, 709 MRB, 3420 N Broad Street, Philadelphia, PA 19140; Department of Microbiology and Immunology, Temple University School of Medicine, 709 MRB, 3420 N Broad Street, Philadelphia, PA 19140; Fax 2157074003; Phone 2157079921; Email: dganea@temple.edu.
Ronald F Tuma, Center for Substance Abuse Research, Temple University School of Medicine, 231 OMS, 3400 N Broad Street, Philadelphia, PA 19140; Department of Physiology, Temple University School of Medicine, 231 OMS, 3400 N Broad Street, Philadelphia, PA 19140; Fax 2157074003; Phone 2157075485; Email tumarf@temple.edu (corresponding author).
References
- Ashton JC, Rahman RM, Nair SM, Sutherland BA, Glass M, Appleton I. Cerebral hypoxia-ischemia and middle cerebral artery occlusion induce expression of the cannabinoid CB2 receptor in the brain. Neurosci Lett. 2007;412:114–117. doi: 10.1016/j.neulet.2006.10.053. [DOI] [PubMed] [Google Scholar]
- Baker D, Pryce G, Croxford JL, Brown P, Pertwee RG, Makriyannis A, Khanolkar A, Layward L, Fezza F, Bisogno T, Di Marzo V. Endocannabinoids control spasticity in a multiple sclerosis model. Faseb J. 2001;15:300–302. doi: 10.1096/fj.00-0399fje. [DOI] [PubMed] [Google Scholar]
- Belayev L, Busto R, Zhao W, Fernandez G, Ginsberg MD. Middle cerebral artery occlusion in the mouse by intraluminal suture coated with poly-L-lysine: neurological and histological validation. Brain Res. 1999;833:181–190. doi: 10.1016/s0006-8993(99)01528-0. [DOI] [PubMed] [Google Scholar]
- Berdyshev EV. Cannabinoid receptors and the regulation of immune response. Chem Phys Lipids. 2000;108:169–190. doi: 10.1016/s0009-3084(00)00195-x. [DOI] [PubMed] [Google Scholar]
- Croxford JL. Therapeutic potential of cannabinoids in CNS disease. CNS Drugs. 2003;17:179–202. doi: 10.2165/00023210-200317030-00004. [DOI] [PubMed] [Google Scholar]
- Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. 2003;62:127–136. doi: 10.1093/jnen/62.2.127. [DOI] [PubMed] [Google Scholar]
- Golech SA, McCarron RM, Chen Y, Bembry J, Lenz F, Mechoulam R, Shohami E, Spatz M. Human brain endothelium: coexpression and function of vanilloid and endocannabinoid receptors. Brain Res Mol Brain Res. 2004;132:87–92. doi: 10.1016/j.molbrainres.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Grundy RI, Rabuffetti M, Beltramo M. Cannabinoids and neuroprotection. Mol Neurobiol. 2001;24:29–51. doi: 10.1385/MN:24:1-3:029. [DOI] [PubMed] [Google Scholar]
- Hata R, Mies G, Wiessner C, Fritze K, Hesselbarth D, Brinker G, Hossmann KA. A reproducible model of middle cerebral artery occlusion in mice: hemodynamic, biochemical, and magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18:367–375. doi: 10.1097/00004647-199804000-00004. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Mishima K, Abe K, Hasebe N, Takamatsu F, Yasuda H, Ikeda T, Inui K, Egashira N, Iwasaki K, Fujiwara M. Cannabidiol prevents infarction via the non-CB1 cannabinoid receptor mechanism. Neuroreport. 2004;15:2381–2385. doi: 10.1097/00001756-200410250-00016. [DOI] [PubMed] [Google Scholar]
- Heinel LA, Rubin S, Rosenwasser RH, Vasthare US, Tuma RF. Leukocyte involvement in cerebral infarct generation after ischemia and reperfusion. Brain Res Bull. 1994;34:137–141. doi: 10.1016/0361-9230(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Zhang JH, Nanda A, Granger DN. Inflammatory responses to ischemia and reperfusion in the cerebral microcirculation. Front Biosci. 2004;9:1339–1347. doi: 10.2741/1330. [DOI] [PubMed] [Google Scholar]
- Jin KL, Mao XO, Goldsmith PC, Greenberg DA. CB1 cannabinoid receptor induction in experimental stroke. Ann Neurol. 2000;48:257–261. [PubMed] [Google Scholar]
- Kanemoto Y, Nakase H, Akita N, Sakaki T. Effects of anti-intercellular adhesion molecule-1 antibody on reperfusion injury induced by late reperfusion in the rat middle cerebral artery occlusion model. Neurosurgery. 2002;51:1034–1041; discussion 1041–1032. doi: 10.1097/00006123-200210000-00033. [DOI] [PubMed] [Google Scholar]
- Kishimoto TK, Rothlein R. Integrins, ICAMs, and selectins: role and regulation of adhesion molecules in neutrophil recruitment to inflammatory sites. Adv Pharmacol. 1994;25:117–169. doi: 10.1016/s1054-3589(08)60431-7. [DOI] [PubMed] [Google Scholar]
- Klein TW, Cabral G. Cannabinoid-Induced Immune Suppression and Modulation of Antigen-Presenting Cells. J Neuroimmune Pharmacol. 2006;1:50–64. doi: 10.1007/s11481-005-9007-x. [DOI] [PubMed] [Google Scholar]
- Klein TW, Newton CA, Friedman H. Cannabinoids and the immune system. Pain Res Manag. 2001;6:95–101. doi: 10.1155/2001/326867. [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Lombard C, Nagarkatti M, Nagarkatti P. CB2 cannabinoid receptor agonist, JWH-015, triggers apoptosis in immune cells: potential role for CB2-selective ligands as immunosuppressive agents. Clin Immunol. 2007;122:259–270. doi: 10.1016/j.clim.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- McCollum L, Howlett AC, Mukhopadhyay S. Anandamide-mediated CB1/CB2 receptor-independent NO production in rabbit aortic endothelial cells. J Pharmacol Exp Ther. 2007 doi: 10.1124/jpet.106.117549. [DOI] [PubMed] [Google Scholar]
- Mishima K, Hayakawa K, Abe K, Ikeda T, Egashira N, Iwasaki K, Fujiwara M. Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism. Stroke. 2005;36:1077–1082. doi: 10.1161/01.STR.0000163083.59201.34. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado E, Vela JM, Arevalo-Martin A, Almazan G, Molina-Holgado F, Borrell J, Guaza C. Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J Neurosci. 2002;22:9742–9753. doi: 10.1523/JNEUROSCI.22-22-09742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthian S, Rademacher DJ, Roelke CT, Gross GJ, Hillard CJ. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience. 2004;129:743–750. doi: 10.1016/j.neuroscience.2004.08.044. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA. Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci. 1999;19:2987–2995. doi: 10.1523/JNEUROSCI.19-08-02987.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni X, Geller EB, Eppihimer MJ, Eisenstein TK, Adler MW, Tuma RF. Win 55212-2, a cannabinoid receptor agonist, attenuates leukocyte/endothelial interactions in an experimental autoimmune encephalomyelitis model. Mult Scler. 2004;10:158–164. doi: 10.1191/1352458504ms1009oa. [DOI] [PubMed] [Google Scholar]
- Nunez E, Benito C, Pazos MR, Barbachano A, Fajardo O, Gonzalez S, Tolon RM, Romero J. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse. 2004;53:208–213. doi: 10.1002/syn.20050. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci. 2002;22:9771–9775. doi: 10.1523/JNEUROSCI.22-22-09771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazos MR, Nunez E, Benito C, Tolon RM, Romero J. Functional neuroanatomy of the endocannabinoid system. Pharmacol Biochem Behav. 2005;81:239–247. doi: 10.1016/j.pbb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter LS, Orozco JA, Coull BM, McDonagh PF, Rosenblum WI. Leukocyte accumulation and hemodynamic changes in the cerebral microcirculation during early reperfusion after stroke. Stroke. 2000;31:1153–1161. doi: 10.1161/01.str.31.5.1153. [DOI] [PubMed] [Google Scholar]
- Rodriguez de Fonseca F, Del Arco I, Bermudez-Silva FJ, Bilbao A, Cippitelli A, Navarro M. The endocannabinoid system: physiology and pharmacology. Alcohol Alcohol. 2005;40:2–14. doi: 10.1093/alcalc/agh110. [DOI] [PubMed] [Google Scholar]
- Sughrue ME, Mehra A, Connolly ES, Jr, D’Ambrosio AL. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004;53:497–508. doi: 10.1007/s00011-004-1282-0. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Tomita M, Fukuuchi Y. Leukocytes, macrophages and secondary brain damage following cerebral ischemia. Acta Neurochir Suppl. 1996;66:32–39. doi: 10.1007/978-3-7091-9465-2_6. [DOI] [PubMed] [Google Scholar]
- Tsuchiya D, Hong S, Kayama T, Panter SS, Weinstein PR. Effect of suture size and carotid clip application upon blood flow and infarct volume after permanent and temporary middle cerebral artery occlusion in mice. Brain Res. 2003;970:131–139. doi: 10.1016/s0006-8993(03)02300-x. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisogno T, Bar PR, Veldink GA, Vliegenthart JF, Di Marzo V, Nicolay K. Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J Neurosci. 2001;21:8765–8771. doi: 10.1523/JNEUROSCI.21-22-08765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasthare US, Heinel LA, Rosenwasser RH, Tuma RF. Leukocyte involvement in cerebral ischemia and reperfusion injury. Surg Neurol. 1990;33:261–265. doi: 10.1016/0090-3019(90)90046-r. [DOI] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. Br J Pharmacol. 2004;141:775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver M, Leshley K, Sands H, Gritman KR, Legos JJ, Tuma RF. LEX032, a novel recombinant serpin, protects the brain after transient focal ischemia. Microvasc Res. 2002;63:327–334. doi: 10.1006/mvre.2002.2405. [DOI] [PubMed] [Google Scholar]
- White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. doi: 10.1016/s0022-510x(00)00386-5. [DOI] [PubMed] [Google Scholar]
- Wiley JL, LaVecchia KL, Karp NE, Kulasegram S, Mahadevan A, Razdan RK, Martin BR. A comparison of the discriminative stimulus effects of delta(9)-tetrahydrocannabinol and O-1812, a potent and metabolically stable anandamide analog, in rats. Exp Clin Psychopharmacol. 2004;12:173–179. doi: 10.1037/1064-1297.12.3.173. [DOI] [PubMed] [Google Scholar]
- Wood PL. Neuroinflammation: mechanisms and management. Humana Press; Totowa, N.J: 2003. [Google Scholar]
- Zhang M, Martin BR, Adler MW, Razdan RK, Jallo JI, Tuma RF. Cannabinoid CB(2) receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J Cereb Blood Flow Metab. 2007;27:1387–1396. doi: 10.1038/sj.jcbfm.9600447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti C, Kipmen-Korgun D, Osibow K, Malli R, Graier WF. Anandamide initiates Ca(2+) signaling via CB2 receptor linked to phospholipase C in calf pulmonary endothelial cells. Br J Pharmacol. 2003;140:1351–1362. doi: 10.1038/sj.bjp.0705529. [DOI] [PMC free article] [PubMed] [Google Scholar]