

Abstract
Tularemia is a zoonotic disease caused by the Gram-negative intracellular pathogen Francisella tularensis. These bacteria evade phagolysosomal fusion, escape from the phagosome and replicate in the host cell cytoplasm. IFNγ has been shown to suppress the intra-macrophage growth of Francisella through both nitric oxide-dependent and -independent pathways. Since Francisella is known to subvert host immune responses, we hypothesized that this pathogen could interfere with IFNγ signaling. Here, we report that infection with Francisella suppresses IFNγ-induced STAT1 expression and phosphorylation in both human and murine mononuclear phagocytes. This suppressive effect of Francisella is independent of phagosomal escape or replication and is mediated by a heat-stable and constitutively-expressed bacterial factor. An analysis of the molecular mechanism of STAT1 inhibition indicated that expression of SOCS3, an established negative regulator of IFNγ signaling, is highly up-regulated during infection and suppresses STAT1 phosphorylation. Functional analyses revealed that this interference with IFNγ signaling is accompanied by the suppression of IP-10 production and iNOS induction resulting in increased intracellular bacterial survival. Importantly, the suppressive effect on IFNγ-mediated host cell protection is most effective when IFNγ is added post infection, suggesting that the bacteria establish a permissive environment within the host cell.
Keywords: Francisella, IFNγ, SOCS3, immune evasion
Introduction
Francisella tularensis is a Gram-negative, intracellular, zoonotic pathogen that causes the disease tularemia. F. tularensis subspecies tularensis (Type A) and subspecies holarctica (Type B) cause disease in humans. F. tularensis has been categorized by the CDC as a Category A Select agent and is perceived as a potential bio-weapon. However little is known about its pathogenesis1. Other subspecies include F. tularensis novicida and mediasiatica. F. novicida is attenuated in humans but highly infectious in mice. Thus it is a widely used experimental model for tularemia.
F. tularensis predominantly infects monocytes and macrophages. The phagosomes containing F. tularensis do not fuse with lysosomes and the bacteria escape into the cytosol where they replicate and subsequently trigger apoptosis of the host cell2,3. Escape of this pathogen into the cytosol requires the expression of bacterial proteins such as IglC, MglA, PmrA and AcpA4–8. Priming of macrophages with IFNγ, however, inhibits this escape9
IFNγ is a cytokine mainly produced by natural killer and T cells10, although we have found that infected monocytes also secrete very low levels of IFNγ. The signaling cascade initiated by engagement of the IFNγ receptor (IFNγR) has been comprehensively examined10. Recruitment and activation of the JAK kinases and STAT1 are a prerequisite for the transcription of IFNγ response genes. IFNγ signaling is negatively regulated by several mechanisms including the internalization of IFNγR, dephosphorylation of JAKs by the tyrosine phosphatase SHP-1 and the induction of the SOCS (Suppressors Of Cytokine Signaling) proteins. SOCS proteins function by binding to JAKs and inhibiting their phosphorylation, and thereby the JAK-dependent phosphorylation of the receptor and STAT112.
IFNγ plays a crucial role in modulating host immune responses. In particular, IFNγ is important for the activation of anti-microbial events such as the production of nitric oxide and reactive oxygen species and the up-regulation of FcγRI, complement receptor CR3 and NRAMP110. Several pathogens such as Leishmania donovani13, Mycobacterium avium14, Mycobacterim tuberculosis15,16, and Listeria monocytogenes17 have developed mechanisms to evade IFNγ-mediated host responses. Since Francisella is often referred to as a ‘stealth pathogen’ that effectively evades host immune responses, it is reasonable to postulate that this organism may also interfere with the host IFNγ response.
In this study we report that Francisella suppresses IFNγ-induced STAT1 expression and tyrosine phosphorylation in both human and murine mononuclear phagocytes. Examination of downstream events shows that IFNγ-induced iNOS expression is reduced, suggesting a mechanism for increased bacterial survival. This signaling interference is independent of phagosomal escape, replication and viability of the pathogen. Further, we demonstrate that the negative regulator SOCS3 is highly up-regulated and accounts, at least in part, to the suppression of STAT1 phosphorylation. Finally, we show that administration of recombinant IFNγ leads to reduced intracellular bacterial survival as previously reported9, but that administration after infection offers little benefit. This suggests that Francisella subverts IFNγ signaling upon infection.
Materials and Methods
Cells, antibodies and reagents
Raw 264.7 and THP-1 cells were obtained from ATCC and maintained in RPMI 1640 with 5% heat-inactivated fetal bovine serum (FBS). Recombinant IFNγ (mouse and human) and mouse IFNβ were purchased from R& D Systems (Minneapolis, MN). Antibodies specific for phospho-STAT1, phospho-JAK2, phospho-JAK1, were purchased from Cell Signaling Technology (Beverly, MA). Actin and SOCS3 antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against STAT1 and iNOS were obtained from BD Biosciences (San Jose, CA). F. novicida U112 (JSG1819), an mglA mutant of F. novicida (JSG2250) and F. tularensis LVS were used in all experiments. The iglD mutant of F. novicida bacteria were a generous gift from Dr. Yousef Abu Kwaik (U. of Louisville, KY). Control siRNA and SOCS3-specific siRNA (On-Targetplus SMARTpool) were obtained from Dharmacon (Lafayette, CO). Francisella strains were streaked and grown overnight on Chocolate II agar plates (Becton Dickinson and company, MD) at 37°C.
Isolation of peripheral blood monocytes
Peripheral blood monocytes (PBMs) were isolated as previously described11. Briefly, PBMCs were first isolated by density gradient centrifugation over Histopaque (Sigma-Aldrich, St Louis, MO) and monocytes were then purified from PBMCs by negative selection using MACS Monocyte Isolation kit (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions. The purity of the monocytes was more than 97% as determined by flow cytometry using CD14 antibodies.
Cell stimulation, lysis, and Western blotting
Macrophages were infected with plate-grown (grown on Chocolate II agar plates for 16–18 h at 37°C) F. novicida as previously described18 at an MOI of 10 unless described otherwise. Briefly, RAW 264.7 cells were plated overnight in 12-well or 6 well plates and allowed to adhere. F. novicida resuspended in RPMI medium containing 5% heat inactivated FBS was added to the adherent macrophages and then incubated at 37°C and 5% CO2 for the indicated time points. In parallel, the viability of bacteria was tested by plating the inoculum on chocolate II agar plates and bacterial numbers in the inoculum were quantified using the Petroff-Hauser chamber. These data indicated that >98% of the bacteria in the inoculum were viable in all cases (wild type and mutants). During the infection in the experiments shown the cells were not washed at any point. However, similar responses were seen when cells were extensively washed 1 hour post infection, treated with gentamicin and allowed to incubate (data not shown). Where indicated, cells were stimulated with 25 ng/ml of IFNγ. In various experiments IFNγ was added at different points -either 8 hours prior to infection (pre), at the time of infection (co) or 8 hours post infection (post). These details are provided in the corresponding Figure legends. In some experiments, bacteria were killed by either heating the bacterial suspension at 98°C for 10 min or by treating the bacteria with 50 μg/ml of gentamicin for 60 min. In some experiments macrophages were treated with vehicle control (DMSO) or 5 μg/ml of cytochalasin D for 30 minutes before infection.
Uninfected and infected cells were lysed in TN1 buffer (50mM Tris pH 8.0, 10mM EDTA, 10mM Na4P2O7, 10mM NaF, 1% Triton-X 100, 125mM NaCl, 10mM Na3VO4, 10μg/ml each aprotinin and leupeptin). Post-nuclear lysates were boiled in Laemmli Sample Buffer and were separated by SDS/PAGE, transferred to nitrocellulose filters, probed with the antibody of interest and developed by enhanced chemiluminescence.
Microarray analysis
PBMs were isolated from 4 separate donors and resuspended in RPMI 1640 with 5% heat-inactivated FBS at 5 million cells per ml. The monocytes were then infected with F. novicida at 100 MOI for 24 hours. Bone marrow-derived macrophages were isolated and differentiated from 3 C57Bl/6 mice as described previously18. Seven days after differentiation, macrophages from each mouse were plated into 2 wells of a 6-well dish at 2.5 million per well in RPMI containing 5% heat-inactivated FBS. F. novicida was added to each well at an MOI of 100. Cells were gently mixed and incubated at 37°C in 5% CO2 for 24 h.
Twenty four hours post-infection, RNA was extracted from both PBMs and BMMs using TRIzol® Reagent (Invitrogen Life Technologies, Carlsbad, CA), column-purified using RNeasy columns (Qiagen, Valencia, CA) and hybridized to Affymetrix GeneChip® Human Genome 133 plus 2.0 and Mouse Genome 430 2.0 Arrays (Affymetrix, Santa Clara, CA). Expression values were calculated using the “gcrma” package in BioConductor (www.bioconductor.org) and the “limma” package (Smyth, 2004) was used to find genes significantly different (p ≤0.05) between infected and uninfected samples.
siRNA transfection
RAW 264.7 cells were transfected with either control siRNA or SOCS3-specific siRNA using the Amaxa Nucleofector (Amaxa biosystems, Germany) as previously described18. Briefly, 7×106 cells were resuspended in 100 μl Nucelofector Solution V and were nucelofected with 10 μl of 100μM siRNA. Immediately post- nucleofection, 500 μl of pre-warmed RPMI was added to the transfection mix before transferring to 12-well plates containing 1.5 ml pre-warmed RPMI per well. Plates were incubated for 16 hours at 37°C before infections were performed.
ELISA measurement of cytokine production
Raw 264.7 cells were infected with F. novicida for varying time points. Cell supernatants were harvested, centrifuged to remove dead cells and analyzed by ELISA using an IP-10 specific kit from R & D Systems (Minneapolis, MN). Data were analyzed using an unpaired Student’s t-test. A p value ≤ 0.05 was considered significant.
Intracellular survival assay
This assay was performed as previously reported with a few modifications18. Briefly, 90 min post-infection with 100 MOI of F. novicida, cells were washed twice with RPMI and further incubated for 22 h. The cell cultures were treated with 50 μg/ml of gentamicin for 60 min at 37°C and 5 % CO2, washed twice with RPMI and subsequently lysed in 0.1 % SDS for 5 minutes. Immediately, 10 fold serial dilutions were made and appropriate dilutions were plated on Chocolate II agar plates. Assays were performed in triplicate for each test group. To maintain constant infection and IFNγ exposure periods in “pre-, co- and post-groups” the following protocol was followed. Briefly, within each group two parallel sets of macrophage cultures were infected for a constant infection period. One set of the samples in each group was exposed to 25 ng/ml of IFNγ for 24 hours to maintain constant IFNγ exposure across the groups. Subsequently, both sets in a group were processed to measure the intra-cellular number of the bacteria, and the bacterial count in IFNγ-treated samples was expressed as a percentage of bacterial number obtained in untreated samples of that group.
Measurement of nitrite by Griess Reagent
The levels of NO were measured by assaying the culture supernatants for NO2−, a stable product of NO with molecular oxygen. The assay was performed as previously described19. Macrophages were plated in RPMI containing no phenol red and no serum. Cells were then infected with 10 MOI of F. novicida for 24 hours and where indicated cells were simultaneously exposed to bacteria and 25 ng/ml of IFNγ. Harvested cell supernatants were centrifuged to clear any dead cells and the concentration of nitrite in the cell supernatants was estimated by Griess reaction. For nitrite assay, 100 μl of supernatant was mixed with 50 μl of Griess Reagent and incubated at room temperature in dark for 30 minutes. The absorbance was measured at 520 nm and nitrite concentrations were calculated from a standard curve obtained using standards containing increasing concentration of NaNO2.
Results
Francisella suppresses IFNγ-induced STAT1 phosphorylation
IFNγ orchestrates diverse immune processes that are critical for efficient pathogen clearance. Recently Santic et al. demonstrated that phagosomes containing Francisella are able to fuse with lysosomes in IFNγ-activated macrophages9. Thus, we hypothesized that Francisella may hinder the IFNγ-mediated host response. To test this hypothesis, we examined tyrosine phosphorylation of the downstream STAT1 (pY STAT1). For this, RAW 264.7 cells were infected with F. novicida in the presence or absence of IFNγ. Unless indicated otherwise both bacteria and IFNγ were added at the same time in all the experiments. Protein-matched lysates were analyzed by Western blotting with antibodies specific for tyrosine phosphorylated STAT1 (Figure 1A, upper panel). IFNγ treatment of the RAW 264.7 cells resulted in a robust induction of STAT1 phosphorylation which was dampened by infection with F. novicida (lane 4). To ensure equal loading of protein in all lanes the same membranes were reprobed with actin antibody. STAT-1 protein expression is upregulated by IFNγ treatment. Interestingly, we also observed that Francisella partially prevented the IFNγ-mediated induction of STAT1 protein itself (Figure 1B). The decrease in the induction of IFNγ-mediated STAT1 protein during Francisella infection is likely a consequence of the suppressed activation of STAT1, which can feed back to up-regulate itself. Thus, hereafter we have assessed the tyrosine phosphorylation levels of STAT1 which reflect the activation status of IFNγ signaling cascade. The reduction in IFNγ-induced STAT1 phosphorylation when challenged with Francisella was also observed in primary bone marrow-derived macrophages (BMM; Figure 1C).
Figure 1. Francisella suppresses IFNγ-induced STAT1 phosphorylation.

A. RAW 264.7 were infected with 10 MOI of F. novicida (FN) in the presence or absence of IFNγ (25 ng/ml) for 24 h. Protein-matched lysates were resolved by SDS PAGE and analyzed by Western blotting with phosphotyrosine STAT1 (pY STAT1) antibody (upper panel). The same membranes were re-probed with actin antibody (lower panel). The results are representative of six independent experiments. B. RAW 264.7 cells were infected with 10 MOI of F. novicida (FN) in the presence or absence of IFNγ (25 ng/ml) for the indicated time points. Protein-matched lysates were resolved by SDS PAGE and analyzed by Western blotting with STAT1 antibody (upper panel) and later with actin antibody (lower panel). These results are representative of three similar experiments. C. BMMs were infected with 10 MOI of F. novicida (FN) in the presence or absence of IFNγ (25 ng/ml) for 24 h and protein-matched lysates were resolved by SDS PAGE and analyzed by Western blotting with pY STAT1 antibody (upper panel). The same membrane was re-probed with actin antibody (lower panel). These results are representative of two independent experiments. D. Raw 264.7 cells were infected with 1, 10 or 100 MOI of FN for 24 hours in the presence or absence of IFNγ and the levels of pY STAT1 were analyzed by Western blotting (upper panel). The lower panel is a reprobe of the same membrane with actin antibody. These results are representative of two independent experiments. E. Raw 264.7 were infected with 10 MOI of F. novicida (FN) or F. tularensis LVS in the presence or absence of IFNγ (25 ng/ml) for 24 h. Protein-matched lysates were analyzed by Western blotting with pY STAT1 antibody (upper panel). The same membranes were reprobed with actin antibody (lower panel). These results are representative of three independent experiments. F. RAW 264.7 cells were infected in the presence or absence of IFNβ (500 IU/ml). 24 hours after the infection, the phosphorylation of STAT1 was assessed by Western blotting. Results are representative of three independent experiments. In all experiments described above, bacteria and IFNγ were added to the macrophages at the same time (co-stimulation). R, resting.
To examine whether the suppressive effect of F. novicida on IFNγ-induced pY STAT1 levels is dose-dependent, RAW 264.7 cells were infected with increasing MOI in the presence or absence of IFNγ. The results indicated that there is a decrease in IFNγ-induced STAT1 phosphorylation at all of the multiplicities of infection tested (Figure 1D). Further characterization revealed that this suppressive effect of F. novicida on IFNγ-induced STAT1 phosphorylation was seen as early as 1 hour (data not shown).
To examine whether F. tularensis LVS (Type B, vaccine strain) could likewise suppress IFNγ-induced STAT1 phosphorylation, similar infection experiments were performed with this latter strain. The results shown in Figure 1E demonstrate that F. tularensis LVS is also capable of suppressing IFNγ-induced STAT1 phosphorylation. A recent study reported that Type 1 IFN is produced and is important for the inflammasome activation during Francisella infection 20. In addition to STAT1-STAT2 heterodimers, STAT1 homodimers are also formed during Type 1 interferon signaling, although to a lesser extent. So we have examined the influence of Francisella on Type 1 interferon signaling as well and observed that Francisella is capable of suppressing STAT1 phosphorylation induced by IFNβ (Figure 1F). Thus, it is possible that Francisella may attempt to interfere with the signaling induced by IFNs in general.
Phagosomal escape, replication and viability of Francisella are dispensable for its suppressive effect on IFNγ-induced STAT1 phosphorylation
We next examined the bacterial factors that may be responsible for Francisella-mediated down-regulation of IFNγ-induced STAT1 phosphorylation. MglA is a global transcriptional regulator of Francisella essential for the expression of many virulence genes. In addition, MglA is critical for the bacterial escape from the phagosome4. To test the possible involvement of any MglA-dependent protein/event in the Francisella-mediated suppression of IFNγ signaling, RAW 264.7 cells were infected with F. novicida or an mglA mutant of F. novicida (FN mglA), in the presence or absence of IFNγ. In parallel, we examined whether bacterial replication is required for the suppression of IFNγ-induced STAT1 phosphorylation. For this, we used an iglD mutant of F. novicida. IglD is an MglA-dependent protein that was recently reported to be critical for the replication of the bacteria within host cells21. Protein-matched lysates were resolved by SDS/PAGE and analyzed by Western blotting with pY STAT1 antibody (Figure 2A, upper panel). Results indicated that both FN mglA and FN iglD effectively suppressed IFNγ-induced STAT1 phosphorylation, indicating that the suppressive effect is not dependent on phagosomal escape nor intracellular replication of Francisella. Of note, we also tested the number of viable bacteria in the inoculum immediately after infection to confirm that a comparable MOI was achieved in these experiments.
Figure 2. Phagosomal escape, replication and viability of Francisella are dispensable for the inhibition of IFNγ signaling.
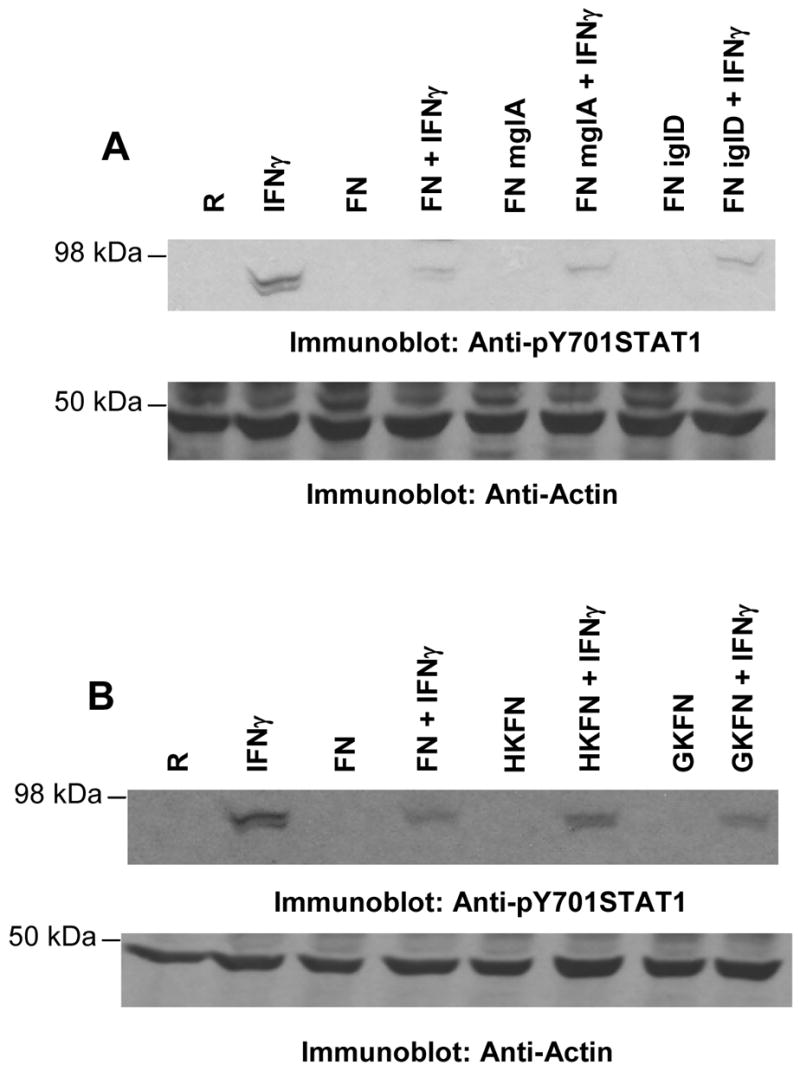
A & B RAW 264.7 cells were infected with wild type (FN), FN mglA or FN iglD (A) and with live (FN), heat-killed (HKFN) or gentamicin-killed (GKFN) bacteria (B) in the presence or absence of IFNγ for 24 h. Protein-matched lysates were analyzed by immunoblotting with pY STAT1 antibody (upper panels). The same membranes were reprobed with actin antibody to ensure equal loading (lower panels). In all experiments described above bacteria and IFNγ were added to the macrophages at the same time (co-stimulation). R, resting. These results are representative of four independent experiments.
To examine the possible involvement of an inducible bacterial factor in the down-regulation of IFNγ-induced STAT1 phosphorylation, macrophages were infected with live or killed bacteria in the presence or absence of IFNγ, and pY STAT1 levels were analyzed by Western blotting (Figure 2B). Bacteria were killed by either heat treatment (HKFN) or exposure to gentamicin (GKFN). Both live and dead bacteria effectively suppressed IFNγ-induced pY STAT1. These results suggest that a constitutively-expressed and heat-stable bacterial factor is involved in the suppression of IFNγ-induced STAT1 phosphorylation.
Francisella-induced SOCS3 expression dampens STAT1 phosphorylation
We next examined the molecular mechanism of Francisella-mediated suppression of IFNγ-induced STAT1 phosphorylation. Here, we considered two possibilities: a) that Francisella down-regulates positive regulators upstream of STAT1 phosphorylation, and/or b) that Francisella up-regulates negative regulators of STAT1 phosphorylation. To address whether positive regulators are down-regulated, Raw 264.7 cells were infected with F. novicida in the presence or absence of IFNγ and the expression levels of JAK1 and JAK2 were assessed by Western blot analysis. In parallel, phosphorylation (indicative of activation) of JAK1 and JAK2 were assessed by Western blotting with phospho-specific antibodies to JAK1 and JAK2. Results indicated that there is no suppressive effect on the expression nor activation of JAK1 and JAK2 during infection (data not shown).
Since the tyrosine phosphorylation level of the JAKs was not suppressed by F. novicida infection, we did not examine the activation of SHP-1, the tyrosine phosphatase that dephosphorylates JAK kinases. However, we examined whether other negative regulators of IFNγ signaling, the SOCS proteins, were up-regulated during infection. SOCS1 and SOCS3 are established negative regulators of the IFNγ signaling pathway. Down-regulation of IFNγ-induced signaling by several pathogens is associated with the up-regulation of SOCS3 protein17,22–24. To this end, we began by examining microarray results from an experiment comparing infected and uninfected BMMs and analyzed the expression of various SOCS proteins. Results shown in Figure 3A indicate that during F. novicida infection SOCS3 but not SOCS1 mRNA was highly up-regulated in murine BMMs. The expression of SOCS3 was confirmed at the level of protein by Western blotting in RAW 264.7 cells (Figure 3B). Infection alone induced the expression of SOCS3 as early as 6h post-infection and the protein levels persisted even at 24h post-infection (Figures 3 and data not shown). In addition, co-stimulation of macrophages with F. novicida and IFNγ resulted in a further increase in the SOCS3 protein levels. Similar results were obtained with F. tularensis LVS (data not shown). Consistently, phagosomal escape, replication and viability of the bacteria, which were not necessary for the suppression of IFNγ-induced STAT1 phosphorylation, were also found to be dispensable for the Francisella-induced up-regulation of SOCS3 protein (data not shown).
Figure 3. Francisella induces the expression of SOCS3.
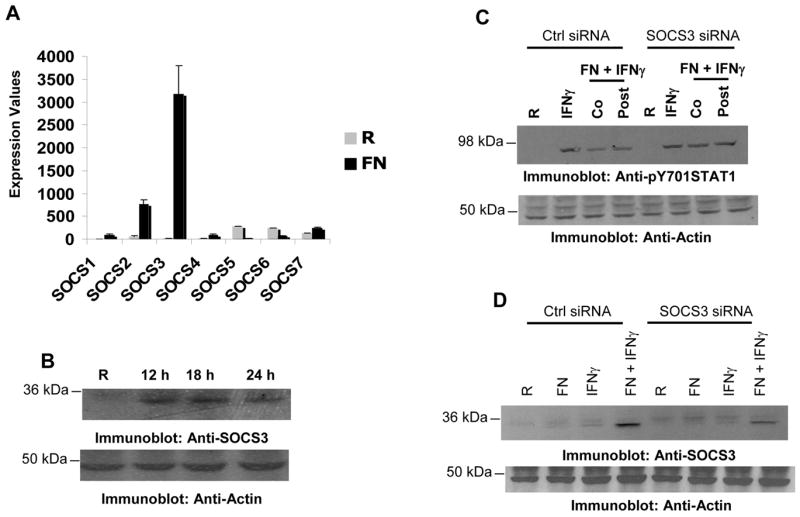
A. Bone marrow-derived macrophages (BMM) were infected with 100 MOI of F. novicida for 24 h. RNA was extracted, purified and microarray analysis was performed. Expression values for SOCS mRNA from three independent infections are shown. B. RAW 264.7 cells were infected with 10 MOI of F. novicida and the expression of SOCS3 protein was analyzed by Western blotting (upper panel). The same membrane was re-probed with actin antibody to ensure equal loading (lower panel). C. RAW 264.7 cells were nucleofected with either control or SOCS3-specific siRNA. 24 hours after transfection, cells were infected with F. novicida in the presence or absence of IFNγ (25 ng/ml) for 16 hours and the tyrosine phosphorylation of STAT1 was assessed by Western blotting. Co, IFNγ was added to the transfectants at the time of infection; Post, IFNγ was added 6 hours after infection; R, resting. These results are representative of three independent experiments. D. RAW 264.7 cells were transfected with control or SOCS3-specific siRNA and 16 hours after transfection, cells were infected with F. novicida in the presence or absence of IFNγ (25ng/ml) for 16 hours and expression of SOCS3 was analyzed by Western blotting (upper panel). The same membrane was reprobed with actin antibody to ensure equal loading. (lower panel). These results are representative of 4 similar and independent experiments.
We next tested whether Francisella-induced SOCS3 may contribute to the suppression of IFNγ-induced STAT1 phosphorylation. For this, RAW 264.7 cells were transfected with either control or SOCS3-specific siRNA. Approximately 16 hours after transfection, cells were infected and the tyrosine phosphorylation of STAT1 was assessed by Western blotting (Figure 3C). When exposed to both IFNγ and F. novicida, SOCS3-specific siRNA transfectants displayed higher STAT1 phosphorylation levels than control transfectants. This suggests that SOCS3 may at least in part contribute to the Francisella-mediated suppression of IFNγ-induced STAT1 phosphorylation. In parallel, we also examined the levels of SOCS3 protein in the transfectants by Western blotting with SOCS3 antibody and found that cells transfected with SOCS3-specific siRNA contained less SOCS3 protein than the control transfectants (Figure 3D).
Francisella suppresses IFNγ-induced STAT1 phosphorylation in human monocytes
To test whether Francisella can also down-regulate IFNγ-mediated STAT1 phosphorylation in human cells, we infected human peripheral blood monocytes (PBM) or THP-1 (human monocytic cell line) with F. novicida in the presence or absence of IFNγ. In all these experiments cells were exposed to IFNγ before infection with Francisella. Similar to the results obtained with murine macrophages (RAW 264.7 and BMMs), we observed that Francisella infection suppressed IFNγ-induced STAT1 phosphorylation in both PBMs and THP-1 cells (Figure 4A and 4B). Similar results were obtained when THP-1 cells were infected with F. tularensis LVS (Figure 4B). Moreover, the reduction in the IFNγ-induced STAT1 phosphorylation was evident as early as 2h post-infection of THP-1 cells (data not shown). Also consistent with the results obtained in RAW 264.7 cells, FN mglA or FN iglD effectively suppressed IFNγ-induced STAT1 phosphorylation (Figure 4B). Finally, SOCS3 gene expression was also found to be up-regulated during F. novicida infection of PBMs (Figure 4C).
Figure 4. Francisella suppresses IFNγ-induced STAT1 phosphorylation in human cells.

A. PBMs were pre-treated with IFNγ for 8 hours and then infected with 10 MOI of F. novicida in the presence or absence of IFNγ for 24 h. Protein-matched lysates were analyzed by Western blotting with pY STAT1 antibody (upper panel). The same membranes were re-probed with actin antibody (lower panel). B. THP-1 cells were pre-treated with IFNγ for 8 hours and then infected with 10 MOI of wild type (FN), FN mglA, FN iglD or F. tularensis LVS bacteria in the presence or absence of IFNγ. Tyrosine phosphorylation of STAT1 was assessed by Western blotting (upper panel). The same membranes were reprobed with actin antibody to ensure equal loading (lower panel). These data are representative of 3 independent experimenta. C. PBMs were infected with 100 MOI of F. novicida for 24 h. RNA was extracted, purified and microarray analysis was performed. Expression values for SOCS mRNA from four monocyte donors are shown. R, resting.
Francisella suppresses STAT1-dependent nitric oxide production
IFNγ treatment of macrophages is reported to induce the expression of iNOS leading to the production of NO, which is essential for killing pathogens25. In agreement with previous reports, we observed that co-stimulation of macrophages with IFNγ and F. novicida resulted in NO production19,26 (Figure 5A). Likewise, co-stimulation of cells with IFNγ and F. novicida resulted in the up-regulation of iNOS (Figure 5B). The observation that Francisella down-regulates IFNγ-induced STAT1 phosphorylation but does not suppress STAT1-dependent iNOS induction appeared paradoxical. We reasoned that Francisella may require some time to establish a permissive environment and be able to counter IFNγ-induced iNOS expression. To test this notion, we compared the induction levels of iNOS under priming, co-stimulatory and post-infection conditions. For this, IFNγ was added to RAW 264.7 cells 8h prior to infection (priming), at the time of infection (co-stimulation) or 8h after infection (post-infection) and the expression of iNOS under these conditions was analyzed by Western blotting (Figure 6A, upper panel). Infection alone or treatment with IFNγ alone induced very low levels of iNOS expression. The induction of iNOS was highly enhanced when IFNγ-primed macrophages were infected with F. novicida. However, when IFNγ was administered at the time of infection (co-stimulation) the induction of iNOS was dramatically reduced, and almost completely abrogated if IFNγ was administered post infection. Likewise, compared to the IFNγ priming condition, co-stimulation of macrophages with IFNγ and F. novicida resulted in lower levels of STAT1 phosphorylation, which is further reduced when cells were exposed to IFNγ after infection (Figure 6A, middle panel). A similar trend was seen with IFNγ-induced IP-10, an IFNγ-dependent chemokine (Figure 6B). Collectively, these data indicate that F. novicida establishes a permissive environment in the host cell over time to counteract the host protective effects of IFNγ.
Figure 5. Co-stimulation of macrophages with Francisella and IFNγ induces iNOS/nitric oxide.
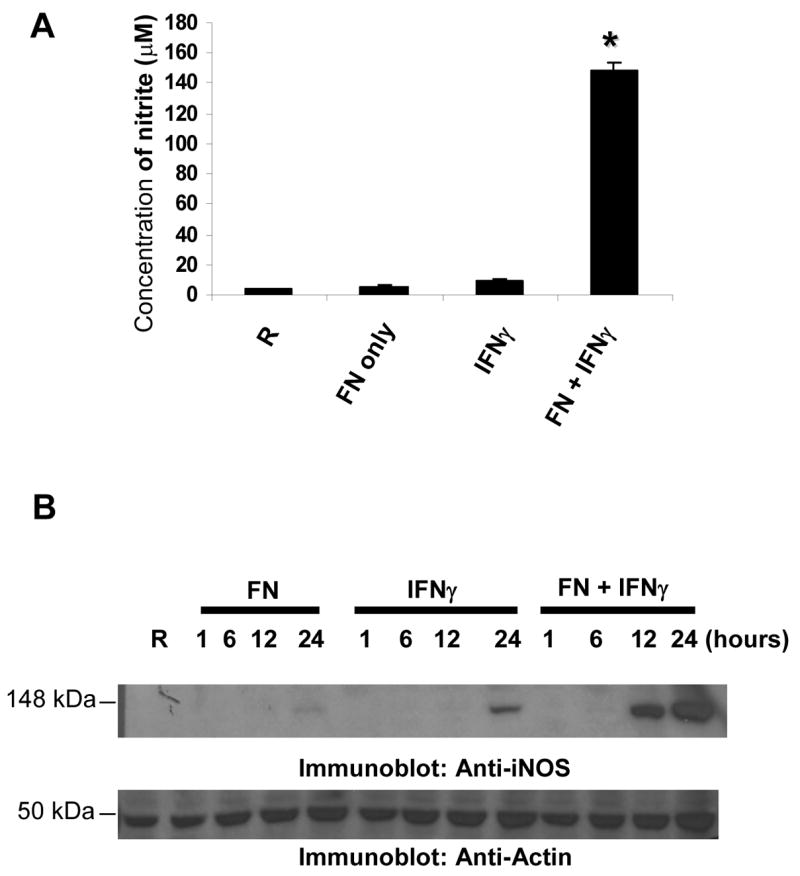
A. RAW 264.7 cells were infected with 10 MOI of F. novicida in the presence or absence of IFNγ (25 ng/ml; co-stimulation) for 24 h and the amount of NO in the cell supernatants was measured by the Griess reagent. The graphs show mean and SD of values obtained from 3 independent experiments. * p<0.05 compared with IFNγ value. B. RAW 264.7 cells were infected in the presence or absence of IFNγ for the indicated time points (co-stimulation) and protein-matched cell lysates were analyzed for the expression of iNOS by Western blotting. R, resting. These results are representative of three independent experiments.
Figure 6. Francisella suppresses IFNγ-induced iNOS leading to enhanced bacterial survival.

A. RAW 264.7 cells were exposed to IFNγ 8 h prior to infection (pre), at the time of infection (co) or 8 h post-infection (post) for a constant IFNγ-exposure period of 24 hours and the cellular levels of iNOS and pY STAT1 were analyzed by Western blotting. The same membranes were re-probed with actin antibody. These data are representative of three independent experiments. B. RAW 264.7 cells were treated as described in A and the amount of IP-10 produced was measured by ELISA. The graph shows mean and SD of values obtained from triplicate samples. These data are representative of 3 independent experiments. * p<0.05 compared with the IFNγ value. R, resting. C. RAW 264.7 cells were treated as described in the Materials and Methods section to maintain a constant infection period within each of the three groups (pre, co and post) and a constant IFNγ exposure period across the three groups. CFUs in samples that were not treated with IFNγ were set as 100%. The CFUs obtained from the samples treated with IFNγ were expressed as a percent of the corresponding non-IFNγ treated sample. The graph shows the mean and SD of values obtained from 3 independent experiments.
Establishment of Francisella infection in macrophages leads to resistance to IFNγ-induced bacterial death
Earlier studies demonstrated that prior exposure of infected cells to IFNγ drastically reduces the intra-macrophage survival of bacteria9. This is consistent with the induction of iNOS in our model. However, our data indicate that if IFNγ is administered either at the time of infection or after infection Francisella can suppress IFNγ-induced iNOS expression. Thus, to test if this reduction in iNOS levels correlates with an increase in bacterial survival, CFUs from infected cell lysates under various conditions of IFNγ exposure (priming, co-stimulatory and post-infection) were compared. The results are shown in Figure 6C. When IFNγ was added 8 hours prior to or at the time of infection, there was ~ 90% decrease in intracellular bacterial survival, compared to the survival in the absence of IFNγ administration. However, when IFNγ was added 8 hours after infection there was only a 30–40% reduction in intracellular bacterial survival. The reduced efficiency of IFNγ to induce bacterial death when added 8 hours post-infection correlates with the decrease in the levels of iNOS under these conditions as shown in Figure 6A.
Together, our results provide evidence that in both mouse and human cells Francisella suppresses IFNγ-induced STAT1 phosphorylation and this Francisella-mediated interference of IFNγ signaling is associated with an induction of SOCS3 (Figure 7). The results also show that SOCS3, at least in part, contributes to the Francisella-mediated suppression of IFNγ-induced STAT1 phosphorylation. Further, Francisella-mediated suppression of IFNγ-induced STAT1 phosphorylation results in a lower induction of STAT1-dependent proteins such as iNOS, leading to enhanced intra-macrophage survival of the bacteria.
Figure 7. Proposed model of Francisella-mediated interference of IFNγ signaling response.
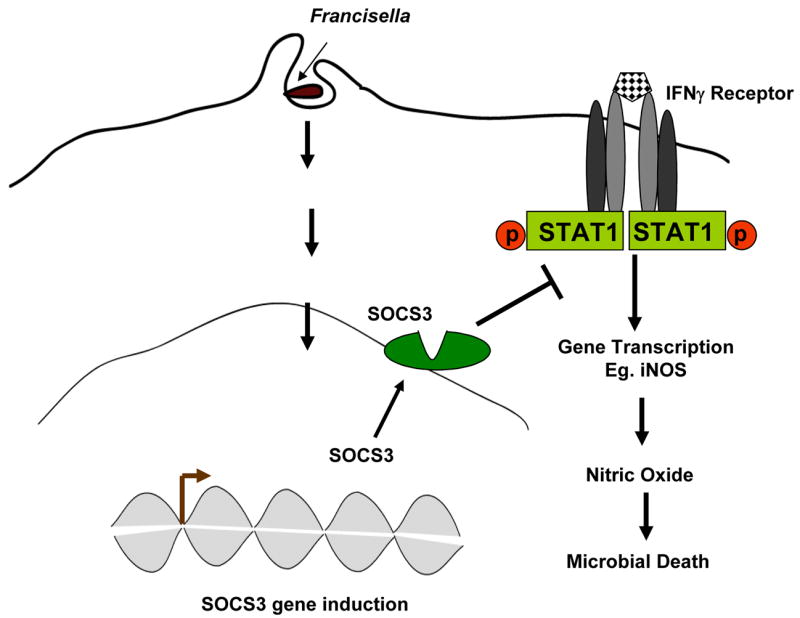
Francisella infection up-regulates SOCS3 expression, which suppresses STAT1 phosphorylation potentially by binding to the IFNγ receptor and dampening subsequent recruitment and activation of STAT1. Ultimately, Francisella-mediated suppression of the IFNγ response leads to the inhibition of IFNγ-induced iNOS and other anti-microbial events, resulting in enhanced bacterial survival in host cells.
Discussion
Phagocytic cells such as monocytes and macrophages ingest and subsequently destroy pathogens through the phago-lysosomal pathway and the production of inflammatory mediators such as cytokines. These microbicidal activities are greatly enhanced by exposure of the cells to IFNγ. Such IFNγ-activated macrophages are often referred to as classically activated or Type I activated macrophages. Highly successful pathogens have developed various strategies to subvert microbicidal responses of the host cell and, instead, create a favorable intracellular environment to suit their needs. Our knowledge regarding the immune evasion mechanisms utilized by Francisella remains limited. These mechanisms have been reported to include evasion of phago-lysosomal fusion2,3, dampening of the inflammatory response upon escape into the cytosol27,28 and abrogation of T cell responses29. One strategy employed by pathogens such as Leishmania donovani13, Mycobacterium avium14, Mycobacterium tuberculosis15,16, and Listeria monocytogenes17 is to interfere with the IFNγ-mediated activation of macrophages. In this process, pathogens such as Leishmania donovani inhibit IFNγ-induced STAT1 activation13 whereas Mycobacterium tuberculosis does not influence STAT1 activation but exerts a gene-selective inhibition of IFNγ-induced transcriptional responses by disrupting its interaction with transcriptional co-activators15. Results from the present study provide evidence that Francisella suppresses the IFNγ-mediated signaling response and gene expression which results in decreased microbicidal activity of the host cell.
The inhibitory effect of Francisella on IFNγ-mediated signaling does not require the bacteria to be metabolically active. Our finding that killed Francisella also downregulates IFNγ signaling is consistent with the findings obtained with other pathogens such as L. donovani24 and Mycobacterium bovis23. Together, these data provide evidence that a heat-stable bacterial component is responsible for the down-regulation of IFNγ-induced STAT1 tyrosine phosphorylation. This observation is in accord with the results obtained in other studies which demonstrate that cells treated with heat-stable bacterial components such as LPS30 or Mycobacterial TDM23 efficiently suppressed IFNγ-induced STAT1 phosphorylation. In addition, we have observed that prevention of bacterial uptake by treating the cells with cytochalasin D did not reverse the Francisella-mediated inhibition of IFNγ signaling, suggesting that bacterial contact itself is sufficient to cause this effect (data not shown). Perhaps the ligation of a host cell receptor by a surface bacterial component such as LPS may contribute to the interference of IFNγ signaling. For example the inhibition of IFNγ-mediated responses by Mycobacterium tuberculosis31–33 and Mycobacterium avium32 is at least partly dependent upon TLR2 engagement. TLR2 is one of the major macrophage receptors involved in the recognition of Francisella34–36. This tempts us to speculate that TLR2 may play a critical role in Francisella-mediated interference of IFNγ signaling as well.
In our murine model, Francisella infection did not reduce the IFNγ-induced phosphorylation of JAK tyrosine kinases, which is in contrast with findings obtained with other intracellular pathogens13,23. Instead, we found that Francisella-mediated suppression of IFNγ signaling is associated with the up-regulation of SOCS3 and that knock-down of SOCS3 partially restored the IFNγ-induced STAT1 phosphorylation during Francisella infection. The interference of IFNγ signaling by several pathogens has been correlated with expression of either SOCS1 or SOCS3 or both. For example, Salmonella enterica serovar Typhi induces both SOCS1 and SOCS3 expression while Burkholderia pseudomallei induces only SOCS3 expression22. SOCS proteins are suppressors of cytokine signaling that form a key part of the negative feedback of cytokine signal transduction. Our microarray data obtained with infected murine BMMs and human PBMs indicate that SOCS3 but not SOCS1 is highly up-regulated during Francisella infection. SOCS1 can directly interact with JAK1 and JAK2 and inhibit their phosphorylation and kinase activity 37. Therefore, the unchanged levels of IFNγ-induced JAK phosphorylation during Francisella infection correlate with the lack of induction of SOCS1 in our experiments.
The mechanism of SOCS3 action is not clear. One study showed that although SOCS3 associated with the JAK kinases, overexpression of SOCS3 did not inhibit the in vitro kinase activity of JAK1 or JAK238. Other studies showed that SOCS3 binds to phosphorylated receptors including the leptin receptor39 and IL-2 receptor β chain40. When bound to the activated receptors, SOCS3 may inhibit JAK2 activation. However, high levels of activated growth hormone receptor were found to be essential for SOCS3-mediated inhibition of growth hormone induced JAK2 activation41.
Our data demonstrate that akin to the suppression of IFNγ-induced STAT1 phosphorylation, a heat-stable bacterial factor(s), but not live bacteria, is essential for the induction of SOCS3. These results are in agreement with results from other studies showing purified LPS30 of other bacteria or mycobacterial TDM23 could activate SOCS3 expression. Ongoing experiments are aimed at understanding the molecular mechanism(s) of Francisella-induced SOCS3 expression. One formal possibility is that the induction of SOCS3 is mediated by feedback signaling initiated by cytokines secreted by infected cells. Since cytokine induction during Francisella infection is dependent on host cell contact with the bacteria via TLR2 but independent of bacterial uptake or viability, this is consistent with the induction of SOCS3 expression.
The observation that iNOS induction is suppressed during Francisella infection is consistent with previous findings that over-expression of SOCS3 results in the suppression of the mouse iNOS gene promoter30. Interestingly, we observed that Francisella requires some time (in the absence of IFNγ exposure) for the establishment of maximal inhibition of host-protective events. Such a requirement was also observed in the case of Burkholderia pseudomallei infection22. This suggests that Francisella regulates the host cell response to protective inflammatory mediators over time, the net result of which is increased intracellular survival. Thus, there continues to be a dynamic interplay between the pathogen and host cell during the course of infection.
Data obtained from various mouse models strongly support the protective role of IFNγ during tularemia42,43. Leiby et al. have shown that intra-peritoneal injections of IFNγ neutralizing antibody at the time of infection resulted in the death of mice even at a very low dose of infection44. On the other hand, administration of the IFNγ neutralizing antibody two days post infection did not significantly influence the progress of the disease, suggesting that the protective effect of IFNγ is most effective at early time points after infection. This is consistent with our infection model where IFNγ is ineffective at protecting host cells if administered after the infection has occurred. Such strict time dependence for IFNγ therapy is also seen with Listeria monocytogenes, a pathogen previously reported to suppress the IFNγ response45. Further, Conlan et al observed that neutralizing antibodies against IFNγ did not alter the progress of primary murine tularemia initiated through intra-nasal administration of Francisella46. Thus, based on our studies it may be speculated that alveolar macrophages may be more effective in suppressing IFNγ responses and may thus contribute to the rapid lethality observed during the pneumonic form of tularemia.
As described above, in mouse models of tularemia, IFNγ is produced but mice still succumb. This could be partly explained by the ability of Francisella to induce SOCS3 and thereby potentially circumvent the IFNγ-mediated survival benefit. To our knowledge, this is the first study to demonstrate Francisella-mediated suppression of IFNγ signaling and provide evidence for the functional consequence of this signaling interference.
Acknowledgments
We thank Dr. Yousef Abu Kwaik for the availability of iglD mutant of Francisella novicida. We thank Thomas Cremer for assistance with the experiments and Dr. Melissa Hunter for kindly providing JAK1, 2 and pJAK2 antibodies. We thank Luke Davis for assistance with blood collection. This work was sponsored by the NIH/NIAID Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (RCE) Program. The authors wish to acknowledge membership within and support from the Region V ‘Great Lakes’ RCE (NIH award 1-U54-AI-057153). This work was also supported by NIH grants R01 AI059406 and P01 CA095426 to ST. JPB is supported by T32CA090223.
Abbreviations used in this paper
- IFNγ
Interferon gamma
- STAT1
signal transducer and activator of transcription 1
- SOCS
suppressor of cytokine signaling
- iNOS
inducible nitric oxide synthase
- BMM
bone marrow-derived macrophages
- PBM
peripheral blood monocytes
- MOI
multiplicity of infection
- HKFN
heat killed Francisella novicida
- GKFN
gentamicin killed Francisella novicida
- CFU
colony forming unit
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol. 2004;2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 2.Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjostedt A. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun. 2003;71:5940–5950. doi: 10.1128/IAI.71.10.5940-5950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clemens DL, Lee BY, Horwitz MA. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun. 2004;72:3204–3217. doi: 10.1128/IAI.72.6.3204-3217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santic M, Molmeret M, Klose KE, Jones S, Kwaik YA. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol. 2005;7:969–979. doi: 10.1111/j.1462-5822.2005.00526.x. [DOI] [PubMed] [Google Scholar]
- 5.Nano FE, Zhang N, Cowley SC, Klose KE, Cheung KK, Roberts MJ, Ludu JS, Letendre GW, Meierovics AI, Stephens G, Elkins KL. A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol. 2004;186:6430–6436. doi: 10.1128/JB.186.19.6430-6436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohapatra NP, Soni S, Bell BL, Warren R, Ernst RK, Muszynski A, Carlson RW, Gunn JS. Identification of an orphan response regulator required for Francisella virulence and transcription of pathogenicity island genes. Infect Immun. 2007 doi: 10.1128/IAI.00351-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohapatra NP, Balagopal A, Soni S, Schlesinger LS, Gunn JS. AcpA is a Francisella acid phosphatase that affects intramacrophage survival and virulence. Infect Immun. 2007;75:390–396. doi: 10.1128/IAI.01226-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindgren H, Golovliov I, Baranov V, Ernst RK, Telepnev M, Sjostedt A. Factors affecting the escape of Francisella tularensis from the phagolysosome. J Med Microbiol. 2004;53:953–958. doi: 10.1099/jmm.0.45685-0. [DOI] [PubMed] [Google Scholar]
- 9.Santic M, Molmeret M, Abu KY. Modulation of biogenesis of the Francisella tularensis subsp novicida-containing phagosome in quiescent human macrophages and its maturation into a phagolysosome upon activation by IFN-gamma. Cell Microbiol. 2005;7:957–967. doi: 10.1111/j.1462-5822.2005.00529.x. [DOI] [PubMed] [Google Scholar]
- 10.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 11.Butchar JP, Rajaram MV, Ganesan LP, Parsa KV, Clay CD, Schlesinger LS, Tridandapani S. Francisella tularensis induces IL-23 production in human monocytes. J Immunol. 2007;178:4445–4454. doi: 10.4049/jimmunol.178.7.4445. [DOI] [PubMed] [Google Scholar]
- 12.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 13.Ray M, Gam AA, Boykins RA, Kenney RT. Inhibition of interferon-gamma signaling by Leishmania donovani. J Infect Dis. 2000;181:1121–1128. doi: 10.1086/315330. [DOI] [PubMed] [Google Scholar]
- 14.Hussain S, Zwilling BS, Lafuse WP. Mycobacterium avium infection of mouse macrophages inhibits IFN-gamma Janus kinase-STAT signaling and gene induction by down-regulation of the IFN-gamma receptor. J Immunol. 1999;163:2041–2048. [PubMed] [Google Scholar]
- 15.Ting LM, Kim AC, Cattamanchi A, Ernst JD. Mycobacterium tuberculosis inhibits IFN-gamma transcriptional responses without inhibiting activation of STAT1. J Immunol. 1999;163:3898–3906. [PubMed] [Google Scholar]
- 16.Kincaid EZ, Ernst JD. Mycobacterium tuberculosis exerts gene-selective inhibition of transcriptional responses to IFN-gamma without inhibiting STAT1 function. J Immunol. 2003;171:2042–2049. doi: 10.4049/jimmunol.171.4.2042. [DOI] [PubMed] [Google Scholar]
- 17.Stoiber D, Stockinger S, Steinlein P, Kovarik J, Decker T. Listeria monocytogenes modulates macrophage cytokine responses through STAT serine phosphorylation and the induction of suppressor of cytokine signaling 3. J Immunol. 2001;166:466–472. doi: 10.4049/jimmunol.166.1.466. [DOI] [PubMed] [Google Scholar]
- 18.Parsa KV, Ganesan LP, Rajaram MV, Gavrilin MA, Balagopal A, Mohapatra NP, Wewers MD, Schlesinger LS, Gunn JS, Tridandapani S. Macrophage pro-inflammatory response to Francisella novicida infection is regulated by SHIP. PLoS Pathog. 2006;2:e71. doi: 10.1371/journal.ppat.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loegering DJ, Drake JR, Banas JA, McNealy TL, Mc Arthur DG, Webster LM, Lennartz MR. Francisella tularensis LVS grown in macrophages has reduced ability to stimulate the secretion of inflammatory cytokines by macrophages in vitro. Microb Pathog. 2006;41:218–225. doi: 10.1016/j.micpath.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henry T, Brotcke A, Weiss DS, Thompson LJ, Monack DM. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J Exp Med. 2007;204:987–994. doi: 10.1084/jem.20062665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santic M, Molmeret M, Barker JR, Klose KE, Dekanic A, Doric M, Kwaik YA. A Francisella tularensis pathogenicity island proteins essential for bacterial proliferation within the host cell cytosol. Cell Microbiol. 2007 doi: 10.1111/j.1462-5822.2007.00968.x. In Press. [DOI] [PubMed] [Google Scholar]
- 22.Ekchariyawat P, Pudla S, Limposuwan K, Arjcharoen S, Sirisinha S, Utaisincharoen P. Burkholderia pseudomallei-induced expression of suppressor of cytokine signaling 3 and cytokine-inducible src homology 2-containing protein in mouse macrophages: a possible mechanism for suppression of the response to gamma interferon stimulation. Infect Immun. 2005;73:7332–7339. doi: 10.1128/IAI.73.11.7332-7339.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imai K, Kurita-Ochiai T, Ochiai K. Mycobacterium bovis bacillus Calmette-Guerin infection promotes SOCS induction and inhibits IFN-gamma-stimulated JAK/STAT signaling in J774 macrophages. FEMS Immunol Med Microbiol. 2003;39:173–180. doi: 10.1016/S0928-8244(03)00231-1. [DOI] [PubMed] [Google Scholar]
- 24.Bertholet S, Dickensheets HL, Sheikh F, Gam AA, Donnelly RP, Kenney RT. Leishmania donovani-induced expression of suppressor of cytokine signaling 3 in human macrophages: a novel mechanism for intracellular parasite suppression of activation. Infect Immun. 2003;71:2095–2101. doi: 10.1128/IAI.71.4.2095-2101.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakravortty D, Hensel M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 2003;5:621–627. doi: 10.1016/s1286-4579(03)00096-0. [DOI] [PubMed] [Google Scholar]
- 26.Lindgren H, Stenman L, Tarnvik A, Sjostedt A. The contribution of reactive nitrogen and oxygen species to the killing of Francisella tularensis LVS by murine macrophages. Microbes Infect. 2005;7:467–475. doi: 10.1016/j.micinf.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 27.Telepnev M, Golovliov I, Sjostedt A. Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mononuclear cells. Microb Pathog. 2005;38:239–247. doi: 10.1016/j.micpath.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Rajaram MV, Ganesan LP, Parsa KV, Butchar JP, Gunn JS, Tridandapani S. Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J Immunol. 2006;177:6317–6324. doi: 10.4049/jimmunol.177.9.6317. [DOI] [PubMed] [Google Scholar]
- 29.Woolard MD, Wilson JE, Hensley LL, Jania LA, Kawula TH, Drake JR, Frelinger JA. Francisella tularensis-infected macrophages release prostaglandin E2 that blocks T cell proliferation and promotes a Th2-like response. J Immunol. 2007;178:2065–2074. doi: 10.4049/jimmunol.178.4.2065. [DOI] [PubMed] [Google Scholar]
- 30.Crespo A, Filla MB, Murphy WJ. Low responsiveness to IFN-gamma, after pretreatment of mouse macrophages with lipopolysaccharides, develops via diverse regulatory pathways. Eur J Immunol. 2002;32:710–719. doi: 10.1002/1521-4141(200203)32:3<710::AID-IMMU710>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 31.Banaiee N, Kincaid EZ, Buchwald U, Jacobs WR, Jr, Ernst JD. Potent inhibition of macrophage responses to IFN-gamma by live virulent Mycobacterium tuberculosis is independent of mature mycobacterial lipoproteins but dependent on TLR2. J Immunol. 2006;176:3019–3027. doi: 10.4049/jimmunol.176.5.3019. [DOI] [PubMed] [Google Scholar]
- 32.Lafuse WP, Alvarez GR, Curry HM, Zwilling BS. Mycobacterium tuberculosis and Mycobacterium avium inhibit IFN- gamma -induced gene expression by TLR2-dependent and independent pathways. J Interferon Cytokine Res. 2006;26:548–561. doi: 10.1089/jir.2006.26.548. [DOI] [PubMed] [Google Scholar]
- 33.Arko-Mensah J, Julian E, Singh M, Fernandez C. TLR2 but not TLR4 signalling is critically involved in the inhibition of IFN-gamma-induced killing of mycobacteria by murine macrophages. Scand J Immunol. 2007;65:148–157. doi: 10.1111/j.1365-3083.2006.01888.x. [DOI] [PubMed] [Google Scholar]
- 34.Katz J, Zhang P, Martin M, Vogel SN, Michalek SM. Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect Immun. 2006;74:2809–2816. doi: 10.1128/IAI.74.5.2809-2816.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malik M, Bakshi CS, Sahay B, Shah A, Lotz SA, Sellati TJ. Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect Immun. 2006;74:3657–3662. doi: 10.1128/IAI.02030-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Nookala S, Bina XR, Bina JE, Re F. Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J Leukoc Biol. 2006;80:766–773. doi: 10.1189/jlb.0406294. [DOI] [PubMed] [Google Scholar]
- 37.Kile BT, Alexander WS. The suppressors of cytokine signalling (SOCS) Cell Mol Life Sci. 2001;58:1627–1635. doi: 10.1007/PL00000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ, Nicola NA. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 1999;18:375–385. doi: 10.1093/emboj/18.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 40.Cohney SJ, Sanden D, Cacalano NA, Yoshimura A, Mui A, Migone TS, Johnston JA. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol. 1999;19:4980–4988. doi: 10.1128/mcb.19.7.4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen JA, Lindberg K, Hilton DJ, Nielsen JH, Billestrup N. Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol Endocrinol. 1999;13:1832–1843. doi: 10.1210/mend.13.11.0368. [DOI] [PubMed] [Google Scholar]
- 42.Anthony LS, Ghadirian E, Nestel FP, Kongshavn PA. The requirement for gamma interferon in resistance of mice to experimental tularemia. Microb Pathog. 1989;7:421–428. doi: 10.1016/0882-4010(89)90022-3. [DOI] [PubMed] [Google Scholar]
- 43.Pammit MA, Raulie EK, Lauriano CM, Klose KE, Arulanandam BP. Intranasal vaccination with a defined attenuated Francisella novicida strain induces gamma interferon-dependent antibody-mediated protection against tularemia. Infect Immun. 2006;74:2063–2071. doi: 10.1128/IAI.74.4.2063-2071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leiby DA, Fortier AH, Crawford RM, Schreiber RD, Nacy CA. In vivo modulation of the murine immune response to Francisella tularensis LVS by administration of anticytokine antibodies. Infect Immun. 1992;60:84–89. doi: 10.1128/iai.60.1.84-89.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakane A, Minagawa T, Kohanawa M, Chen Y, Sato H, Moriyama M, Tsuruoka N. Interactions between endogenous gamma interferon and tumor necrosis factor in host resistance against primary and secondary Listeria monocytogenes infections. Infect Immun. 1989;57:3331–3337. doi: 10.1128/iai.57.11.3331-3337.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Conlan JW, KuoLee R, Shen H, Webb A. Different host defences are required to protect mice from primary systemic vs pulmonary infection with the facultative intracellular bacterial pathogen, Francisella tularensis LVS. Microb Pathog. 2002;32:127–134. doi: 10.1006/mpat.2001.0489. [DOI] [PubMed] [Google Scholar]