

Abstract
Estrogen hormone (E2) signaling is primarily conveyed by the estrogen receptors (ER) α and β. ERs are encoded by two distinct genes and share varying degrees of domain-specific structural/functional similarities. ERs mediate a complex array of nuclear and non-nuclear events critical for the homeodynamic regulation of various tissue functions. The canonical nuclear signaling involves the interaction of ERα and ERβ with specific DNA sequences, the so-called estrogen responsive elements (EREs). This interaction constitutes the initial step in ERE-dependent signaling in which ERβ is a weaker transcription factor than ERα in response to E2. However, it remains unclear why transactivation potencies of ER subtypes differ. Studies suggest that the amino-terminus, the least conserved structural region, of ERβ, but not that of ERα, impairs the ability of the receptor to bind to ERE independent of E2. Although the impaired ERβ-ERE interaction contributes, it is not sufficient to explain the weak transactivation potency of the receptor. It appears that the lack of transactivation ability and of the capability of the amino-terminus of ERβ, as opposed to that of ERα, to functionally interact with the carboxyl-terminal hormone-dependent activation domain is also critical for the receptor-specific activity. Thus, the structurally distinct amino-termini of ERs are important determinants in defining the function of ER-subtypes in the ERE-dependent pathway. This could differentially affect the physiology and pathophysiology of E2 signaling.
Keywords: Estrogen, Estrogen Receptor, ERE, ER-ERE Interaction, Transcription
Introduction
The estrogen hormone, primarily 17β-estradiol (E2), information is primarily conveyed by the members of a nuclear receptor superfamily, estrogen receptor (ER) α and β [1, 2]. ERα and ERβ are distinct gene products and expressed in the same tissue as well as in different tissues at varying levels [1, 2]. ERα is the dominant species expressed in uterus, liver, adipose, skeletal muscle, pituitary and hypothalamus, whereas ERβ is the major form in ovary, testis and prostate, as well as some brain regions including the limbic system, cerebellum and cerebral cortex [2]. ERα and ERβ are co-expressed in breast tissue, the urogenital tract, bone and cardiovascular system within the same cell-type as well as in different cell populations [2].
ERs are modular in nature in that isolated structural domains display subsets of the functional activities of the intact receptor [1, 2]. The distinct amino terminal A/B domains of ERs share 17% amino-acid identity. The A/B domain of ERα contains a ligand-independent activation function (AF1), while the function of the amino-terminus of ERβ is unclear. The central C region of ERs is the DNA binding domain (DBD) and shows a near identical (97%) amino-acid homology. The flexible hinge, or D, domain contains a nuclear localization signal and links the C domain to the multi-functional carboxyl terminal (E/F) domain. E/F, which shares 56% amino-acid identity between ERs, is involved in ligand binding, dimerization, and ligand-dependent activation function (AF2). The E/F domain is also referred to as the ligand binding domain (LBD).
The effects of E2-ER are exerted through a complex array of convergent and divergent signaling pathways that mediate genomic and non-genomic events [1, 2]. The interaction of E2-ER with specific DNA sequences, the so-called estrogen responsive elements (EREs), constitutes one primary genomic signaling pathway. EREs are permutations of a palindromic DNA sequence with three central non-specific nucleotides, 5’-GGTCAnnnTGACC-3', and are primarily located at the promoter regions of E2 responsive genes [3].
Despite comparable ERE and ligand binding properties [4–6] arising from structural similarities between DBDs and LBDs, studies have indicated that E2-ERβ is a weaker transactivator than E2-ERα in the ERE-dependent signaling [4, 7–10]. We aim here to review recent findings to comparatively assess the functional differences between two ER subtypes in order to present a perspective about the mechanism of ERβ action in the ERE-dependent signaling pathway.
ApoERα-mediated nuclear signaling
Recent studies using fluorescence resonance energy transfer (FRET) approaches [11] show that apoERα, as well as apoERβ, dimerize and translocate to the nucleus, likely as a part of large protein complexes [12], independent of E2. Moreover, fluorescence recovery after photobleaching (FRAP) indicates that the nuclear apoERα is highly mobile molecules dynamically partitioned between nuclear matrix and target sites on chromatin [13].
Expression of the pS2 gene in cells derived from breast neoplasm expressing ER endogenously or exogeneously is augmented by E2 through an imperfect ERE [14, 15]. Chromatin immunoprecipitation (ChIP) approaches of the pS2 gene promoter as an ER-responsive model indicated that apoERα can be detected as the ERE-bound [16–18]. However, various coregulatory proteins, including histone acetylases (HATs), histone methytransferases (HMTs), chromatin remodelers as well as basal transcription machinery and active RNA polymerase II (Pol II) are also present on the pS2 promoter [17, 18]. This obscures whether the interaction of apoERα with the ERE initiates a sequence of events critical for basal transcription or poises the promoter for E2-augmented gene expression.
Detailed kinetic studies utilizing ChIP-based assays of the pS2 promoter previously cleared of transcription factors by α-amanitin treatment suggest that apoERα interacts with the ERE of the promoter cyclically [17, 18]. This episodic engagement involves both activating and repressing epigenetic processes that provide a mechanism enabling a rapid adaptation of transcription to E2 [17, 18]. It appears that the binding of apoERα to ERE initiates the association of chromatin remodeling complexes with the promoter. Additional recruitment, albeit inefficiently, likely by the ERE-bound-apoERα through both the amino- and carboxyl-termini [4, 19], of limited number of HMTs and HATs further modifies the local chromatin [17, 18]. These coregulator complexes alter the nucleosome structure by modifying the histone-DNA interface exposing the TATA box previously occluded by the histone core [17]. Some members of basal transcription machinery are subsequently recruited to the promoter. However, the absence of Pol II renders the complex transcriptionally silent. Subsequent ubiquitination of apoERα and associated factors disassemble the complex for proteasomal degradation [20]. This promoter clearance is also coupled with nucleosomal modifications as a result of the recruitment of protein complexes, exemplified by histone deacetylases (HDACs) [17, 18]. This remodeling provides a chromatin environment restrictive for transcription until the commencement of the next cycle.
E2-mediated ERα-ERE interactions
The LBD of ERs displays a fold with 12 α-helices (numbered H1–H12) arranged into three layers. The two parallel outer layers sandwich a central layer. This arrangement of helices forms the ligand-binding cavity which is flanked by the carboxyl terminal H12 [21, 22]. The binding of E2 to apoERα is accompanied by a major structural reorganization of the LBD. E2 binding realigns H12 across the LBD fold and buries the hormone within the cavity. This H12 repositioning together with residues from H3, H4 and H5 creates a shallow hydrophobic groove that serves as the docking site for nuclear receptor coregulators through one or more copies of an α-helix motif with the consensus sequence of LXXLL (L denotes lysine residues, X refers to any amino-acid) [22].
Studies have indicated that the binding of E2 dramatically enhances the affinity of the AF2 domain for coregulators [1, 2, 10]. FRET approaches in vitro [23] and in situ [11] show that the binding of E2 is also associated with the stability of the ERα dimer mediated by an extensive interface formed by H8 /H11 layer of the LBD fold [24].
More importantly, E2 binding enhances the association of ERα with ERE in situ as demonstrated by promoter interference [25], chromatin modeling [26] and ChIP [16, 27] assays. Although the mechanism is unclear, pre- and post-ERE binding events could participate in the E2-mediated augmentation of ERα-ERE interactions. One possible pre-ERE binding event involves allosteric alteration of the folding, or the stability of, the DBD of ERα upon binding to E2. This could lead to an increase in the population of the receptor capable of interacting with ERE. Alternatively, E2 mediates the dissociation of ERα from chaperones/nuclear matrix-associated proteins bound to the DBD, or to other regions that sterically block the DBD [28, 29]. This unmasks the DBD thereby allowing the interaction of ERα with ERE. E2 could also influence the intermolecular association of ERα with protein complexes to enhance the stability of ERα-ERE interactions [30–32]. Pre-ERE binding events could affect the partitioning of E2-ERα to chromatin from nuclear matrix reflected as a decrease in the mobility of the nuclear E2-ERα complex compared to apoERα [13].
Since the cyclic promoter interaction comprises assembly and disassembly of the transcription complex, post-ERE binding events could also contribute to the E2-mediated increase in ERα-ERE interaction. ChIP approaches further demonstrated that the binding of E2-ERα to the ERE of the pS2 gene promoter initiates a series of interdependent events that result in an extended periodicity of cyclic promoter engagement [17, 18, 33]. Following an initial transcriptionally silent cycle, analogous to that mediated by apoERα in the α-amanitin synchronized pS2 promoter, E2-ERα recruits many multisubunit coactivator complexes, enzymes of the ubiquitin-proteasome pathway, and the basal transcription machinery together with Pol II to initiate transcription. In addition to the ability of the ERα amino-terminus to interact with various coregulators independent of E2 [19, 34–36], the binding of E2 dramatically enhances the affinity of the LBD for coregulators [10]. An effective recruitment of coregulators by both the AF1 and AF2 domains of ERα could form a stable platform necessary for subsequent ordered and combinatorial recruitment of complexes for transcription. These events could lead to an increase in the duration of promoter occupancy of E2-ERα.
Kinetic ChIP analysis also indicates that at the end of a transcriptionally productive cycle, HDAC complexes are recruited by the activated Pol II in association with chromatin remodelers to modify local chromatin structure. Activities of these complexes restrict transcriptional engagement by repositioning nucleosomes to occlude ERE and the TATA box sequences. This leads to the dissociation of associated factors from the promoter and to transcription termination [17, 18, 33].
Although the formation of a stable and transcriptionally productive complex is required, it may not be sufficient to explain the E2-mediated increase in ERα-ERE interaction. The recruitment of ubiquitin-proteasome enzymes to the pS2 promoter and the prevention of transcription by the inhibition of proteasome function imply that transcription and degradation processes are inherently linked [17]. However, studies also showed that transcriptionally impaired ER variants with abrogated AF1 and/or AF2 functions display an E2-mediated increase in ERE binding and cyclical promoter occupancy that are similar to those observed with E2-ERα [37, 38]. ApoERα and E2-ERα are degraded through the ubiquitin-proteasome pathway by utilizing different mechanisms [37, 38]. Since variant ERs also undergo distinct proteasome-mediated degradations [38, 39], a delay in the disassembly of transcription complexes could also extend the duration of promoter engagement of E2-ERα. Post-translational modifications including phosphorylation, acetylation, sumoylation and/or ubiquitination could influence the periodicity of the promoter occupancy of ERα by providing unique target surfaces for the recruitment of distinct coregulatory complexes that differentially modify the amplitude of transcription, and also differently affect the degradation of ERα independently from transcription. Since E2 dramatically enhances the ubiquitination of ERα [37, 38] it is possible that differences in the sequence of events leading to poly-ubiquitination could delay the dissociation of E2-ERα from the promoter. For example, lysine residues serve as common attachment sites for acetylation and sumoylation of the hinge domain of ERα, the latter of which is strictly dependent upon E2 binding [40, 41]. Since post-translational processing is a reversible and dynamic process, sumoylation, or acetylation, prior to poly-ubiquitination could modify the transactivity of ERα and could also disguise the receptor recognition as a proteolytic substrate for degradation, extending the promoter occupancy. Similarly, phosphorylation status of ERα could increase the duration of promoter engagement by uncoupling transactivation from degradation through the repression of poly-ubiquitination and turnover of ERα [38].
Thus, it appears that the E2-mediated increase in ERα-ERE interaction involves both pre- and post-ERE binding events that are manifested as increases in the population of ERα capable of interacting with ERE and in the periodicity of cyclic engagement of ERα with estrogen responsive promoters. These events are anticipated to affect the transcription potency of ERα from the ERE-dependent signaling pathway.
ERβ-mediated nuclear signaling
Crystallographic studies showed that the DBD of ERα interacts with one face of the palindromic sequence in adjacent major grooves of DNA [42, 43]. The ERα-ERE interaction is mediated by the binding of the first zinc-finger motif of each DBD that makes base-specific contacts within the major groove of the DNA helix, while the second zinc-finger motif forms a dimer interface between the two DBDs [42, 43]. These interactions determine the specificity of the response element recognition. Studies using various in vitro approaches indicated that the nearly identical amino acid sequence of the DBD of ERβ to that of ERα allows the receptor to bind to the same spectrum of DNA sequences with similar affinities [4, 5]. Moreover, approaches using a hydroxyl radical cleavage assay, which assesses the protein-DNA interactions at single residue resolution, demonstrated that the structurally homologous DBDs of ERs also account for the abilities of receptors to interact with an ERE utilizing the same nucleotides [4, 44].
Crystallographic studies also indicated that the LBDs of ERα and ERβ display similar tertiary and quaternary architecture [24, 45]. These comparable structural features are responsible for comparable binding affinities of both ER subtypes to E2 [6].
In spite of the fact that ERs display similar ERE and E2 binding properties in vitro, numerous studies have established that E2-ERβ is less effective than E2-ERα in inducing transcription from the ERE-dependent signaling pathway [1, 2]. The mechanism of ER subtype-specific transactivation is, however, unclear. Since the ER-ERE interaction is the pivotal step in transcription, differences in the abilities of ERs to interact in situ with an ERE could be one mechanism that contributes to subtype-specific transcriptional responses. To address this issue, we utilized a novel in situ ERE competition and ChIP assays [46] (Fig 1).
Figure 1. ERE binding activator and in situ competition assay.
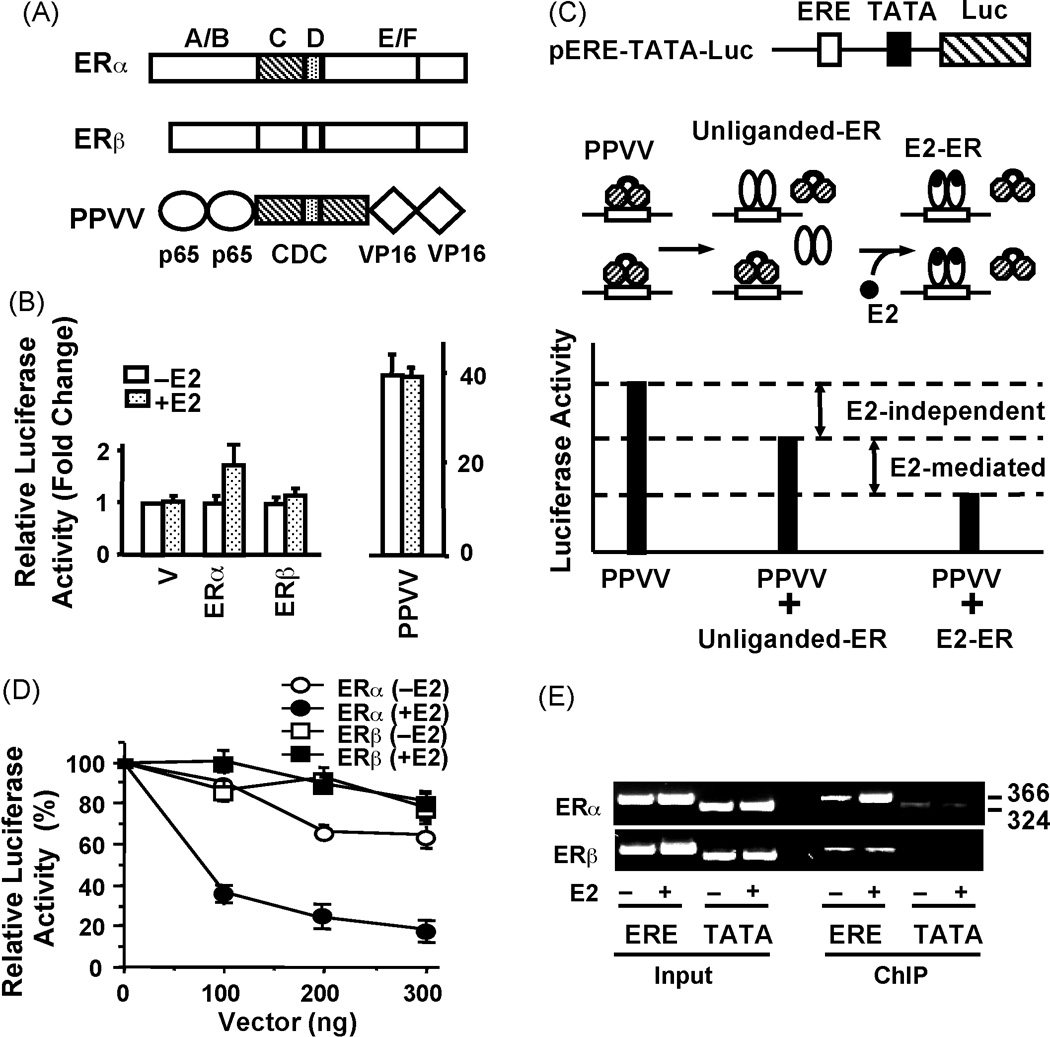
(A) Schematics of ERα, ERβ and PPVV, all of which contain an amino-terminus Flag epitope. Two C domains of ERα were co-joined by the D domain to generate the ERE binding module, CDC. The designer transactivator PPVV was engineered by genetically fusing two tandem activation domains of p65 and VP16 to the amino- and carboxyl-termini of CDC, respectively. (B) Comparative transcriptional responses to PPVV and ERs in CHO cells. Cells were co-transfected with 300 ng expression plasmid bearing none (Vector, V), ERα, ERβ or PPVV cDNA and 125 ng the TATA box promoter with one ERE that drives the expression of the firefly luciferase cDNA as a reporter enzyme in the absence or presence of E2 (10−9) M for 24h. The normalized luciferase activities are presented as fold change compared to the control (V) without E2, which was set to one. The mean ± SEM indicates three independent experiments performed in duplicate. (C) Schematic of the in situ ERE competition assay. (D) The differential effect of E2 on in situ ERE binding of ERα and ERβ in CHO cells. Cells were transfected with 125 ng reporter TATA box promoter bearing one ERE and 300 ng expression plasmid for PPVV, together with varying concentration of expression vector bearing ERα or ERβ cDNA as indicated in the absence or (−E2) or presence (+E2) of 10−9 M E2 for 24h. The luciferase activity is presented as percentage (%) change compared to control (PPVV alone in the absence of E2, which was set to 100%). The mean ± SEM are three independent experiments performed in duplicate. (E) Chromatin immunoprecipitation (ChIP) assay. CHO cells were transiently transfected with the ERα or ERβ expression vector together with a reporter vector bearing none (TATA) or one ERE (ERE) TATA box promoter. Cells were treated with 10−7 M E2 for 1h prior to ChIP using a Flag antibody. Sizes of DNA fragments in base-pair are indicated. Shown are modified figures from Huang et al. [46] and used with permission from The Endocrine Society, Copyright 2005.
Taking advantage of the modular nature of ERs, we engineered a monomeric ERE binding module, CDC, by genetically joining two DBDs (C domains) of ERα with the hinge domain (D domain) [47]. The monomer CDC binds to ERE in a manner similar to the dimer ERα. Moreover, CDC effectively competes with ERα for binding to ERE. Since ERs share a 97% amino-acid homology in their DBDs, CDC also represents an ER subtype-independent ERE binder. Integration of strong activation domains from other transcription factors into this CDC module generated ERE binding transactivators [47]. These designer proteins specifically target and potently regulate ERE-driven gene transcription independent of dimerization, ER-subtype, ligand, promoter- and cell-type. One of the ERE-binding transactivators designated as PPVV (Fig. 1A) contains two tandem activation domains of the p65 subunit of the nuclear factor κ B, NFκB, protein (residues 416–550) [48] and of the viral protein 16, VP16 (residues 403–490) [49], genetically fused to the amino and carboxyl termini of CDC, respectively.
Studies have established that ERα and ERβ in response to E2 have minimal effects on transcription from a single ERE placed upstream of a simple TATA box promoter that drive the expression of a reporter enzyme cDNA. Both receptors require tandem ERE sequences to significantly induce transcription, the extent of which depends on ER-subtype and cell-context [10, 50, 51]. PPVV, on the other hand, dramatically increases reporter enzyme activity compared to E2-ER from the TATA box promoter bearing one or tandem EREs in transiently transfected model cells [47] (Fig. 1B).
These observations prompted us to establish a sensitive ERE competition assay [46] in order to assess the effects of E2 on ER-ERE interaction in situ (Fig. 1C). We reasoned that if ER interacts with ERE in the absence of E2, the ERE bound apoER should decrease the reporter enzyme activity compared to the activity induced by PPVV alone. Furthermore, if E2 were to augment the ERE binding of ER, E2-ER would be expected to compete with PPVV more effectively than apoER. Therefore, a further decrease in the reporter enzyme activity should be observed. Therefore, interference of activator-mediated transcription by unliganded or E2 bound ERs could be taken as an indication of ER-ERE interaction. Results from transiently transfected mammalian cells revealed that the apoERs decrease the PPVV-mediated reporter enzyme activity comparably (Fig. 1D). This suggests that apoERs interact similarly with ERE in situ. The treatment of transfected cells with E2, on the other hand, further augmented the ERα, but not ERβ, mediated decrease in enzyme activity induced by PPVV. Thus, E2 enhances ERα-ERE interaction without altering the binding of ERβ to ERE. ChIP assays further corroborated this conclusion. We found that E2 enhanced the binding of ERα, but not that of ERβ, to the ERE of the simple TATA box (Fig. 1E) and the pS2 promoter construct in transiently transfected mammalian cells, or of the endogenous pS2 gene promoter in adenovirus infected breast cancer cells [46]. These results indicate that although apoERs interact with ERE similarly, E2 enhances ERα-ERE interactions without affecting the binding of ERβ to ERE. This finding is consistent with a previous conclusion that ERβ interacts in situ with ERE independent of E2 [9]. This was based on the observation that the transactivation capacity of a constitutively active chimeric ERβ is not altered by E2, whereas E2 further enhances the activity of the chimeric ERα.
Structural studies further revealed that the A/B domain of ERβ impairs the ability of the receptor to interact in situ with ERE independent of E2, in contrast to the A/B domain of ERα that does not affect the interaction of the receptor with ERE [47]. We found that progressive truncations of the A/B domain of ERβ (Fig. 2A) increased the ability of ERβ species to interact with ERE in situ (Fig. 2B) [47]. Thus, the amino-terminus of ERβ adversely affects receptor-ERE interactions. Although it is not clear, the inter- and/or intramolecular interactions of the ERβ amino-terminus could sterically mask, or allosterically affect the folding of, the DBD. This could limit the population of ERβ capable of interacting with ERE. Additionally, or alternatively, a differential interaction of ERβ with coregulatory proteins could contribute to the stability of the receptor-ERE interaction.
Figure 2. Structural regions in the amino-terminus of ERβ involved in the impaired receptor-ERE interaction.

(A) Schematics of amino-terminal truncations of ERβ. (B) The evaluation of the in situ ERE binding of ERβ variants with the in situ competition assay in CHO cells, which was carried out as described in legend of Fig. 1. Relative ERE binding of receptor species using 300 ng expression vector is depicted as percent (%) decrease in luciferase activity induced by PPVV at 300 ng. (C) The transactivation capacities of ERβ variants in CHO cells. An expression vector bearing none (Vector, V) or an ER variant cDNA was co-transfected with a reporter vector bearing three-tandem consensus EREs upstream of a TATA box promoter driving the expression of the firefly luciferase cDNA (3XERE-TATA-Luc). The mean ± SEM indicates three independent experiments performed in duplicate. Shown are modified figures from Huang et al. [46] and used with permission from The Endocrine Society, Copyright 2005.
The in situ ERE competition assay further revealed that gradual increases in variant ERβ-ERE interactions correlate with enhanced transcription potencies of receptors in transiently transfected cells (Fig. 2C). The extent of transactivation mediated by ERβ variants remained, however, significantly lower compared to that observed with ERα (Fig. 2C). Thus, the impairment of ERβ-ERE interactions by the A/B domain contributes, but is not sufficient, to explain transcription inefficiency of the receptor.
It is well documented that the amino terminal A/B domain of ERα contains an activation function that operates independently as well as in cooperation with the carboxyl-terminus in a cell and promoter context-dependent manner [52–56]. Moreover, the A/B domain is a target for post-translational processing by various signaling pathways that affect regulatory potential of the receptor in the absence or presence of E2 [57, 58]. The ability of the A/B domain to recruit coregulatory proteins [4, 19] is critical for not only AF1 but also the functional integration of both AF1 and AF2 for ERα to mediate transcription at full capacity [10, 53, 59].
Studies using a mammalian one-hybrid system in which the A/B domain is genetically fused to the DBD of the transcription factor Gal4 to assess the intrinsic activation potential showed that the A/B domain of ERβ lacks the ability to induce transcription (Ref. [7] and Fig. 3B). This contrasts to the A/B domain of ERα that significantly enhances gene expression. Moreover, a two-hybrid system that evaluates protein-protein interactions to mediate gene expression further demonstrated that in contrast to ERα, the A/B domain of ERβ is incapable of functionally integrating with AF2 to augment transcription in response to E2 (Ref [10] and Fig. 3C).
Figure 3. Mammalian one- or two-hybrid assays.

(A) Schematic of pGal4-TATA-Luc reporter vector, which bear five Gal4 response elements (5xGal4) juxtaposed to the simple TATA box promoter driving the expression of the firefly luciferase cDNA as the reporter. (B) One-hybrid assay was used to assess the intrinsic activation function of the amino-terminus of ERα (amino acids 1–180) or ERβ (amino acids 1–148), which is genetically fused to the Gal4 DNA-binding domain (Gal4). Constructs (300 ng) were transfected into HepG2 cells together with the pGal4-TATA-Luc reporter vector (500 ng). (B) Two-hybrid assay was used to assess the functional interaction between the amino- and carboxyl-termini of ERs. The carboxyl-terminal E/F domain of ERα (amino acids 301–595) or ERβ (amino acids 287–530) was genetically fused to Gal4 DBD, while the amino-terminal A/B region of ERα or ERβ was conjugated to the activation domain (AD) of viral protein 16 (VP16). The proteins were expressed in COS-1 cells together with pGal4-TATA-Luc reporter vector in the presence of 10−8 M E2 for 48h. The mean ± SEM depicts three independent experiments performed in duplicate. Panel C is a modified figure from Yi et al. [10] and used with permission from The Endocrine Society, Copyright 2005.
Thus, the structurally distinct A/B domain is critical in defining the function of receptor-subtype in the ERE-dependent signaling pathway.
Is there an ER subtype-specific coregulator exchange mechanism that could contribute to differences in transregulatory potentials of ERs?
Structural analysis of ERβ showed that the binding of the amino-terminally truncated ERβ variant to ERE remains independent of E2 unless AF2 is also prevented [46]. On the other hand, the amino-terminally truncated ERα with or without functional AF2 shows an E2-mediated increase in ERE binding similar to the parent ERα. These findings suggest that although the amino-terminus of ERβ is a dominant region to impair ERβ-ERE interaction, the structural basis for the differential effect of E2 on ER-ERE interactions resides in the carboxyl-termini of ERs.
The apparent requirement of the abrogation of AF2 function for the manifestation of E2-mediated increase in ERβ-ERE interaction implies that the binding of a factor(s) to the cofactor interaction surface on the LBD renders the interaction of ERβ with ERE independent of E2. This putative factor could affect pre- and/or post-ERE binding events of ERβ that contribute to the transactivation capacity of the receptor. Possible candidates are the closely related corepressor proteins, Silencing Mediator of Retinoid and Thyroid Responsive Transcription and Nuclear Receptor CoRepressor (SMRT/N-CoR).
Studies indicate that retinoic acid and thyroid hormone receptors can act as ligand-independent repressors or ligand-dependent activators depending on an exchange of SMRT/N-CoR-containing corepressor complexes for coactivators in response to cognate ligands [60, 61]. SMRT/N-CoR has been found as a component of holocorepressor complexes that also include histone and chromatin remodelers, TGFβ-activated kinase 1 binding protein 2 (TAB2) as a sensory protein as well as transducin-β-like (TBL1) and TBL1-related protein (TBLR1) that act as adapter proteins for the recruitment of the ubiquitin/proteasome enzymes [36, 62–65].
It appears that the molecular basis of SMRT/N-CoR recruitment is similar to that of coactivator recruitment that involves cooperative binding of two helical interaction motifs within the N-CoR carboxyl terminus to nuclear receptors. The receptor interaction motifs exhibit a consensus sequence of LXXI/HIXXXI/L (L, I, H refer to leucine, isoleucine and histidine, respectively; while X denotes any amino-acid), or CoRNR box, representing an extended helix compared to the coactivator LXXLL motif [66, 67]. This motif interacts with specific residues in the same receptor pocket required for coactivator binding. The use of a common binding pocket by many coactivators and corepressors indicates that corepressor/coactivator exchange mechanisms are critical for the responsive gene expression [66, 67].
Studies showed that a region in the carboxyl-terminus that encompasses AF2 of apoERα interacts in vitro and in situ [18, 68–70] with a sequence in the carboxyl-terminus receptor of N-CoR that resembles the coactivator consensus LXXLL motif [70]. Importantly, the binding of E2 to ERα releases corepressors from the receptor. In contrast, the E2 binding does not promote the dissociation of corepressors from ERβ [70].
Although E2 binding to ERα appears to be sufficient in vitro to dissociate N-CoR from the receptor, in situ studies suggest that an active coregulator exchange mechanism is involved. ChIP assays showed that apoERα bound to ERE-bearing promoters is associated with complexes containing SMRT/N-CoR [18]. A recent elegant study demonstrated that an evolutionarily conserved amino-terminal L/HX7LL motif (between residues 5 and 15 in the A domain) is required for the interaction of ERα with TAB2 as a component of an N-CoR corepressor complex [36]. This interaction could be critical for basal transcriptional levels of responsive genes as suggested by the observations that the removal of the amino-terminal A domain increases transcriptional responses to apoERα [71, 72].
Moreover, the binding of E2 augments the interaction of TAB2 with the amino-terminus of ERα [36]. Although the mechanism is unclear, it could involve a transconformational change induces by the E2 binding to LBD or an E2-induced functional integration of the carboxyl- and amino-termini. It appears that TAB2 allows TBL1 and TBLR1 to recruit the ubiquitin/proteasome enzymes to the holocorepressor for dismissal and subsequent degradation [36, 65]. This TBL1/TBLR1-mediated N-CoR removal is apparently required for the subsequent productive transcriptional cycle of E2-ERα mediated by the recruitment of coactivator containing complexes [63].
Studies in vitro also indicated that a region in the carboxyl-terminus that encompasses AF2 of ERβ, analogous to ERα, also interact with N-CoR [73]. However in contrast to ERα, the E2 binding does not promote the dissociation of corepressors from ERβ [73]. Although it remains to be explored in situ, the lack of a conserved L/HX7LL motif in ERβ for binding to TAB2 could be one underlying mechanism for the inability of E2 to dissociate corepressor complexes from the receptor. Since E2 is necessary for transactivation, however E2 must also convert the inactive ERE-bound ERβ to a transcriptionally active state. This could involve a concurrent recruitment of coactivators through a distinct cofactor interacting surface(s). This possibility is in concordance with an observation that the regulation of the IκBα gene expression by NFκB in response to tumor necrosis factor-α is accomplished by a dynamic interplay between corepressor and coactivator complexes that are simultaneously recruited to the promoter [74]. The presence of both coactivators and corepressors could also contribute to the weak activity of E2-ERβ in the ERE-dependent signaling.
Conclusion
Comparative analysis of structure/function studies indicates that the amino-terminus of ERα is a multi-functional domain involved in ligand-independent transactivation, coregulator exchange, and functional integration of AF2 critical for the regulatory potential of the receptor. This contrasts to the amino-terminus of ERβ that impairs the receptor-ERE interactions, lacks an activation function and is incapable of interacting with the carboxyl-terminus. We therefore suggest that the amino-terminus is an inhibitory region for the activity of ERβ in the ERE-dependent signaling pathway.
Although the amino-termini of ERs are important determinants in defining ER-subtype functions, alterations in pre- and post-ERE binding of ERβ mediated by, for example, heterodimerization with ERα [44, 75] and/or post-translational modifications [76], could alter the receptor activity, thereby contributing to receptor action. Moreover, the integration of various E2-ER signaling pathways [77–79] is ultimately responsible for the physiological regulation of responsive tissue functions in which aberrations lead to malignancies. A better understanding of the mechanism of receptor action through the use of structural and molecular approaches would propel discovery of various aspects of E2 signaling as a basis to broaden the pharmacological possibilities to medicine.
Acknowledgment
This work in the laboratory is supported by grants from NIH (RO1 CA113682) and the Susan G. Komen Foundation to MM. We thank Patrick Brandt for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 2.Nilsson S, Gustafsson JA. Estrogen receptor action. Crit Rev Eukaryot Gene Expr. 2002;12:237–257. doi: 10.1615/critreveukaryotgeneexpr.v12.i4.10. [DOI] [PubMed] [Google Scholar]
- 3.Klinge CM. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001;29:2905–2919. doi: 10.1093/nar/29.14.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi P, Driscoll MD, Huang J, Bhagat S, Hilf R, Bambara RA, Muyan M. The effects of estrogen-responsive element- and ligand-induced structural changes on the recruitment of cofactors and transcriptional responses by ERα and ERβ. Mol Endocrinol. 2002;16:674–693. doi: 10.1210/mend.16.4.0810. [DOI] [PubMed] [Google Scholar]
- 5.Loven MA, Wood JR, Nardulli AM. Interaction of estrogen receptors α and β with estrogen response elements. Mol Cell Endocrinol. 2001;181:151–163. doi: 10.1016/s0303-7207(01)00491-9. [DOI] [PubMed] [Google Scholar]
- 6.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors α and β. Endocrinology. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 7.Cowley SM, Parker MG. A comparison of transcriptional activation by ERα and ERβ. J Steroid Biochem Mol Biol. 1999;69:165–175. doi: 10.1016/s0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 8.McInerney EM, Weis KE, Sun J, Mosselman S, Katzenellenbogen BS. Transcription activation by the human estrogen receptor subtype β (ERβ) studied with ERβ and ERα receptor chimeras. Endocrinology. 1998;139:4513–4522. doi: 10.1210/endo.139.11.6298. [DOI] [PubMed] [Google Scholar]
- 9.Hall JM, McDonnell DP. The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 10.Yi P, Bhagat S, Hilf R, Bambara RA, Muyan M. Differences in the abilities of estrogen receptors to integrate activation functions are critical for subtype-specific transcriptional responses. Mol Endocrinol. 2002;16:1810–1827. doi: 10.1210/me.2001-0323. [DOI] [PubMed] [Google Scholar]
- 11.Bai Y, Giguere V. Isoform-selective interactions between estrogen receptors and steroid receptor coactivators promoted by estradiol and ErbB-2 signaling in living cells. Mol Endocrinol. 2003;17:589–599. doi: 10.1210/me.2002-0351. [DOI] [PubMed] [Google Scholar]
- 12.Zheng FF, Wu RC, Smith CL, O'Malley BW. Rapid estrogen-induced phosphorylation of the SRC-3 coactivator occurs in an extranuclear complex containing estrogen receptor. Mol Cell Biol. 2005;25:8273–8284. doi: 10.1128/MCB.25.18.8273-8284.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stenoien DL, Patel K, Mancini MG, Dutertre M, Smith CL, O'Malley BW, Mancini MA. FRAP reveals that mobility of oestrogen receptor-α is ligand- and proteasome-dependent. Nat Cell Biol. 2001;3:15–23. doi: 10.1038/35050515. [DOI] [PubMed] [Google Scholar]
- 14.Nunez AM, Berry M, Imler JL, Chambon P. The 5' flanking region of the pS2 gene contains a complex enhancer region responsive to oestrogens, epidermal growth factor, a tumour promoter (TPA), the c-Ha-ras oncoprotein and the c-jun protein. EMBO J. 1989;8:823–829. doi: 10.1002/j.1460-2075.1989.tb03443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berry M, Nunez AM, Chambon P. Estrogen-responsive element of the human pS2 gene is an imperfectly palindromic sequence. Proc Natl Acad Sci U S A. 1989;86:1218–1222. doi: 10.1073/pnas.86.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 17.Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERα on responsive promoters is an integral feature of estrogen signaling. Mol Cell. 2003;11:695–707. doi: 10.1016/s1097-2765(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 18.Metivier R, Penot G, Carmouche RP, Hubner MR, Reid G, Denger S, Manu D, Brand H, Kos M, Benes V, Gannon F. Transcriptional complexes engaged by apo-estrogen receptor-α isoforms have divergent outcomes. EMBO J. 2004;23:3653–3666. doi: 10.1038/sj.emboj.7600377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Webb P, Nguyen P, Shinsako J, Anderson C, Feng W, Nguyen MP, Chen D, Huang SM, Subramanian S, McKinerney E, Katzenellenbogen BS, Stallcup MR, Kushner PJ. Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol Endocrinol. 1998;12:1605–1618. doi: 10.1210/mend.12.10.0185. [DOI] [PubMed] [Google Scholar]
- 20.Lonard DM, Nawaz Z, Smith CL, O'Malley BW. The 26S proteasome is required for estrogen receptor-α and coactivator turnover and for efficient estrogen receptor-α transactivation. Mol Cell. 2000;5:939–948. doi: 10.1016/s1097-2765(00)80259-2. [DOI] [PubMed] [Google Scholar]
- 21.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nature Str Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 22.Pike AC. Lessons learnt from structural studies of the oestrogen receptor. Best Pract Res Clin Endocrinol Metab. 2006;20:1–14. doi: 10.1016/j.beem.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Tamrazi A, Katzenellenbogen JA. Site-specific fluorescent labeling of estrogen receptors and structure-activity relationships of ligands in terms of receptor dimer stability. Methods Enzymol. 2003;364:37–53. doi: 10.1016/s0076-6879(03)64003-6. [DOI] [PubMed] [Google Scholar]
- 24.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 25.Reese JC, Katzenellenbogen BS. Examination of the DNA-binding ability of estrogen receptor in whole cells: implications for hormone-independent transactivation and the actions of antiestrogens. Mol Cell Biol. 1992;12:4531–4538. doi: 10.1128/mcb.12.10.4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nye AC, Rajendran RR, Stenoien DL, Mancini MA, Katzenellenbogen BS, Belmont AS. Alteration of large-scale chromatin structure by estrogen receptor. Mol Cell Biol. 2002;22:3437–3449. doi: 10.1128/MCB.22.10.3437-3449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen HW, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. doi: 10.1016/s0092-8674(00)80054-9. [DOI] [PubMed] [Google Scholar]
- 28.Pratt WB, Gehring U, Toft DO. Molecular chaperoning of steroid hormone receptors. Exs. 1996;77:79–95. doi: 10.1007/978-3-0348-9088-5_6. [DOI] [PubMed] [Google Scholar]
- 29.Oesterreich S. Scaffold attachment factors SAFB1 and SAFB2: Innocent bystanders or critical players in breast tumorigenesis? J Cell Biochem. 2003;90:653–661. doi: 10.1002/jcb.10685. [DOI] [PubMed] [Google Scholar]
- 30.Romine LE, Wood JR, Lamia LA, Prendergast P, Edwards DP, Nardulli AM. The high mobility group protein 1 enhances binding of the estrogen receptor DNA binding domain to the estrogen response element. Mol Endocrinol. 1998;12:664–674. doi: 10.1210/mend.12.5.0111. [DOI] [PubMed] [Google Scholar]
- 31.Loven MA, Muster N, Yates JR, Nardulli AM. A novel estrogen receptor α-associated protein, template-activating factor Iβ, inhibits acetylation and transactivation. Mol Endocrinol. 2003;17:67–78. doi: 10.1210/me.2002-0280. [DOI] [PubMed] [Google Scholar]
- 32.Loven MA, Davis RE, Curtis CD, Muster N, Yates JR, Nardulli AM. A novel estrogen receptor α-associated protein alters receptor-deoxyribonucleic acid interactions and represses receptor-mediated transcription. Mol Endocrinol. 2004;18:2649–2659. doi: 10.1210/me.2003-0195. [DOI] [PubMed] [Google Scholar]
- 33.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 34.Endoh H, Maruyama K, Masuhiro Y, Kobayashi Y, Goto M, Tai H, Yanagisawa J, Metzger D, Hashimoto S, Kato S. Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor α. Mol Cell Biol. 1999;19:5363–5372. doi: 10.1128/mcb.19.8.5363. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Warnmark A, Wikstrom A, Wright AP, Gustafsson JA, Hard T. The N-terminal regions of estrogen receptor α and β are unstructured in vitro and show different TBP binding properties. J Biol Chem. 2001;276:45939–45944. doi: 10.1074/jbc.M107875200. [DOI] [PubMed] [Google Scholar]
- 36.Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 37.Wijayaratne AL, McDonnell DP. The human estrogen receptor-α is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem. 2001;276:35684–35692. doi: 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- 38.Valley CC, Metivier R, Solodin NM, Fowler AM, Mashek MT, Hill L, Alarid ET. Differential regulation of estrogen-inducible proteolysis and transcription by the estrogen receptor alpha N terminus. Mol Cell Biol. 2005;25:5417–5428. doi: 10.1128/MCB.25.13.5417-5428.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tateishi Y, Kawabe Y, Chiba T, Murata S, Ichikawa K, Murayama A, Tanaka K, Baba T, Kato S, Yanagisawa J. Ligand-dependent switching of ubiquitin-proteasome pathways for estrogen receptor. EMBO J. 2004;23:4813–4823. doi: 10.1038/sj.emboj.7600472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sentis S, Le Romancer M, Bianchin C, Rostan MC, Corbo L. Sumoylation of the estrogen receptor α hinge region regulates its transcriptional activity. Mol Endocrinol. 2005;19:2671–2684. doi: 10.1210/me.2005-0042. [DOI] [PubMed] [Google Scholar]
- 41.Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, Fuqua SA, Lopez GN, Kushner PJ, Pestell RG. Direct acetylation of the estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem. 2001;276:18375–18383. doi: 10.1074/jbc.M100800200. [DOI] [PubMed] [Google Scholar]
- 42.Schwabe JW, Neuhaus D, Rhodes D. Solution structure of the DNA-binding domain of the oestrogen receptor. Nature. 1990;348:458–461. doi: 10.1038/348458a0. [DOI] [PubMed] [Google Scholar]
- 43.Schwabe JW, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell. 1993;75:567–578. doi: 10.1016/0092-8674(93)90390-c. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M. Single-chain estrogen receptors (ERs) reveal that the ERα/β heterodimer emulates functions of the ERα dimer in genomic estrogen signaling pathways. Mol Cell Biol. 2004;24:7681–7694. doi: 10.1128/MCB.24.17.7681-7694.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M. Structure of the ligand-binding domain of oestrogen receptor β in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–4618. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang J, Li X, Maguire CA, Hilf R, Bambara RA, Muyan M. Binding of estrogen receptor β to estrogen response element in situ is independent of estradiol and impaired by its amino terminus. Mol Endocrinol. 2005;19:2696–2712. doi: 10.1210/me.2005-0120. [DOI] [PubMed] [Google Scholar]
- 47.Huang J, Li X, Yi P, Hilf R, Bambara RA, Muyan M. Targeting estrogen responsive elements (EREs): design of potent transactivators for ERE-containing genes. Mol Cell Endocrinol. 2004;218:65–78. doi: 10.1016/j.mce.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 48.Moore PA, Ruben SM, Rosen CA. Conservation of transcriptional activation functions of the NF-κB p50 and p65 subunits in mammalian cells and Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:1666–1674. doi: 10.1128/mcb.13.3.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Triezenberg SJ, Kingsbury RC, McKnight SL. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 1988;2:718–729. doi: 10.1101/gad.2.6.718. [DOI] [PubMed] [Google Scholar]
- 50.Klein-Hitpass L, Kaling M, Ryffel GU. Synergism of closely adjacent estrogen-responsive elements increases their regulatory potential. J Mol Biol. 1988;201:537–544. doi: 10.1016/0022-2836(88)90635-3. [DOI] [PubMed] [Google Scholar]
- 51.Tyulmenkov VV, Jernigan SC, Klinge CM. Comparison of transcriptional synergy of estrogen receptors α and β from multiple tandem estrogen response elements. Mol Cell Endocrinol. 2000;165:151–161. doi: 10.1016/s0303-7207(00)00250-1. [DOI] [PubMed] [Google Scholar]
- 52.Beekman JM, Allan GF, Tsai SY, Tsai MJ, O'Malley BW. Transcriptional activation by the estrogen receptor requires a conformational change in the ligand binding domain. Mol Endocrinol. 1993;7:1266–1274. doi: 10.1210/mend.7.10.8264659. [DOI] [PubMed] [Google Scholar]
- 53.Kraus WL, McInerney EM, Katzenellenbogen BS. Ligand-dependent, transcriptionally productive association of the amino- and carboxyl-terminal regions of a steroid hormone nuclear receptor. Proc Natl Acad Sci U S A. 1995;92:12314–12318. doi: 10.1073/pnas.92.26.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tora L, White J, Brou C, Tasset D, Webster N, Scheer E, Chambon P. The human estrogen receptor has two independent nonacidic transcriptional activation functions. Cell. 1989;59:477–487. doi: 10.1016/0092-8674(89)90031-7. [DOI] [PubMed] [Google Scholar]
- 55.Tzukerman MT, Esty A, Santiso-Mere D, Danielian P, Parker MG, Stein RB, Pike JW, McDonnell DP. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol. 1994;8:21–30. doi: 10.1210/mend.8.1.8152428. [DOI] [PubMed] [Google Scholar]
- 56.McInerney EM, Tsai MJ, O'Malley BW, Katzenellenbogen BS. Analysis of estrogen receptor transcriptional enhancement by a nuclear hormone receptor coactivator. Proc Natl Acad Sci U S A. 1996;93:10069–10073. doi: 10.1073/pnas.93.19.10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ali S, Metzger D, Bornert JM, Chambon P. Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J. 1993;12:1153–1160. doi: 10.1002/j.1460-2075.1993.tb05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 59.Benecke A, Chambon P, Gronemeyer H. Synergy between estrogen receptor α activation functions AF1 and AF2 mediated by transcription intermediary factor TIF2. EMBO Rep. 2000;1:151–157. doi: 10.1093/embo-reports/kvd028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 61.Zamir I, Harding HP, Atkins GB, Horlein A, Glass CK, Rosenfeld MG, Lazar MA. A nuclear hormone receptor corepressor mediates transcriptional silencing by receptors with distinct repression domains. Mol Cell Biol. 1996;16:5458–5465. doi: 10.1128/mcb.16.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jepsen K, Rosenfeld MG. Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci. 2002;115:689–698. doi: 10.1242/jcs.115.4.689. [DOI] [PubMed] [Google Scholar]
- 63.Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- 64.Perissi V, Rosenfeld MG. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol. 2005;6:542–554. doi: 10.1038/nrm1680. [DOI] [PubMed] [Google Scholar]
- 65.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 66.Hu X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 67.Perissi V, Staszewski LM, McInerney EM, Kurokawa R, Krones A, Rose DW, Lambert MH, Milburn MV, Glass CK, Rosenfeld MG. Molecular determinants of nuclear receptor-corepressor interaction. Genes Dev. 1999;13:3198–3208. doi: 10.1101/gad.13.24.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith CL, Nawaz Z, O'Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 69.Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, Del-Rio AL, Ricote M, Ngo S, Gemsch J, Hilsenbeck SG, Osborne CK, Glass CK, Rosenfeld MG, Rose DW. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci U S A. 1998;95:2920–2925. doi: 10.1073/pnas.95.6.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Webb P, Nguyen P, Kushner PJ. Differential SERM Effects on Corepressor Binding Dictate ERα Activity in Vivo. J Biol Chem. 2003;278:6912–6920. doi: 10.1074/jbc.M208501200. [DOI] [PubMed] [Google Scholar]
- 71.Metivier R, Petit FG, Valotaire Y, Pakdel F. Function of N-terminal transactivation domain of the estrogen receptor requires a potential alpha-helical structure and is negatively regulated by the A domain. Mol Endocrinol. 2000;14:1849–1871. doi: 10.1210/mend.14.11.0546. [DOI] [PubMed] [Google Scholar]
- 72.Sathya G, Yi P, Bhagat S, Bambara RA, Hilf R, Muyan M. Structural regions of ERα critical for synergistic transcriptional responses contain co-factor interacting surfaces. Mol Cell Endocrinol. 2002;192:171–185. doi: 10.1016/s0303-7207(01)00673-6. [DOI] [PubMed] [Google Scholar]
- 73.Webb P, Valentine C, Nguyen P, Price RH, Jr, Marimuthu A, West BL, Baxter JD, Kushner PJ. ERβ Binds N-CoR in the Presence of Estrogens via an LXXLL-like Motif in the N-CoR C-terminus. Nucl Recept. 2003;1:4. doi: 10.1186/1478-1336-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao Z, Chiao P, Zhang X, Zhang X, Lazar MA, Seto E, Young HA, Ye J. Coactivators and corepressors of NF-κB in IκB α gene promoter. J Biol Chem. 2005;280:21091–21098. doi: 10.1074/jbc.M500754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tremblay GB, Tremblay A, Labrie F, Giguere V. Dominant activity of activation function 1 (AF-1) and differential stoichiometric requirements for AF-1 and -2 in the estrogen receptor α-β heterodimeric complex. Mol Cell Biol. 1999;19:1919–1927. doi: 10.1128/mcb.19.3.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tremblay A, Tremblay GB, Labrie F, Giguere V. Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol Cell. 1999;3:513–519. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 77.Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74:311–317. doi: 10.1016/s0960-0760(00)00108-4. [DOI] [PubMed] [Google Scholar]
- 78.Safe S. Transcriptional activation of genes by 17β-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001;62:231–252. doi: 10.1016/s0083-6729(01)62006-5. [DOI] [PubMed] [Google Scholar]
- 79.Levin ER. Cellular functions of plasma membrane estrogen receptors. Steroids. 2002;67:471–475. doi: 10.1016/s0039-128x(01)00179-9. [DOI] [PubMed] [Google Scholar]