

Summary
Antibody-antigen complex mediated inflammation is integral to the pathogenesis of many autoimmune diseases. Mice deficient in the γ-chain of Fc-receptors are protected in IgG-mediated glomerulonephritis and the Arthus reaction and FcR-bearing mast cells and macrophages have been assigned primary roles in these processes. Here we demonstrate that neutrophil selective transgenic expression of the two uniquely human activating FcγRs, FcγRIIA and FcγRIIIB was sufficient to restore susceptibility to progressive anti-glomerular basement membrane (GBM) nephritis and the cutaneous Reverse Passive Arthus (RPA) reaction in γ-chain deficient mice. Both FcγRIIA and FcγRIIIB mediated robust neutrophil accumulation in tissues suggesting direct roles for these human receptors in IC-induced neutrophil recruitment, while FcγRIIA alone mediated organ injury. In an acute model of anti-GBM nephritis, both FcγRIIIB and FcγRIIA promoted initial neutrophil recruitment to glomerular immune-complexes (ICs) accessible to circulating cells, while FcγRIIA further sustained accumulation. In a model of soluble ICs deposited strictly within the post-capillary venules of the cremaster muscle, FcγRIIIB was solely responsible for converting initial selectin-dependent tethers to slow rolling and adhesion. However, in the cremaster RPA reaction, dependent on vascular and tissue accumulation of soluble ICs, FcγRIIA predominated in neutrophil recruitment that was dependent on G-protein coupled receptor activation. Thus, human FcγRs on neutrophils serve as the primary molecular links between ICs and immunological disease with FcγRIIA promoting tissue injury, and FcγRIIIB and FcγRIIA displaying specialized context-dependent functions in IC-induced neutrophil recruitment.
Introduction
Deposition of antigen-antibody complexes in tissues is a hallmark of human diseases from autoimmune disorders and early transplant rejection to rheumatic fever. IgG-mediated diseases are produced either by the binding of pathogenic antibody to self or foreign antigens on host cells or the deposition of circulating antigen-antibody complexes in tissue. Cell surface receptors that bind IgG-immune complexes (ICs), known collectively as FcγRs, play essential roles in diseases initiated by antibodies (Schmidt and Gessner, 2005). In particular, mice deficient in the common γ-chain (γ−/−) required for expression of the murine activating FcγRs are protected in acute and progressive glomerulonephritis, autoimmune skin diseases, arthritis, systemic lupus erythematosus nephritis, and Reverse Passive Arthus (RPA) reaction (Ji et al., 2002; Trcka et al., 2002). In addition to FcγRs, C5aR binding to complement C5a activated by ICs may modulate disease pathogenesis by regulating the balance of FcγR expression and inducing chemokine release (Ravetch and Bolland, 2001). Despite the importance of FcγRs and complement in IC-mediated inflammation, mechanisms downstream of their activation and the relevant Fc-bearing cell type involved still remain largely unresolved. The present view is that tissue resident mast cells and macrophages sense ICs through FcγRs and complement receptors, and subsequently release inflammatory mediators that recruit effector cells through the well-described multistep process of endothelial activation, selectin-dependent rolling and integrin mediated adhesion (Ley et al., 2007; Schmidt and Gessner, 2005; Skokowa et al., 2005).
Neutrophils are key effector cells in innate immune responses yet FcγRs specifically on neutrophils have not been implicated as initial mediators of cellular activation in IgG-disease models in mice. Given the structural differences between murine and human low affinity FcγRs, it is not clear how well studies mediated by murine receptors accurately reflect human inflammation. Murine neutrophils express FcγRIII and FcγRIV (Nimmerjahn and Ravetch, 2006) that are transmembrane receptors relying on a common ITAM-containing γ-chain for expression and signaling. In contrast, human neutrophils express a unique glycosyl-phosphatidyl-inositol (GPI)-anchored FcγRIIIB and a single polypeptide ITAM-containing FcγRIIA for which there are no genetic equivalents in mice or other mammals (Hogarth, 2002). Thus the low affinity human receptors are single polypeptide molecules with FcγRIIA containing its own signaling domain while the murine counterparts function as multi-protein complexes, with ligand binding and signaling functions present on separate polypeptides.
The biological role of the two unique human neutrophil FcγRs, FcγRIIA and FcγRIIIB remains largely unclear. Genetic evidence indicates that polymorphisms in FcγRIIA and IIIB correlate with autoimmune disease in patients (van Sorge et al., 2003) and copy number polymorphisms of FcγRIIIB is associated with increased susceptibility to glomerulonephritis (Aitman et al., 2006). In vitro, engagement of FcγRIIA promotes phagocytosis, degranulation and reactive oxygen species generation. FcγRIIIB’s function remains elusive despite its 4–5 fold higher surface expression levels than FcγRIIA in human neutrophils (Selvaraj et al., 1988; Unkeless et al., 1995). FcγRIIIB cross-linking induces calcium mobilization, triggers degranulation and leukotriene release (Crockett-Torabi et al., 1992; Unkeless et al., 1995). In vitro, human FcγRIIIB preferentially tethers neutrophils to immobilized ICs under physiological flow conditions (Coxon et al., 2001; Florey et al., 2007; Luscinskas and Mayadas, 2007) while FcγRIIA, in cooperation with chemokine receptors, was recently shown to enhance leukocyte adhesion to IgG bound to activated endothelial cells (Florey et al., 2007).
To examine the relative contribution of human neutrophil activating receptors, FcγRIIA and FcγRIIIB in effector responses to IgG in vivo we generated transgenic mice that express one or both of these receptors selectively in neutrophils using a myeloid restricted promoter (Lagasse and Weissman, 1994; Lagasse and Weissman, 1997). These mice were crossed to γ−/− mice to eliminate endogenous murine activating receptors. We show that expression of both FcγRIIA and IIIB in neutrophils was sufficient to restore disease in γ−/− mice subjected to a model of progressive glomerulonephritis or the Reverse Arthus reaction (RPA). These are prototypic models of Type II and Type III autoimmunity induced by in situ or soluble ICs respectively. FcγRIIIB and FcγRIIA promoted neutrophil accumulation in both models while FcγRIIA alone was required for tissue injury. The observed neutrophil recruitment in the absence of FcγR expression in macrophages and mast cells suggested a direct role for neutrophil FcγRs in neutrophil recruitment. We provide evidence that FcγRIIA and FcγRIIIB tether to in situ formed glomerular ICs, and play distinct context-dependent roles in soluble IC-induced slow leukocyte rolling, adhesion and transmigration. Together our work suggests a new paradigm in human IgG mediated diseases wherein neutrophils are recruited, and promote tissue injury through their own FcγRs. Further, our data indicate that each of the FcγRs specializes in separate steps leading to organ injury.
Results
Generation of mice with neutrophil selective expression of human FcγRIIA and FcγRIIIB and analysis of receptor expression
Human neutrophil FcγRs were placed under the control of the human myeloid-related protein 8 (hMRP8) promoter (Figure 1A), which drives expression primarily in the myeloid lineage (Lagasse and Weissman, 1994). The two human FcγR transgenics, and a third line generated by breeding mice expressing the single transgenes, were bred to Fcγ-chain deficient (γ−/−) mice to produce animals that express the activating human FcγRs on neutrophils in the absence of endogenous murine activating FcγRs (tg/γ−/−). These transgenic lines are referred to as IIAtg/γ−/−, IIIBtg/γ−/− and IIA+IIIB tg/γ−/−. Flow cytometric analysis revealed that the human proteins were present on greater than 85–95% of peripheral blood transgenic neutrophils and on a population of monocytes (FcγRIIA on 20%, and FcγRIIIB on 70% of cells) (Figure 1B). Both receptors were largely absent on macrophages, mast cells, CD3+ T cells, platelets (Figure 1C) and B cells (data not shown). PMA stimulation resulted in FcγRIIIB shedding from the surface of activated transgenic neutrophils (Figure 1D) as reported for human neutrophils (Huizinga et al., 1988) indicating similar regulation of the receptor in transgenic murine and human cells.
Figure 1.

Neutrophil specific expression of human Fcγ receptors in transgenic mice. (A) Transgenic constructs were generated by inserting human FcγR cDNAs into the human MRP8 promotor cassette as shown. (B–C) hFcγR expression was evaluated by flow cytometry on γ−/− (dotted line) and RIIA+RIIIBtg/γ−/−(solid line) mice. hFcγRIIA and IIIB expression was analyzed on (B) peripheral blood neutrophils (Gr-1high/mCD11b+) and monocytes (mCD115+/mCD11b+), and FcγRIIA expression was assessed on (C) F4/80+ resident peritoneal and bone marrow derived macrophages, peritoneal mast cells, CD3+ T cells and platelets. The percentages of FcγR positive cells are indicated. (D) Cell surface expression of mouse CD11b, hFcγRIIIB and mouse CD62L (L-selectin) in BMNs from RIIIBtg/γ+/+ mice stimulated without (dotted line) or with 100 ng/ml of PMA for 10 min (solid line). hFcγRIIIB shedding (middle panel) is associated with the CD11b upregulation and L-selectin shedding, hallmarks of neutrophil activation. (E) Levels of messenger RNA transcripts of endogenous mouse FcγRIII (open bar) and transgenic human FcγRs (filled bars) were measured by quantitative RT-PCR in RIIAtg/γ+/+ or RIIIBtg/γ+/+ mouse BMN and reported relative to β-actin. The results are shown as average ± SD of n=4 per group. (F) Comparison of FcγRs on human PMN (hPMN) and mouse bone marrow neutrophils (mBMN) from RIIA+RIIIBtg/γ−/− animals by flow cytometry analysis. Solid lines indicate staining for hFcγRIIA or FcγRIIIB; the mean fluorescent intensity is given. Dotted line shows the cell populations stained with isotype IgG control.
The relative level of the human transgenic versus endogenous mouse FcγRs was determined by quantitating message levels of the proteins in neutrophils expressing FcγRIIA or FcγRIIIB on a wild-type background (i.e. tg/γ+/+). This approach rather than the antibody mediated detection of protein levels was pursued as different antibodies may have differing affinities for their targets and therefore cannot be directly compared. Importantly, mRNA transcript levels of the two human FcγR transgenes and the endogenous murine FcγRIII were similar as detected by quantitative RT-PCR (Figure 1E). To determine how closely the level of expression of human FcγRs on murine neutrophils recapitulates that observed in human neutrophils, human FcγR surface expression in murine transgenic and human neutrophils was compared by FACs analysis. Transgenic FcγRIIA protein expression was equivalent, while FcγRIIIB was reduced compared to human neutrophils (Figure 1F).
FcγRIIA but not FcγRIIIB engagement results in reactive oxygen species generation
The contribution of the human FcγRs to IC mediated adhesion and the release of reactive oxygen species (ROS) was evaluated. Similar numbers of isolated tg/γ−/− and wild-type neutrophils adhered to immobilized BSA-anti-BSA ICs. Despite this, the morphology and F-actin distribution in IIIBtg/γ−/− neutrophils was significantly altered. That is, IIAtg/γ−/−, RIIA+IIIBtg/γ−/− and wild-type neutrophils contained distinct lamellipodia enriched in cortical actin while these structures were largely absent in IIIBtg/γ−/ cells (Figure 2A). Adhesion dependent H2O2 production was comparable in the wild-type, IIAtg/γ−/− and RIIA+IIIBtg/γ−/− but was minimal in the IIIBtg/γ−/− transgenic neutrophils. γ−/− neutrophils exhibited no significant binding to ICs or ROS production thus showing the dependence of the assay on activating FcγRs (Figure 2A,B). To bypass any effects of defective adhesion on ROS generation, we examined FcγR cross-linking induced ROS generation in cells in suspension. FcγRIIA cross-linking on IIAtg/γ−/− or IIA+IIIBtg/γ−/− neutrophils resulted in robust ROS generation (Figure 2C). In contrast, FcγRIIIB cross-linking resulted in minimal ROS generation in IIIBtg/γ−/− that was only marginally increased in IIA+IIIBtg/γ−/− neutrophils (Figure 2C). FcγRIIA induced ROS generation was inhibited with pharmacological inhibitors of src, syk kinase and phosphatidylinositol 3-kinase (Figure 2C) and was associated with tyrosine phosphorylation (data not shown) as is expected for ITAM-based signal transduction (Underhill and Goodridge, 2007). Thus FcγRIIA links to the expected ITAM based signalling machinery in murine neutrophils to trigger ROS generation while FcγRIIIB does not engage this effector response.
Figure 2.

In vitro analysis of neutrophil adhesion to immune complexes and reactive oxygen species generation. Neutrophils isolated from bone marrow of the indicated mouse strains, were placed on plates coated with BSA-anti-BSA ICs or BSA alone. (A) The average number of adherent neutrophils was quantitated (left). All data are mean ± SEM of 4 independent experiments. Representative pictures of neutrophils adherent to immobilized-IC for 30 min taken at low (top) and high power (bottom) are shown to the right. RIIAtg/γ−/− neutrophils spread with prominent cortical F-actin (arrow heads) while RIIIBtg/γ−/− neutrophils failed to do so and remained retracted. (B) H2O2 concentration in culture supernatant harvested from neutrophils adherent to ICs or BSA alone. All data are mean ± SEM of 3–4 independent experiments. (C) Cross-linking induced ROS generation. Upper panel; Neutrophils from indicated mice were preincubated with mouse anti-human FcγRIIA (IV.3) or anti-FcγRIII (3G8) following GM-CSF priming. Real-time generation of ROS was monitored upon addition of F(ab′)2 goat anti-mouse IgG (GAM) using a luminol-based assay. The ROS profile of γ−/− neutrophils following FcγRIIA cross-linking is also shown as a control. Lower panel; Real-time generation of ROS was evaluated in RIIAtg/γ−/− neutrophils treated with Piceatannol (Syk inhibitor), PP2 (Src inhibitor) or LY294002 (PI3K inhibitor), primed with GM-CSF and subjected to FcγRIIA cross-linking. One of 3 representative experiments is shown.
Human neutrophil FcγRs are sufficient to restore progressive glomerulonephritis in γ−/− mice
Progressive nephrotoxic serum (NTS) nephritis in mice is a prototypic Type II hypersensitivity response in the kidney induced by antibody directed against the glomerular basement membrane (GBM), which is exposed to circulating blood through open endothelial fenestrae. Presensitization of mice with rabbit IgG prior to challenge with rabbit NTS results in glomerular injury and renal dysfunction that resembles aspects of Goodpasture syndrome in humans (Neale and Wilson, 1982), and relies entirely on heterologous (rabbit or sheep) and not autologous mouse IgG (Dean et al., 2005; Li et al., 1997; Rosenkranz et al., 1999). While γ−/− mice subjected to progressive NTS nephritis failed to develop disease (Figure 3A–E) as expected (Park et al., 1998), RIIAtg/γ−/− exhibited mortality, renal dysfunction (elevation of urine albumin and serum creatinine) and histopathologic evidence of significant glomerular and interstitial damage (Figure 3A–E). Renal injury was absent in RIIIBtg/γ−/− mice (Figure 3A–E). However, mice expressing both FcγRIIA and FcγRIIIB exhibited considerably more disease compared to animals expressing FcγRIIA alone indicating cooperation between FcγRIIA and FcγRIIIB. Compared to wild-type animals, IIAtg/γ−/− and RIIA+RIIIBtg/γ−/− mice had earlier onset of disease, greater disease severity and ultimately mortality (Figure 3A–E). These data suggest a primacy of human neutrophil FcγRs in glomerular inflammation and pathology.
Figure 3.

Analysis of progressive NTS nephritis. Mice were pre-immunized with rabbit IgG in incomplete Freud’s adjuvant on day -3, and injected intravenously with NTS on day 0. (A) Survival of five different strains of mice after induction of disease. RIIAtg/γ−/− mice (n=23) and RIIA+RIIIBtg/γ−/− mice (n=8) showed high mortality. All RIIIBtg/γ−/−, γ−/− and wild-type (γ+/+) mice (n=12–16 per group) survived the entire experimental time period. (B–C) Analysis of renal function. Albuminuria (n=8–23 per group) (B), and serum creatinine at day 14 (n=6–11 for each group) (C), were significantly elevated in RIIAtg/γ−/− and RIIA+RIIIBtg/γ−/− mice compared to γ−/− and RIIIBtg/γ−/−, and disease was accelerated in RIIAtg/γ−/−, RIIA+RIIIBtg/γ−/− compared to wild-type mice. (D) Representative pictures of Periodic acid-Schiff (PAS)-stained sections of kidney harvested on day 21. Deposition of PAS positive material (indicative of glomerular damage), occlusion of the glomerular capillary lumen and adhesion to Bowman’s capsule as well as interstitial damage including tubular dilation, severe tubular cell atrophy and cast formation were observed only in RIIAtg/γ−/− mice. Bars: 200 μm (upper) and 50 μm (lower). (E) A quantitation of glomerular PAS deposits of indicated strains at day 7 is shown (n=6–10 per group). All data are mean±SEM. p<0.001 by ANOVA among all strains of mice. Tukey/Kramer was done for comparison between two mouse strains at 5% significant level. *vs γ−/− and RIIIBtg/γ−/−, #vs RIIAtg/γ−/−, §vs C57Bl/6.
NTS nephritis is associated with glomerular inflammatory cell infiltration as a result of antibody deposition (Fries et al., 1988) and secondary interstitial leukocyte accumulation as a consequence of damage to the glomerulus (Tipping and Holdsworth, 2006). Glomerular neutrophil accumulation was minimal in γ−/− mice (Figure 4A) as previously shown (Suzuki et al., 1998). On the contrary, a striking increase in neutrophil influx was observed in RIIAtg/γ−/− and RIIA+IIIBtg/γ−/− at day 7 after disease induction (Figure 4A). The apparent decrease at later timepoints, likely reflecting the destruction or clearance of neutrophils in the inflamed tissue. Significant glomerular neutrophil accumulation was also detected in RIIIBtg/γ−/− mice. However, this was not accompanied by renal injury in these animals (Figure 3), implying that the recruited neutrophils are not activated. Elevated glomerular neutrophil counts were observed in RIIA+RIIIBtg/γ−/− compared to mice expressing either transgene alone and this far exceeded that observed in wild-type mice (Figure 4A). In the renal interstitium, infiltration of neutrophils, macrophages and CD4+ T cells was significant in both RIIAtg/γ−/− and RIIA+RIIIBtg/γ−/− mice (Figure 4B, C) which correlated with the severity of glomerular injury observed (see Figure 3). Interstitial leukocytic infiltration was minimal in RIIIBtg/γ−/−(Figure 4B), which correlates with a lack of glomerular injury in this group of animals (Figure 3A–E). CD3+ T cells and F4/80 positive macrophages in renal infiltrates remained negative for hFcγR expression (data not shown).
Figure 4.

Analysis of renal neutrophil and macrophage accumulation in progressive NTS nephritis (A) Glomerular neutrophil accumulation was evaluated at the indicated time points (n=5–14 per group) following induction of progressive NTS nephritis. Representative pictures of sections with neutrophil specific esterase stain are shown from day 7 samples. Arrowheads indicate esterase positive neutrophils (B–C) Analysis of interstitial infiltrates of kidneys harvested at the indicated days after NTS injection by FACs analysis of renal single cell suspensions. Percentages of interstitial neutrophils (Gr-1high/F4/80−) and monocytes/macrophages (F4/80+) (B) are given. Dotted line indicates baseline levels of infiltrates present in non-treated γ−/− mice. In (C), the ratio of CD4+/CD8+ T cells in renal cell infiltrates and peripheral blood is given. Representative renal sections from RIIAtg/γ−/− mice immunohistochemically stained for CD4+ and CD8+ T cells revealed significant periglomerular infiltration of CD4+ T cells while CD8+ T cells were primarily restricted to the interstitium. N= 7–14 animals per group. All data are mean±SEM. Statistics are as described in Figure 3.
Human neutrophil FcγRs are sufficient to restore the Reverse Passive Arthus reaction in γ−/− mice
The cutaneous RPA reaction is elicited by soluble ICs (Sylvestre and Ravetch, 1994). Neutrophil accumulation and edema were significantly attenuated in γ−/− mice compared to wild-type mice (Figure 5A,C) as reported (Sylvestre and Ravetch, 1994). In contrast, robust edema was observed in RIIAtg/γ−/− and RIIA+RIIIBtg/γ−/− mice that was associated with significant dermal neutrophil accumulation (Figure 5A, C). Mice expressing both human FcγRs exhibited disease indices in excess of wild-type mice. In contrast, edema was largely absent in RIIIBtg/γ−/− mice (Figure 5A,B) despite neutrophil accumulation that was comparable to that seen in wild-type animals (Figure 5C). Thus, as observed with NTS nephritis, human neutrophil FcγRs expression was sufficient to support the RPA reaction: FcγRIIA and FcγRIIIB mediated neutrophil accumulation, FcγRIIA elicited tissue injury, and together with FcγRIIIB promoted disease that was in excess of that observed in wild-type animals expressing endogenous murine FcγRs.
Figure 5.

Analysis of the Reverse Passive Arthus reaction. (A) The Reverse Passive Arthus reaction was induced by the subcutaneous administration of anti-OVA antibody and the intravenous injection of OVA with (upper panels) or without (lower panels) Evans blue dye. Representative pictures of tissues harvested 4 hrs after anti-OVA injection are shown. Evans blue leakage, a measurement of edema, is prominent in RIIAtg/γ−/− but not γ−/− and RIIIBtg/γ−/− mice. Esterase-stained sections show neutrophil accumulation in the subcutaneous layers of tg/γ−/− mice while this was minimal in γ−/− animals. Bar: 100μm. (B) Evans blue in skin was quantitated by blue dye extraction in dimethyformamide and measurement of absorbance at O.D595. (C) Neutrophil accumulation was assessed on esterase-stained skin sections harvested 4 hrs after disease induction. Average cell number±SEM is given. N= 8–11 mice per group. Statistics are as described in Figure 3.
Evidence for human neutrophil FcγRIIA and FcγRIIIB tethering to immune complexes formed in situ
A widely held view of antibody-mediated disease is that FcγR bearing resident tissue cells promote inflammatory cell infiltration. Our results indicate that neutrophil FcγRs, in the absence of FcγR expressing mast cells and macrophages, support neutrophil recruitment. To explore whether direct tethering of neutrophil FcγRs to ICs deposited in the vessel wall promotes neutrophil accumulation, we evaluated the tg/γ−/− in an acute model of NTS. In this model, NTS delivered in the absence of IgG presensitization results in rapid, transient glomerular neutrophil influx. The relative absence of tissue injury in this model minimizes secondary effects in neutrophil accumulation. Glomerular neutrophil influx was minimal in γ−/− mice (Figure 6) as reported (Coxon et al., 2001; Suzuki et al., 2003). In contrast, neutrophil recruitment was observed in RIIIBtg/γ−/− and wild-type mice that peaked at 1hr after NTS injection. Neutrophil accumulation in RIIAtg/γ−/− was also prominent at the 1hr timepoint but this was sustained and enhanced compared to wild-type and RIIIBtg/γ−/− animals at timepoints up to 4hrs after NTS injection. This response was further increased in RIIIB+RIIAtg/γ−/− mice compared to either of the single transgenics (Figure 6). These results provide evidence that the neutrophil FcγRIIA and FcγRIIIB directly tether to in situ formed glomerular ICs.
Figure 6.
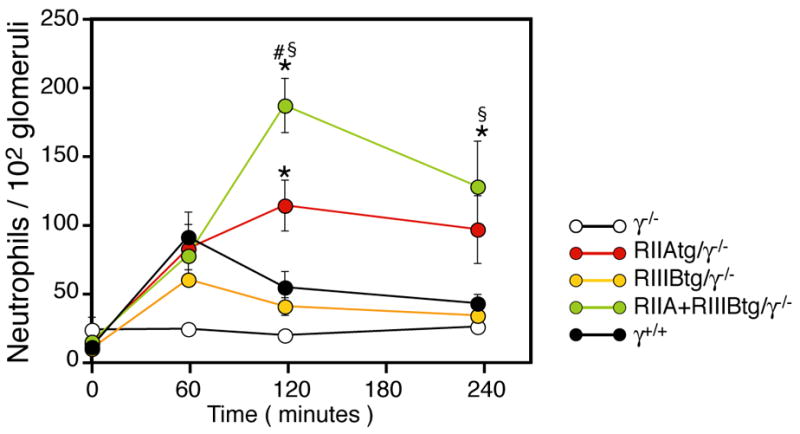
Analysis of neutrophil influx following acute NTS nephritis. Mice were injected with NTS, kidneys were harvested at the indiated timepoints and neutrophil accumulation was enumerated in neutrophil specific esterase stained tissue sections. Comparable neutrophil recruitment was observed in wild-type mice and all three transgenic animals at the 1hr timepoint. At later time points neutrophil accumulation was further increased in IIAtg/γ−/− and IIA+IIIBtg/γ−/− but declined in IIIBtg/γ−/− and WT mice. N=6–12 per group. All data are mean ± SEM. Statistics are as described in Figure 3.
Differential contributions of FcγRs in neutrophil slow rolling, adhesion and transmigration in models of soluble IC deposition
Here we examined the contribution of human FcγRs to neutrophil recruitment in response to soluble ICs. For this, two independent models of soluble IC deposition amenable to intravital microscopy were exploited. In the first model the RPA was induced in the cremaster muscle by the intrascrotal injection of anti-OVA and the intravenous delivery of OVA. Neutrophil recruitment in this model requires complement and TNF as well as cellular responses of mast cells, platelets and activated endothelial cells (Lister et al., 2007; Norman et al., 2005; Norman et al., 2003). When applied to the skin, the RPA results in IC deposition within the vessel, and in the perivascular and extravascular space (Cream et al., 1971; Jancar and Sanchez Crespo, 2005). The RPA did not increase the rolling flux fraction in wild-type mice (data not shown) but did induce significantly slow leukocyte rolling velocities, and enhanced adhesion and transmigration compared to animals treated with OVA alone (Figure 7A–C). The rolling flux fraction was similar between wild-type, tg/γ−/− andγ−/− mice (data not shown). However, leukocyte rolling velocities were significantly reduced in RIIAtg/γ−/− and RIIIB+RIIAtg/γ−/− mice compared to WT and γ−/− animals suggesting that human FcγRIIA promotes slow rolling (Figure 7A). The transition to slow rolling in other models of inflammation is associated with firm arrest (Ley et al., 2007). Consistent with this, slow rolling was a reliable predictor of IC-mediated adhesion as the latter was significantly elevated in RIIAtg/γ−/− and RIIIB+RIIAtg/γ−/− mice compared to γ−/− animals (Figure 7B). Transmigration, which is a time dependent variable of adhesion, was also elevated in RIIAtg/γ−/− and RIIIB+RIIAtg/γ−/− mice compared to γ−/− mice (Figure 7C). G-protein coupled receptors (GPCR) play major roles in leukocyte interactions with the vessel wall in non-immune complex mediated inflammation models (Ley et al., 2007). An intravenous injection of pertussis toxin, a GPCR inhibitor, prior to the induction of the RPA, blocked GPCR function as assessed by the inhibition of fmlp-mediated ROS generation in peripheral blood neutrophils sampled at the end of the intravital microscopy procedure (data not shown). Under these treatment conditions, pertussis toxin completely abrogated FcγRIIA mediated adhesion and transmigration (Figure 7B–C), but had no effect on slow rolling (Figure 7A), rolling flux fractions or peripheral blood leukocyte counts (data not shown). In stricking contrast to FcγRIIA, FcγRIIIB failed to significantly support neutrophil recruitment as slow rolling and adhesion was not observed in RIIIBtg/γ−/− mice (Figure 7A,B). Thus in this model, FcγRIIA cooperate with GPCRs to enhance soluble IC-induced leukocyte adhesion and transmigration, while FcγRIIIB does not significantly participate in this process.
Figure 7.

Intravital microscopic analysis of neutrophil recruitment following deposition of soluble ICs. In panels A–C, the RPA was induced in the cremaster of the indicated mice by an intrascrotal injection of anti-OVA followed by an intravenous injection with OVA. Leukocyte rolling velocity (A), adhesion (B) and transmigration (C) were evaluated in each group. The same was evaluated in wild-type mice (γ+/+) following injection with OVA alone and the average is presented as a dashed line. IIAtg/γ−/− mice pretreated with pertussis toxin (+PTx) are indicated. Following IC injection, IIAtg/γ−/− and IIA+IIIBtg/γ−/− exhibited slow rolling while IIIBtg/γ−/− failed to do so (A). Compared to γ−/−, adhesion (B) and transmigration (C) were significantly increased in IIAtg/γ−/− and IIA+IIIBtg/γ−/− but not in IIIBtg/γ−/−. Representative pictures of a postcapillary venule from γ−/− IIAtg/γ−/− and IIIBtg/γ−/− mice with rolling (arrow), adherent (arrowhead) and transmigrated (asterisk) neutrophils are shown. Pertussis toxin pre-treatment (PTx) completely suppressed the adhesion and transmigration of leukocytes in IIAtg/γ−/−. In panels D–F, the indicated mice were injected intravenously with preformed soluble BSA-anti-BSA ICs and the cremaster was exteriorized for intravital microscopy. IIIBtg/γ−/− mice given pertussis toxin (+PTx) is indicated. Leukocyte rolling velocity (D), adhesion (E) and transmigration (F) were evaluated in each group. The same was evaluated in wild-type mice following injection with BSA alone, and the average is presented as a dashed line. Slow leukocyte rolling was observed in wild-type (γ+/+) mice injected with ICs compared to mice given BSA alone while this behavior was absent in γ−/− mice (D). Following IC injection, IIIBtg/γ−/− and IIA+IIIBtg/γ−/−, but not IIAtg/γ−/− exhibited significant slow rolling (D) and adhesion (E). Transmigration was prominent in all groups of animals except γ−/− mice (F). Representative pictures of neutrophils interacting with postcapillary venules from γ−/−, IIAtg/γ−/− and IIIBtg/γ−/− mice are shown. Data is mean± SEM. *p<0.05, n=5 per group.
Next, mice were evaluated following the deposition of preformed anti-BSA/BSA soluble ICs. In this model, intravenously delivered ICs rapidly deposit within the vessel wall as a result of changes in vascular permeability induced by cremaster exteriorization (Stokol et al., 2004). IC deposition promotes neutrophil slow rolling on P-selectin, and adhesion and transmigration that are dependent on murine activating FcγRs (Stokol et al., 2004). Rolling flux fractions did not increase in mice given preformed ICs compared to mice given BSA alone as described (Stokol et al., 2004) and rolling flux fractions were similar between all animal groups (data not shown). ICs induced slow-rolling, adhesion and transmigration in wild-type mice that were significantly attenuated in γ−/− mice (Figure 7D–F) as reported (Stokol et al., 2004). However, in contrast to the cremaster RPA, FcγRIIIB was solely required for slow-rolling, adhesion and transmigration in this model as only RIIIBtg/γ−/− and IIA+IIIBtg/γ−/− and not RIIAtg/γ−/− supported these processes compared to γ−/− animals (Figure 7D–F, Supplemental Movies S1–3). RIIA/γ−/− mice had measurable numbers of extravascular neutrophils (Figure 7F) suggesting that the small number of adherent FcγRIIA expressing neutrophils efficiently transmigrate. To examine the potential contribution of GPCR binding chemokines to FcγRIIIB mediated neutrophil interactions with the vessel wall, FcγRIIIBtg/γ−/− mice were pre-treated with pertussis toxin prior to the injection of preformed soluble ICs. Pertussis toxin had no effect on FcγRIIIB mediated slow rolling, adhesion or transmigration. Thus under conditions of primarily intravascular ICs, and in the absence of GPCR activation, FcγRIIIB specializes in neutrophil recruitment.
Discussion
Our data demonstrate that expression of human FcγRs selectively on neutrophils is sufficient to induce virtually all aspects of Type II and Type III autoimmune responses and hence may provide critical molecular links between antibodies and immunological injury in a range of IgG mediated disorders. Neutrophils were recruited via FcγRIIA and FcγRIIIB, while FcγRIIA alone signalled tissue injury. The individual neutrophil human FcγRs appear to play separate roles in IC-induced neutrophil recruitment both in response to ICs formed in situ as well as soluble ICs deposited in the vessel wall. In the case of soluble ICs, FcγRIIIB specialized in neutrophil interactions in the context of strictly intravascular ICs while FcγRIIA predominated in the RPA reaction, which is a more complex response of vascular and tissue ICs and GPCR binding chemokines. In the case of in situ generated ICs, FcγRIIIB and FcγRIIA initiated recruitment while FcγRIIA was required for sustained neutrophil accumulation. Neutrophil recruitment and organ injury in the transgenic line expressing the human complement of FcγRs (FcγRIIA and FcγRIIIB), far exceeded that observed in wild-type mice (FcγRIII and FcγRIV) despite equivalent expression levels of the human and murine activating receptors. This provides evidence that the human and mouse neutrophil FcγRs are not functionally equivalent and in humans may play primary roles in initiating IC-mediated diseases.
The greater tissue damage observed in mice expressing the human FcγRs compared to the murine FcγRs (i.e. wild-type mice) may be the result of intrinsic differences such as more effective ITAM based signal transduction leading to neutrophil cytotoxicity (Van den Herik-Oudijk et al., 1995) or a differential capacity of the human and murine FcγRs to support neutrophil recruitment to ICs under conditions of limiting amounts of ICs in vivo. However, we cannot rule out the possibility that extrinsic factors such as the relative degree of interactions between the activating FcγRs with murine FcγRIIB, and/or a potential downregulatory role for murine FcγRs in other cell types in disease progression accounts for these differences. Implicit in our results is that the contribution of neutrophil-expressed murine FcγRs may have been underestimated in past studies. This may indeed be the case as mast cells and macrophages are only partially responsible for the RPA reaction and progressive NTS nephritis respectively (Aitman et al., 2006; Bergtold et al., 2006; Sylvestre and Ravetch, 1996).
Our studies suggest that the fundamental assumptions of the pathogenesis of hypersensitivity disease may require reevaluation in the case of human inflammation. Our finding that neutrophils are sufficient to promote Type II and Type III hypersensitivity requires modification of the current paradigm primarily deduced from studies in knock-out mice, which suggest that FcγR expressing tissue resident cells (mast cells and macrophages) initiate IC-mediated inflammatory reactions (Schmidt and Gessner, 2005). Our finding that human FcγRs on neutrophils play a primary role in progressive NTS nephritis suggests the possibility that neutrophils, and in particular FcγRs on neutrophils may play a dominant role in the pathogenesis of IC-mediated glomerulonephritides. This suggests a broader significance for neutrophils in these conditions than previously anticipated. Importantly, neutrophils have been documented in renal biopsies from patients with membranoproliferative, lupus and crescentic glomerulonephritis (Camussi et al., 1980; Hooke et al., 1987; Segerer et al., 2006). Human FcγRs were also observed in a subpopulation of monocytes in our transgenic lines. Therefore FcγRs on monocytes may be required for the full expression of renal disease. However, it is noteworthy that γ−/− mice with transgenic re-expression of the γ-chain and thus activating FcγRs in monocytes and macrophages, with no detectable expression in neutrophils, continued to exhibit a 70% decrease in indices of glomerular damage compared to wild-type counterparts (Bergtold et al., 2006). This argues that FcγR expression in monocytes is not sufficient for disease induction. Glomerular injury associated proteinuria itself as well as chemokines secreted by the glomerulus, stimulate tubular epithelial cells to secrete chemokines that support interstitial leukocyte infiltration (Anders et al., 2003). Our data indicate that FcγRIIA-mediated glomerular neutrophil recruitment and proteinuria promotes subsequent interstitial influx of macrophage and T cells, which are effector populations known to contribute to disease pathogenesis and end-stage renal failure (Duffield et al., 2005; Hooke et al., 1987; Tipping and Holdsworth, 2003). Of interest, the highest number of neutrophils in human renal biopsies was observed in glomerulonephritides with prominent recruitment of monocytes/macrophages (Segerer et al., 2006).
FcγRIIA alone was sufficient for mediating immunological injury in vivo. The demonstration that reconstitution with an FcγR is critical in disease models shown previously to be dependent on the γ-chain is noteworthy as this ITAM-based adaptor is also central to the regulation of receptors important in MHC-I recognition and myeloid cell and platelet activation raising the possibility that some of the phenotypes in γ−/− mice may be attributed to deficiency in signaling through these receptors (Fodor et al., 2006; Mocsai et al., 2006; Underhill and Goodridge, 2007). The differential cytotoxic activity of the ITAM-containing FcγRIIA versus the GPI-linked FcγRIIIB is likely related to a requirement for ITAM-based signal transduction in generation of neutrophil effector functions such as ROS generation, as demonstrated by our in vitro assays on transgenic neutrophils and published reports on human neutrophils (Underhill and Goodridge, 2007). Furthermore, overexpression of ITAM-containing human FcγRIIA or FcγRI on monocytes/macrophages in wild-type mice has been reported to aggravate arthritis or glomerulonephritis development (Kanamaru et al., 2007; Tan Sardjono et al., 2005). On the other hand, FcγRIIIB may fail to exhibit cytotoxicity in vivo because it is shed from the surface of neutrophils accumulated in the inflamed tissue.
An important finding of our studies was that expression of human FcγRs on neutrophils was sufficient to elicit neutrophil recruitment. This is a complex process previously attributed to endothelial cell adhesion molecule upregulation by cytokines released either from activated endothelial cells and/or by other cells within the vessel wall to which antibody is bound (Mayadas et al., 1996; Nikolic-Paterson et al., 1994; Norman et al., 2003; Schmidt and Gessner, 2005). The human FcγRs transgenics exhibited significant neutrophil accumulation in tissues despite the absence of these receptors on mast cells and macrophages, which indicates a primary role for the FcγRs on neutrophils in IC-induced neutrophil recruitment. FcγRIIA and FcγRIIIB played equivalent roles in initial neutrophil recruitment in response to antibody-antigen complexes that are formed in situ within the glomerular capillaries. As the ICs are accessible to circulating neutrophils through endothelial fenestrae (Fries et al., 1988), our results strongly suggest that neutrophil FcγRs directly tether to ICs. The enhanced accumulation in mice expressing FcγRIIA observed at later time points suggests an additional role for this receptor in sustaining neutrophil accumulation perhaps by signaling the release of leukotrienes and prostaglandins (Jancar and Sanchez Crespo, 2005). Analysis of mice in intravital models of soluble IC deposition indicated novel, nonredundant functions for the two human FcγRs. FcγRIIIB was specialized for slow rolling and adhesion induced by intravascular soluble ICs in a model where neutrophil accumulation is complement C3 and C5 independent, does not require the function of mast cells and is not associated with platelet accumulation (Stokol et al., 2004). However, in a more complex environment of intravascular, perivascular and tissue ICs generated by the RPA (Cochrane, 1963; Cream and Turk, 1971; Movat and Fernando, 1963), FcγRIIA predominated. FcγRIIA dependent adhesion and transmigration were GPCR dependent. We postulate that engagement of GPCR(s) on neutrophils and/or other cell types may directly or indirectly modulate FcγRIIA mediated adhesion by affecting its affinity for deposited IgG (Nagarajan et al., 2000) or upregulating neutrophil CD18 integrins known to support FcγR function (Jones and Brown, 1996).
The assignment of an important physiological role for FcγRIIIB in neutrophil recruitment is particularly significant as the function of this GPI-anchored receptor remains largely unknown. Furthermore, our results ascribe a function to FcγRIIIB that doesn’t require FcγRIIA, a finding unanticipated from all previous work (Unkeless et al., 1995). What is the physiological role of FcγRIIIB in neutrophil tethering to intravascular ICs? We propose that FcγRIIIB may clear ICs from the glomerulus which is a frequent site of IC trapping and thus aid in the maintenance of homeostasis (Nangaku and Couser, 2005). Indeed glomerular ICs can trigger a transient accumulation of neutrophils with their return to the circulation and a complete clearance of the immune deposits and restoration of the glomerular structural integrity within 24 hrs of IC deposition (Fries et al., 1988). The high density of FcγRIIIB with a GPI anchor nearly the size of an Ig domain that could permit it to protrude further than FcγRIIA, its predicted fast mobility in the membrane bilayer (Selvaraj et al., 1988), its presence on microvilli (Coxon et al., 2001) coupled with its weak signaling capacity may suit FcγRIIIB for efficient capture and internalization of ICs with minimal neutrophil activation. The higher expression of FcγRIIIB in human versus our murine transgenic neutrophils, predicts a more significant role for this receptor in IC-induced neutrophil accumulation in humans. FcγRIIIB is normally present in multiple gene copies usually positively correlates with its expression and low gene copy numbers of FcγRIIIB was associated with nephritis in systemic lupus erythematosus (Stranger et al., 2007). We speculate that inefficient FcγRIIIB mediated recruitment and clearance of ICs under homeostatic conditions may enhance susceptibility to IC mediated diseases in patients. Pathogenic IC accumulation may increase the local generation of inflammatory mediators that promote FcγRIIIB shedding, increase the capacity of FcγRIIA to bind ligand and signal and thus promote tissue damage.
In summary, our studies in FcγR humanized mouse models have provided evidence that human FcγR expression on neutrophils is sufficient for the initiation of IC-induced inflammatory disease and thus redefines the currently accepted models of Type II and III autoimmune responses developed in mice. Neutrophils are recruited via their own FcγRs to ICs with FcγRIIA and FcγRIIIB playing novel and distinct context-dependent roles in this process, while FcγRIIA alone is responsible for tissue injury. We anticipate that transgenic mice expressing the human repertoire of neutrophil FcγRs will continue to serve as important tools for investigating the physiological function of the individual receptors, their signaling capacity and the mechanisms by which they promote tissue injury in inflammatory and autoimmune disorders relevant to human disease.
Experimental Procedures
Generation of human FcγR transgenic mice
The 0.95-kb of human FcγRIIA cDNA and 0.7-kb of FcγRIIIB cDNA were subcloned into the BglII site of the hMRP8 promoter (Lagasse and Weissman, 1994). Natural polymorphisms in the FcγRIIA and IIIB have been reported; of these cDNAs encoding NA2 FcγRIIIB and R131 FcγRIIA (van Sorge et al., 2003) were utilized to generate the transgenics. R131 FcγRIIA is a natural variant of FcγRIIA in the human population that is highly responsiveness to mouse IgG (Haagen et al., 1995). Each transgene was released from pUC18-hMRP8 vector by digestion with HindIII and EcoRI and injected into zygotes from C57Bl/6J mice. Transgenic mice were generated in the transgenic facility of Brigham & Women’s Hospital transgenic facility (Boston, MA). A high-expressing founder transgenic line of FcγRIIA transgenic mice (RIIAtg/γ+/+) were crossed with γ−/− mice on a C57Bl/6J-F12 background and bred to be hemizygous for the FcγRIIA transgene and γ-chain deficient, as assessed by PCR of genomic DNA and flow cytometry analysis. RIIIBtg/γ−/− mice were similarly generated. Mice expressing both FcγRIIA and FcγRIIIB (RIIA+IIIBtg/γ−/−) were generated by breeding RIIAtg/γ−/− and RIIIBtg/γ−/− mice. Mice were maintained in a virus- and antibody-free facility at the New Research Building animal housing facility at Harvard Medical School. Mice used for each experiment were between 6–8 weeks of age and sex matched. The Harvard Medical School Animal Care and Use Committee approved all procedures in this study.
Isolation and treatment of leukocyte populations
Peripheral blood sampled from the retroorbital plexus of mice was collected in EDTA containing tubes. Human peripheral neutrophils and murine bone marrow neutrophils were isolated as previously described (Coxon et al., 2001; Hirahashi et al., 2006). For the assessment of FcγR shedding, mouse bone marrow neutrophils were stimulated with or without 100 ng/ml of PMA for 10 min at 37°C, and then fixed with 4% paraformaldehyde. Bone marrow derived macrophages were generated by culturing cells harvested from the tibia and femurs of mice in DMEM supplemented with 10% FCS and 20% L929 conditioned supernatant for 5 days. Peritoneal cells for FACs analysis were harvested by lavaging the peritoneum with cold PBS and plating for 18 hrs on polystyrene dishes in 10% FCS in DMEM. For peritoneal mast cells, peritoneal cells were immediately used for FACs analysis.
Flow cytometry analysis
All antibodies were from BD Biosciences-Pharmingen unless otherwise indicated. Human FcγRIIA or FcγIIIB expression on peripheral blood leukocytes were characterized by using FITC or APC-mouse anti-human FcγRIIA (clone FLI8.26, mouse IgG2κ) or FITC-mouse anti-human FcγRIIIB (clone 3G8, mouse IgG1). Cell populations were identified by PE-rat anti-Gr-1 (Ly-6G and Ly-6C, clone RB6-8C5) for neutrophils, PE-rat anti-CD115(clone AFS98; eBioscience) for monocytes, PE-Cy7-hamster anti-mouse CD3ε (clone 145-2C11) for T cells, allophycocyanin (APC)-rat anti-mouse CD11b (Integrin αM chain, clone M1/70) for neutrophils and monocytes and FITC-rat anti-mouse CD62P (RB40.34) for platelets. As a positive control for receptor shedding, APC-rat anti-mouse CD62L (clone MEL-14) was used.
For analysis of resident peritoneal and bone marrow derived macrophages, cells were stained with PE-mouse anti-human FcγRIIA or FcγRIII antibody amd APC-rat anti-F4/80 (clone A3-1; Caltag Laboratories). Mature mast cell populations were identified using FITC-hamster anti-mouse FcεRIa (clone MAR-1; eBioscience) and PE-rat anti-mouse c-Kit (clone 2B8) antibody. FcγRIa negative, but c-Kit positive cells in tg/γ−/− and γ−/− mice were analyzed for human FcγR expression.
Infiltrating renal neutrophils, macrophages and T cells were quantitated by three-color flow cytometry. Single-cell suspensions from individual kidneys were prepared as described (Vielhauer et al., 2003). For analysis of leukocyte surface markers, cells were incubated with PE-anti-Gr-1, APC-anti-CD3ε or APC-anti-F4/80. The amounts of positively stained neutrophils (Gr-1high/F4/80−), T cells (Gr-1−/CD3ε+), and macrophages (F4/80+) were expressed as percentage of total renal cells. Renal cell suspensions were also stained with APC-anti-CD3ε, FITC-anti-CD4(clone RM4-5) and PE-anti-CD8a(clone 53-6.7) to determine the CD4+/CD8+ ratio in the CD3+ T cell population.
Quantitative real time PCR
Complementary DNAs were synthesized from total RNA of bone marrow neutrophils using a cDNA synthesis kit (Invitrogen). RT-PCR by Taq DNA polymerase (New England Biolabs) was performed using the following primer sets: mouse FcγRIIIA (Qiagen, QT00117803); human FcγRIIA (Qiagen, QT00042826); human FcγRIIIB (5′-CGTGCTTGAGAAGGACAGTG-3′, 5′-CTGGCTTGAGATGAGGCTCT-3′); mouse β-actin (5′-CCTGAGCGCAAGTACTCTGTGT-3′, 5′-GCTGATCCACATCTGCTGGAA-3′). Each RT-PCR product was inserted into pGEMT-Easy vector (Promega) for a reference standard. Quantitative real time PCR on cDNA samples by SYBR green RT-PCR method (Bio-Rad, Hercules, CA, USA) was conducted using the indicated primer sets. Actual transcript levels of mouse FcγRIIIA and human FcγRs genes were determined against the reference standard made by serial dilution of the pGEMT-Easy vector containing FcγR templates. The relative expression of each gene was normalized against β-actin.
Neutrophil adhesion, F-actin staining and determination of cellular H2O2 production on IC
Experimental details for adhesion assay and F-actin staining were previously described (Tang et al., 1997). Adherent cell numbers were quantitated in 3 independent fields at magnification x200 and the average cell number per field was determined. Neutrophils were plated on glass cover slips coated with ICs formed by BSA and polyclonal rabbit anti-BSA antibody (Tang et al., 1997) or BSA alone in 24 well plates. The experimental protocol for cellular H2O2 production was essentially as described by Werner et al., 2003 (Werner, 2003) with some modifications. 1.0×106 bone marrow neutrophils from each mouse strain in 1.0 mL of assay mix solution (100 μM homovanillic acid (Sigma) in HBSS, 5 U/mL horseradish peroxidase type VI (Sigma), and 1 mM Hepes pH 7.5) were incubated at 37°C for indicated time periods. The plate was then centrifuged and cell-free supernatant was collected and further incubated at 37°C for 1 hr. The reaction was terminated by the addition of 0.1 M glycine solution. The fluorescence in supernatants was measured by a fluorometer (excitation 321 nm, emission 421 nm) and the H2O2 concentration in each sample was determined relative to the calibration curve made by an H2O2 standard.
Human FcγR cross-linking-mediated oxidative burst in neutrophils
Bone marrow neutrophils were incubated with 10 μg/ml mouse anti-hFcγRIIA (clone: IV.3, StemCell Technologies) or anti-hFcγRIIIB (clone 3G8, Caltag Laboratories) on ice for 30 min. Cells (2.5×106/sample) were washed, and then incubated with 1 μg/ml of mouse GM-CSF for 30 min following by incubation without or with piceatannol (20 μM, Sigma), PP2 (10 μM, Calbiochem) or LY294002 (20 μM, Calbiochem) at 37°C for 30 min. FcγR cross-linking was initiated by adding goat anti-mouse F(ab′)2 (100 μg/ml, Jackson ImmunoResearch Laboratories) and ROS generation was continuously monitored using a luminol based assay as previously described (Utomo et al., 2006).
Nephrotoxic serum nephritis
Experimental nephrotoxic serum (NTS) nephritis was induced in 8-weeks-old male as previously reported (Rosenkranz et al., 2000) with the following modifications. Briefly, mice were preimmunized subcutaneously in the right footpad with 0.05 mg rabbit IgG (Jackson ImmunoResearch Laboratories Inc.) in Freund incomplete adjuvant and nonviable desiccated Mycobacterium tuberculosis H37Ra (Difco, Michigan). Three days later, mice were injected intravenously with 50 μL heat-inactivated, filter-sterilized nephrotoxic serum. Spot urine samples and peripheral blood were collected at indicated time points after NTS injection. Both kidneys from euthanized mice were harvested for histological analysis and flow cytometry. Acute NTS nephritis was induced in 7-weeks-old male mice by the intravenous injection of 300 μL of nephrotoxic serum without prior preimmunization.
Functional assessment of renal injury
Urine albumin concentrations and creatinine levels in urine and serum were determined by ELISA (Rosenkranz et al., 2000) and Creatinine Assay Kit (Cayman Chemical Company), respectively. Albuminuria was expressed as milligrams albumin per milligrams urinary creatinine to standardize urine albumin excretion for glomerular filtration rate and urinary concentration.
Histological assessment of renal injury, renal neutrophil and T cell accumulation
The presence of PAS-positive deposits within glomeruli was graded semiquantitatively as previously described (Rosenkranz et al., 2000). Glomerular PMN infiltration was assessed by the chloroacetate esterase reaction as reported (Coxon et al., 2001). For each animal, glomerular neutrophil counts in more than 100 glomeruli/kidney section were made. For the histological assessment of T cell accumulation, renal cryostat sections were stained with unlabeled anti-CD4 (clone RM4-5) or anti-CD8a (clone 53-6.7).
Reverse Passive Arthus reaction (RPA)
Rabbit anti-chicken egg albumin IgG (60 μg/30 μL; Cappel, Aurora, OH) or PBS alone were injected subcutaneously (s.c.) in the left or right portion of the dorsal skin in 6 to 8-weeks-old female mice, followed immediately by the intravenous (i.v.) injection of chicken egg ovalbumin (400 μg/mouse; Sigma-Aldrich, St. Louis, MO). 4hrs later, the skin containing the injection site was removed from euthanized mouse. In cases where edema was measured, the solution of chicken egg albumin contained 0.15% Evans blue dye (Sigma-Aldrich). Measurement of Evans blue leakage and neutrophil influx was conducted as described (Utomo et al., 2008).
Intravital microscopy
Soluble IC were prepared as previously described (Stokol et al., 2004). Leukocyte recruitment in cremaster muscle venules was evaluated in mice within 60 min of a single i.v. injection of ICs or BSA. For the RPA in the scrotum, rabbit IgG anti-chicken egg albumin antibody (100 μg/100 μL) was injected intrascrotally, followed by the i.v. injection of chicken egg ovalbumin (240 μg/240 μL; Sigma-Aldrich, St. Louis, MO). Leukocyte recruitment in the cremaster was evaluated 2h after the injection. In some cases, mice were pre-treated i.v. with 4 μg of pertussis toxin (Sigma) 4h prior to injection of preformed soluble ICs or induction of the RPA.
The procedures for preparation of the cremaster of anesthetized mice and subsequent intravital microscopy were essentially as described (Lauterbach et al., 2008). Four venules a mouse were analyzed over a 20 min time period. At the completion of the intravital microcopy experiment, blood was collected from the retro-orbital plexus to measure total leukocyte counts. Leukocyte rolling velocities were measured by tracking single leukocytes (10/venule) over several frames and calculating distance moved per unit time (μm/s). Adherent leukocytes were defined as cells remaining stationary for 30s and were expressed as the number of cells/mm2 of venule. Transmigrated leukocytes were defined as cells outside of the venule and were expressed as the number of cells/mm2.
Statistical analysis
In nephrotoxic anti-GBM nephritis and RPA, data was analyzed by ANOVA among 5 strains, γ−/−, RIIAtg/γ−/−, RIIIBtg/γ−/−, RIIA+IIIBtg/γ−/− and C57Bl/6 mouse except for day 21 samples when 4 strains were analyzed due to low survival in RIIA+RIIIBtg/γ−/− animals. Data, in which significant difference (p<0.001) was shown by ANOVA, was subjected to Tukey/Kramer for comparison between two mouse strains at 5% significant level. In intravital microscopy, Mann-Whitney U test was used for analysis, and statistical significance was accepted at p<0.05.
Supplementary Material
Acknowledgments
The human MRP8 promoter construct was kindly provided by Dr. Eric Lagasse (Univ. of Pittsburgh, PA), and Dr. Irving L. Weissman (Stanford University, CA). We are grateful to Manabu Minami (Brigham and Women’s Hospital) for assistance with quantitative PCR analysis. We thank Ling Xiao and George Stavrakis for excellent technical assistance. This work was supported by a Arthritis Foundation postdoctoral fellowship (N.T.), and NIH RO1 HL065095 and AR050800 (T.N.M). Author contributions: NT and KA designed and performed the research, analyzed the data and wrote the paper, ML performed the research and analyzed the data, TNM designed the research and wrote the paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- Anders HJ, Vielhauer V, Schlondorff D. Chemokines and chemokine receptors are involved in the resolution or progression of renal disease. Kidney Int. 2003;63:401–415. doi: 10.1046/j.1523-1755.2003.00750.x. [DOI] [PubMed] [Google Scholar]
- Bergtold A, Gavhane A, D’Agati V, Madaio M, Clynes R. FcR-bearing myeloid cells are responsible for triggering murine lupus nephritis. J Immunol. 2006;177:7287–7295. doi: 10.4049/jimmunol.177.10.7287. [DOI] [PubMed] [Google Scholar]
- Camussi G, Cappio FC, Messina M, Coppo R, Stratta P, Vercellone A. The polymorphonuclear neutrophil (PMN) immunohistological technique: detection of immune complexes bound to the PMN membrane in acute poststreptococcal and lupus nephritis. Clin Nephrol. 1980;14:280–287. [PubMed] [Google Scholar]
- Cochrane CG. Studies on the Localization of Circulating Antigen-Antibody Complexes and Other Macromolecules in Vessels. I. Structural Studies. J Exp Med. 1963;118:489–502. doi: 10.1084/jem.118.4.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxon A, Cullere X, Knight S, Sethi S, Wakelin MW, Stavrakis G, Luscinskas FW, Mayadas TN. Fc gamma RIII mediates neutrophil recruitment to immune complexes. a mechanism for neutrophil accumulation in immune-mediated inflammation. Immunity. 2001;14:693–704. doi: 10.1016/s1074-7613(01)00150-9. [DOI] [PubMed] [Google Scholar]
- Cream JJ, Bryceson AD, Ryder G. Disappearance of immunoglobulin and complement from the Arthus reaction and its relevance to studies of vasculitis in man. Br J Dermatol. 1971;84:106–109. doi: 10.1111/j.1365-2133.1971.tb06851.x. [DOI] [PubMed] [Google Scholar]
- Cream JJ, Turk L. A review of the evidence for immune-complex deposition as a cause of skin diseases in man. Clin Allergy. 1971;1:235–247. [PubMed] [Google Scholar]
- Crockett-Torabi E, Smith CW, Kateley JR, Patterson R, Tsai P, Fantone JC. Insoluble immune complex-stimulated neutrophil leukotriene B4 production is dependent on Fc gamma RII and Fc gamma RIII and independent of pertussis toxin-sensitive signal transduction pathways. Am J Pathol. 1992;140:613–620. [PMC free article] [PubMed] [Google Scholar]
- Dean EG, Wilson GR, Li M, Edgtton KL, O’Sullivan KM, Hudson BG, Holdsworth SR, Kitching AR. Experimental autoimmune Goodpasture’s disease: a pathogenetic role for both effector cells and antibody in injury. Kidney Int. 2005;67:566–575. doi: 10.1111/j.1523-1755.2005.67113.x. [DOI] [PubMed] [Google Scholar]
- Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, Bonventre JV, Hughes J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey OJ, Johns M, Esho OO, Mason JC, Haskard DO. Antiendothelial cell antibodies mediate enhanced leukocyte adhesion to cytokine-activated endothelial cells through a novel mechanism requiring cooperation between Fc{gamma}RIIa and CXCR1/2. Blood. 2007;109:3881–3889. doi: 10.1182/blood-2006-08-044669. [DOI] [PubMed] [Google Scholar]
- Fodor S, Jakus Z, Mocsai A. ITAM-based signaling beyond the adaptive immune response. Immunol Lett. 2006;104:29–37. doi: 10.1016/j.imlet.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Fries JW, Mendrick DL, Rennke HG. Determinants of immune complex-mediated glomerulonephritis. Kidney Int. 1988;34:333–345. doi: 10.1038/ki.1988.186. [DOI] [PubMed] [Google Scholar]
- Haagen IA, Geerars AJ, Clark MR, van de Winkel JG. Interaction of human monocyte Fc gamma receptors with rat IgG2b. A new indicator for the Fc gamma RIIa (R-H131) polymorphism. J Immunol. 1995;154:1852–1860. [PubMed] [Google Scholar]
- Hirahashi J, Mekala D, Van Ziffle J, Xiao L, Saffaripour S, Wagner DD, Shapiro SD, Lowell C, Mayadas TN. Mac-1 signaling via Src-family and Syk kinases results in elastase-dependent thrombohemorrhagic vasculopathy. Immunity. 2006;25:271–283. doi: 10.1016/j.immuni.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Hogarth PM. Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr Opin Immunol. 2002;14:798–802. doi: 10.1016/s0952-7915(02)00409-0. [DOI] [PubMed] [Google Scholar]
- Hooke DH, Gee DC, Atkins RC. Leukocyte analysis using monoclonal antibodies in human glomerulonephritis. Kidney Int. 1987;31:964–972. doi: 10.1038/ki.1987.93. [DOI] [PubMed] [Google Scholar]
- Huizinga TW, van der Schoot CE, Jost C, Klaassen R, Kleijer M, von dem Borne AE, Roos D, Tetteroo PA. The PI-linked receptor FcRIII is released on stimulation of neutrophils. Nature. 1988;333:667–669. doi: 10.1038/333667a0. [DOI] [PubMed] [Google Scholar]
- Jancar S, Sanchez Crespo M. Immune complex-mediated tissue injury: a multistep paradigm. Trends Immunol. 2005;26:48–55. doi: 10.1016/j.it.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, et al. Arthritis critically dependent on innate immune system players. Immunity. 2002;16:157–168. doi: 10.1016/s1074-7613(02)00275-3. [DOI] [PubMed] [Google Scholar]
- Jones S, Brown E. Functional cooperation between Fcg receptors and complement receptors in phagocytes. In: van de Winkel JGJ, Capel PJA, editors. Human IgG Fc Receptors. R.G Landes Company; 1996. pp. 149–163. [Google Scholar]
- Kanamaru Y, Arcos-Fajardo M, Moura IC, Tsuge T, Cohen H, Essig M, Vrtovsnik F, Loirat C, Peuchmaur M, Beaudoin L, et al. Fc alpha receptor I activation induces leukocyte recruitment and promotes aggravation of glomerulonephritis through the FcR gamma adaptor. Eur J Immunol. 2007;37:1116–1128. doi: 10.1002/eji.200636826. [DOI] [PubMed] [Google Scholar]
- Lagasse E, Weissman IL. bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J Exp Med. 1994;179:1047–1052. doi: 10.1084/jem.179.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagasse E, Weissman IL. Enforced expression of Bcl-2 in monocytes rescues macrophages and partially reverses osteopetrosis in op/op mice. Cell. 1997;89:1021–1031. doi: 10.1016/s0092-8674(00)80290-1. [DOI] [PubMed] [Google Scholar]
- Lauterbach M, O’Donnell P, Mayadas T. Role of Fc gamma receptors and the CD18 integrin, Mac-1 in neutrophil recruitment to intravascular immune complexes. J Leukoc Biol. 2008 in press. [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Li S, Holdsworth SR, Tipping PG. Antibody independent crescentic glomerulonephritis in mu chain deficient mice. Kidney Int. 1997;51:672–678. doi: 10.1038/ki.1997.97. [DOI] [PubMed] [Google Scholar]
- Lister KJ, James WG, Hickey MJ. Immune complexes mediate rapid alterations in microvascular permeability: roles for neutrophils, complement, and platelets. Microcirculation. 2007;14:709–722. doi: 10.1080/10739680701404879. [DOI] [PubMed] [Google Scholar]
- Luscinskas FW, Mayadas T. FcgRs join in the cascade. Blood. 2007;109:3615–3616. [Google Scholar]
- Mayadas TN, Mendrick DL, Brady HR, Tang T, Papayianni A, Assmann KJ, Wagner DD, Hynes RO, Cotran RS. Acute passive anti-glomerular basement membrane nephritis in P-selectin-deficient mice. Kidney Int. 1996;49:1342–1349. doi: 10.1038/ki.1996.190. [DOI] [PubMed] [Google Scholar]
- Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movat HZ, Fernando NV. Allergic inflammation. I. The earliest fine structural changes at the blood-tissue barrier during antigen-antibody interaction. Am J Pathol. 1963;42:41–59. [PMC free article] [PubMed] [Google Scholar]
- Nagarajan S, Venkiteswaran K, Anderson M, Sayed U, Zhu C, Selvaraj P. Cell-specific, activation-dependent regulation of neutrophil CD32A ligand-binding function. Blood. 2000;95:1069–1077. [PubMed] [Google Scholar]
- Nangaku M, Couser WG. Mechanisms of immune-deposit formation and the mediation of immune renal injury. Clin Exp Nephrol. 2005;9:183–191. doi: 10.1007/s10157-005-0357-8. [DOI] [PubMed] [Google Scholar]
- Neale TJ, Wilson CB. Glomerular antigens in glomerulonephritis. Springer Semin Immunopathol. 1982;5:221–249. doi: 10.1007/BF01892087. [DOI] [PubMed] [Google Scholar]
- Nikolic-Paterson DJ, Main IW, Lan HY, Hill PA, Atkins RC. Adhesion molecules in glomerulonephritis. Springer Semin Immunopathol. 1994;16:3–22. doi: 10.1007/BF00196710. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity. 2006;24:19–28. doi: 10.1016/j.immuni.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Norman MU, Lister KJ, Yang YH, Issekutz A, Hickey MJ. TNF regulates leukocyte-endothelial cell interactions and microvascular dysfunction during immune complex-mediated inflammation. Br J Pharmacol. 2005;144:265–274. doi: 10.1038/sj.bjp.0706081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman MU, Van De Velde NC, Timoshanko JR, Issekutz A, Hickey MJ. Overlapping roles of endothelial selectins and vascular cell adhesion molecule-1 in immune complex-induced leukocyte recruitment in the cremasteric microvasculature. Am J Pathol. 2003;163:1491–1503. doi: 10.1016/S0002-9440(10)63506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Ueda S, Ohno H, Hamano Y, Tanaka M, Shiratori T, Yamazaki T, Arase H, Arase N, Karasawa A, et al. Resistance of Fc receptor- deficient mice to fatal glomerulonephritis. J Clin Invest. 1998;102:1229–1238. doi: 10.1172/JCI3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- Rosenkranz AR, Knight S, Sethi S, Alexander SI, Cotran RS, Mayadas TN. Regulatory interactions of alphabeta and gammadelta T cells in glomerulonephritis. Kidney Int. 2000;58:1055–1066. doi: 10.1046/j.1523-1755.2000.00263.x. [DOI] [PubMed] [Google Scholar]
- Rosenkranz AR, Mendrick DL, Cotran RS, Mayadas TN. P-selectin deficiency exacerbates experimental glomerulonephritis: a protective role for endothelial P-selectin in inflammation. J Clin Invest. 1999;103:649–659. doi: 10.1172/JCI5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100:56–67. doi: 10.1016/j.imlet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Segerer S, Henger A, Schmid H, Kretzler M, Draganovici D, Brandt U, Noessner E, Nelson PJ, Kerjaschki D, Schlondorff D, Regele H. Expression of the chemokine receptor CXCR1 in human glomerular diseases. Kidney Int. 2006;69:1765–1773. doi: 10.1038/sj.ki.5000337. [DOI] [PubMed] [Google Scholar]
- Selvaraj P, Rosse WF, Silber R, Springer TA. The major Fc receptor in blood has a phosphatidylinositol anchor and is deficient in paroxysmal nocturnal haemoglobinuria. Nature. 1988;333:565–567. doi: 10.1038/333565a0. [DOI] [PubMed] [Google Scholar]
- Skokowa J, Ali SR, Felda O, Kumar V, Konrad S, Shushakova N, Schmidt RE, Piekorz RP, Nurnberg B, Spicher K, et al. Macrophages induce the inflammatory response in the pulmonary Arthus reaction through G alpha i2 activation that controls C5aR and Fc receptor cooperation. J Immunol. 2005;174:3041–3050. doi: 10.4049/jimmunol.174.5.3041. [DOI] [PubMed] [Google Scholar]
- Stokol T, O’Donnell P, Xiao L, Knight S, Stavrakis G, Botto M, von Andrian UH, Mayadas TN. C1q governs deposition of circulating immune complexes and leukocyte Fcgamma receptors mediate subsequent neutrophil recruitment. J Exp Med. 2004;200:835–846. doi: 10.1084/jem.20040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Gomez-Guerrero C, Shirato I, Lopez-Franco O, Gallego-Delgado J, Sanjuan G, Lazaro A, Hernandez-Vargas P, Okumura K, Tomino Y, et al. Pre-existing glomerular immune complexes induce polymorphonuclear cell recruitment through an Fc receptor-dependent respiratory burst: potential role in the perpetuation of immune nephritis. J Immunol. 2003;170:3243–3253. doi: 10.4049/jimmunol.170.6.3243. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Shirato I, Okumura K, Ravetch JV, Takai T, Tomino Y, Ra C. Distinct contribution of Fc receptors and angiotensin II-dependent pathways in anti-GBM glomerulonephritis. Kidney Int. 1998;54:1166–1174. doi: 10.1046/j.1523-1755.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefining the inflammatory cascade. Science. 1994;265:1095–1098. doi: 10.1126/science.8066448. [DOI] [PubMed] [Google Scholar]
- Sylvestre DL, Ravetch JV. A dominant role for mast cell Fc receptors in the Arthus reaction. Immunity. 1996;5:387–390. doi: 10.1016/s1074-7613(00)80264-2. [DOI] [PubMed] [Google Scholar]
- Tan Sardjono C, Mottram PL, van de Velde NC, Powell MS, Power D, Slocombe RF, Wicks IP, Campbell IK, McKenzie SE, Brooks M, et al. Development of spontaneous multisystem autoimmune disease and hypersensitivity to antibody-induced inflammation in Fcgamma receptor IIa-transgenic mice. Arthritis Rheum. 2005;52:3220–3229. doi: 10.1002/art.21344. [DOI] [PubMed] [Google Scholar]
- Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, Cotran RS, Mayadas TN. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186:1853–1863. doi: 10.1084/jem.186.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping PG, Holdsworth SR. T cells in glomerulonephritis. Springer Semin Immunopathol. 2003;24:377–393. doi: 10.1007/s00281-003-0121-7. [DOI] [PubMed] [Google Scholar]
- Tipping PG, Holdsworth SR. T cells in crescentic glomerulonephritis. J Am Soc Nephrol. 2006;17:1253–1263. doi: 10.1681/ASN.2005091013. [DOI] [PubMed] [Google Scholar]
- Trcka J, Moroi Y, Clynes RA, Goldberg SM, Bergtold A, Perales MA, Ma M, Ferrone CR, Carroll MC, Ravetch JV, Houghton AN. Redundant and alternative roles for activating Fc receptors and complement in an antibody-dependent model of autoimmune vitiligo. Immunity. 2002;16:861–868. doi: 10.1016/s1074-7613(02)00327-8. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Goodridge HS. The many faces of ITAMs. Trends Immunol. 2007;28:66–73. doi: 10.1016/j.it.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Unkeless JC, Shen Z, Lin CW, DeBeus E. Function of human Fc gamma RIIA and Fc gamma RIIIB. Semin Immunol. 1995;7:37–44. doi: 10.1016/1044-5323(95)90006-3. [DOI] [PubMed] [Google Scholar]
- Utomo A, Cullere X, Glogauer M, Swat W, Mayadas TN. Vav proteins in neutrophils are required for FcgammaR-mediated signaling to Rac GTPases and nicotinamide adenine dinucleotide phosphate oxidase component p40(phox) J Immunol. 2006;177:6388–6397. doi: 10.4049/jimmunol.177.9.6388. [DOI] [PubMed] [Google Scholar]
- Utomo A, Hirahashi J, Mekala D, Asano K, Glogauer M, Cullere X, Mayadas T. Requirement for Vav proteins in post-recruitment neutrophil cytotoxicity in IgG but not complement C3 dependent injury. J Immunol. 2008 doi: 10.4049/jimmunol.180.9.6279. in press. [DOI] [PubMed] [Google Scholar]
- Van den Herik-Oudijk IE, Ter Bekke MW, Tempelman MJ, Capel PJ, Van de Winkel JG. Functional differences between two Fc receptor ITAM signaling motifs. Blood. 1995;86:3302–3307. [PubMed] [Google Scholar]
- van Sorge NM, van der Pol WL, van de Winkel JG. FcgammaR polymorphisms: Implications for function, disease susceptibility and immunotherapy. Tissue Antigens. 2003;61:189–202. doi: 10.1034/j.1399-0039.2003.00037.x. [DOI] [PubMed] [Google Scholar]
- Vielhauer V, Anders HJ, Perez de Lema G, Luckow B, Schlondorff D, Mack M. Phenotyping renal leukocyte subsets by four-color flow cytometry: characterization of chemokine receptor expression. Nephron Exp Nephrol. 2003;93:e63. doi: 10.1159/000068517. [DOI] [PubMed] [Google Scholar]
- Werner E. Determination of cellular H2O2 production. Sci STKE 2003. 2003:PL3. doi: 10.1126/stke.2003.168.pl3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.