

Abstract
New sequencing technologies promise a new era in the use of DNA sequence. However, some of these technologies produce very short reads, typically of a few tens of base pairs, and to use these reads effectively requires new algorithms and software. In particular, there is a major issue in efficiently aligning short reads to a reference genome and handling ambiguity or lack of accuracy in this alignment. Here we introduce the concept of mapping quality, a measure of the confidence that a read actually comes from the position it is aligned to by the mapping algorithm. We describe the software MAQ that can build assemblies by mapping shotgun short reads to a reference genome, using quality scores to derive genotype calls of the consensus sequence of a diploid genome, e.g., from a human sample. MAQ makes full use of mate-pair information and estimates the error probability of each read alignment. Error probabilities are also derived for the final genotype calls, using a Bayesian statistical model that incorporates the mapping qualities, error probabilities from the raw sequence quality scores, sampling of the two haplotypes, and an empirical model for correlated errors at a site. Both read mapping and genotype calling are evaluated on simulated data and real data. MAQ is accurate, efficient, versatile, and user-friendly. It is freely available at http://maq.sourceforge.net.
The advent of novel sequencing technologies such as 454 Life Sciences (Roche) (Margulies et al. 2005), Illumina (formerly known as Solexa sequencing), and Applied Biosystems SOLiD opens opportunities to a variety of biological applications, including resequencing (Bentley, 2006; Hillier et al. 2008), ChIP-seq (Barski et al. 2007; Johnson et al. 2007; Robertson et al. 2007), gene expression, miRNA discovery, DNA methylation study, cancer genome research, and whole-transcriptome sequencing. Most of these applications rely on fast and accurate read mapping, and some of them, in particular resequencing, require reliable SNP calling. Meeting these requirements is essential to realize the strength of the new sequencing technologies.
Several of these technologies produce tens of millions of short reads of currently typically 30–40 bp in a single run. Mapping the enormous numbers of short reads to the reference genome poses serious challenges to alignment programs. These challenges come not only from the requirement of highly efficient algorithms but also from the need of accuracy. Whereas existing alignment algorithms (Altschul et al. 1997; Buhler 2001; Ning et al. 2001; Kent 2002; Schwartz et al. 2003; Wu and Watanabe 2005) can be effectively adapted to achieve efficiency, the requirement of accuracy is subtle. Most genomes contain at least some sequence that is repetitive or close to repetitive on the length scale of the reads. As a consequence, some reads will map equally well to multiple positions. Furthermore, one or two mutations or sequencing errors in a short read may lead to its mapping to the wrong location. It is possible to act conservatively by discarding reads that map ambiguously at some level, but this leaves no information in the repetitive regions and it also discards data, reducing coverage in an uneven fashion, which may complicate the calculation of coverage.
An alternative solution to handling these ambiguities is to keep all the reads that can be mapped and to evaluate for each read the likelihood it has been wrongly positioned. Poor alignments can still be discarded later. This strategy essentially resembles phred’s (Ewing et al. 1998; Ewing and Green 1998) strategy for base-calling from capillary reads. In a capillary read, there are frequently low-quality regions. Phred does not discard these regions in the first instance. Instead, it calls each base as best as it can, and assigns a quality score that encodes the probability that the base is wrongly called. This per-base quality score is more informative and helpful than simply discarding poor data (Durbin and Dear 1998). Similarly, if the posterior error probability of each read alignment can be calculated, more information will be retained than if all poor data are discarded. Here, we show how to calculate the error probability of a read mapping.
We also introduce a new statistical model for consensus genotype calling and subsequent SNP calling. For capillary reads, two different approaches have previously been taken to calling SNPs. The first type of approach works on PCR resequencing data from diploid samples. These algorithms directly examine chromatogram trace files and detect variants by extracting or comparing signals in the peaks of traces. The most widely used software includes PolyPhred (Stephens et al. 2006), SNPdetector (Zhang et al. 2005), and novoSNP (Weckx et al. 2005), each of which can call the genotype of the sample as well as detect variants. The second type of approach works for clone-based data. They are usually built upon phred base calls and detect variants by detecting base-pair differences between a read from a single haplotype and the reference sequence. Two representative software of this type are ssahaSNP (Ning et al. 2001) and PolyBayes (Marth et al. 1999). While ssahaSNP uses a heuristic rule known as the neighborhood quality standard (NQS) (Altshuler et al. 2000), PolyBayes develops an explicit statistical framework to model variants.
All new sequencing technologies are shotgun methods that give sequences derived from a single molecule sampled from a larger population. (Current methods amplify the starting template by some form of PCR, but true single molecule methods are expected in the future.) This means the methods for calling variants from new technology data are most closely related to the second group described above, including ssahaSNP and PolyBayes. However, because of sampling and error rate, we need to combine data from multiple reads. In practice, errors at a particular site are correlated, and we must take this correlation into account. This is analogous to calling a consensus from a sequence assembly, and we propose a Bayesian approach to this issue that is related to that used in assembly software CAP3 (Huang and Madan 1999).
In summary, this article presents methods and software for mapping short sequence reads to a reference genome, calculating the probability of a read alignment being correct, and consensus genotype calling with a model that incorporates correlated errors and diploid sampling. The applicability and accuracy of the methods are evaluated based on both real data from the bacterium Salmonella paratyphi and simulated data from the diploid human X chromosome.
Results
Overview of MAQ algorithms
MAQ is a program that rapidly aligns short reads to the reference genome and accurately infers variants, including SNPs and short indels, from the alignment.
At the alignment stage, MAQ first searches for the ungapped match with lowest mismatch score, defined as the sum of qualities at mismatching bases. To speed up the alignment, MAQ only considers positions that have two or fewer mismatches in the first 28 bp (default parameters). Sequences that fail to reach a mismatch score threshold but whose mate pair is mapped are searched with a gapped alignment algorithm in the regions defined by the mate pair. To evaluate the reliability of alignments, MAQ assigns each individual alignment a phred-scaled quality score (capped at 99), which measures the probability that the true alignment is not the one found by MAQ. MAQ always reports a single alignment, and if a read can be aligned equally well to multiple positions, MAQ will randomly pick one position and give it a mapping quality zero. Because their mapping score is set to zero, reads that are mapped equally well to multiple positions will not contribute to variant calling. However, they do give information on copy number of repetitive sequences and on the fraction of reads that can be aligned to the genome, and can easily be filtered out for downstream analysis if desired. Mapping quality scores and mapping all reads that match the genome even if repetitive are where MAQ differs from most other alignment programs.
MAQ fully utilizes the mate-pair information of paired reads. It is able to use this information to correct wrong alignments, to add confidence to correct alignments, and to accurately map a read to repetitive sequences if its mate is confidently aligned. With paired-end reads, MAQ also finds short insertions/deletions (indels) from the gapped alignment described above.
At the SNP calling stage, MAQ produces a consensus genotype sequence from the alignment. The consensus sequence is inferred from a Bayesian statistical model, and each consensus genotype is associated with a phred quality that measures the probability that the consensus genotype is incorrect. Potential SNPs are detected by comparing the consensus sequence to the reference and can be further filtered by a set of predefined rules. These rules are designed to achieve the best performance on deep human resequencing data and aim to compensate for simplifications and assumptions used in the statistical model (e.g., treating neighbor positions independently).
Implementation
We implemented the software MAQ to align short reads and call genotypes based on the algorithm described in the Methods section. MAQ consists of a set of related programs that are compiled into a single binary executable. It is able to map reads, call consensus sequences including SNP and indel variants, simulate diploid genomes and read sequences, and post-process the results in various ways. MAQ also has an option to process Applied Biosystems SOLiD data that uses two base “color-space” encoding. Further details are available from the documentation at the MAQ website.
MAQ is easy to use. For bacterial genomes, alignments and variant calling can be done with a single command line, taking a few minutes on a laptop. In addition, MAQ comes with a compact and fast OpenGL-based read alignment viewer, MAQview, which shows the read alignments, base qualities, and mapping qualities in a graphical interface.
Both MAQ and MAQview are designed with genome-wide human resequencing in mind. First, the read alignment, which is the slowest step in the whole pipeline, can be divided into small tasks and parallelized on a modern computer cluster using less than 1 GB of memory for each processor core. The separate subparts of the alignment can then be merged together to give the final alignment. Second, the read alignments are stored in a binary compressed file. Text-based information is only extracted when necessary. This strategy saves disk space by a factor of three to five. Third, a novel technique is implemented to index the compressed alignment file, which enables swift retrieval of reads in any region of the reference sequence. Viewing the alignments of a human-sized genome is as fast as viewing those of a single BAC sequence. As a whole, MAQ and MAQview provide an efficient suite for managing data from Illumina sequencing.
MAQ and MAQview are implemented in C/C++ with auxiliary tools in Perl. They have been extensively evaluated on large-scale simulated data and real data and have been tested by users from various research groups. MAQ software is freely distributed under the GNU General Public License (GPL). The project home page is at http://maq.sourceforge.net.
SNP calling for large-scale simulated data
Although it is always good to look at real data, it is impossible to assess read alignment accuracy on real data, because in a shotgun sample we cannot know where the reads come from.
We simulated a diploid sequence (two haploid sequences) from the human reference chromosome X, as described in the Methods: 136,012 substitutions, 7377 1-bp insertions, and 7589 1-bp deletions were added to the diploid genome, giving an overall polymorphism rate of 0.001. From this mutated diploid genome, we simulated 100 million pairs of 35-bp mate-pair reads with errors (∼45.2-fold coverage on chromosome X). The average insert size is 170 bp with a standard deviation of 20 bp. Statistics on base qualities were estimated from real data where base qualities have been calibrated.
With the default MAQ options, we aligned the simulated reads against the whole human reference genome excluding Y and unassembled contigs. It took 1100 CPU hours to do this alignment, and 97.44% of reads get mapped. Figure 1B shows the distribution of mapping qualities (red curve) and the mapping error rate (blue curve) in each 10-based quality interval. If the mapping quality were estimated precisely, we would expect to see a straight blue line between (“0–9,” 100) and (“≥90,” 10−9). MAQ qualities appear to be overestimated; in other words, the true alignment error rate is higher than what mapping quality predicts to be. To investigate whether the overestimation is due to the fact that we did not consider mutations and indels in the model, we also simulated reads without introducing any mutations. For these data, the mapping quality could be estimated more accurately (data not shown), which confirms that mutations and indels may interfere with the calculation of mapping qualities. We see in Figure 1 that this effect is greatest for mapping quality ∼70–80. However, even these reads have accuracy better than 10−4, which is sufficient for most mapping based applications, including structural variant calling and SNP calling.
Figure 1.

Distribution of mapping qualities, consensus qualities, true alignment error rate, and true consensus error rate. The red line shows the fraction of reads whose mapping qualities fall in each interval. (Pink line) The fraction of consensus genotypes whose consensus qualities fall in each interval; (blue line) the true alignment error rate of reads in each interval; (green line) the true consensus error rate of reads in each interval. (A) Reads are aligned without using mate-pair information. Single-end alignments do not contain enough information for MAQ to assign mapping quality larger than 90; therefore, the data in the top bin are missing. (B) Reads are aligned using mate-pair information.
We called the consensus sequence from the MAQ alignment. The pink curve in Figure 1B shows that most of the consensus bases have a quality over 60. About 5% of the consensus bases have a quality smaller than 10. They are in repetitive regions where read alignment is not reliable. We then compared the consensus to the diploid sequence from which reads were generated, and calculated the error rate of the consensus. The green curve indicates that the consensus quality also roughly agrees with the true error rate. We called indels using paired-end indel detection methods described in the Methods section, and required at least two reads to support the indel.
After MAQ’s substitution calling, we further filtered the substitutions based on five rules: (1) discard SNPs within the 3-bp flanking region around a potential indel; (2) discard SNPs covered by three or fewer reads; (3) discard SNPs covered by no read with a mapping quality higher than 60; (4) in any 10-bp window, if there are three or more SNPs, discard them all; and (5) discard SNPs with consensus quality smaller than 10. MAQ provides a Perl script maq.pl to achieve all these filters.
To see how well MAQ calls SNPs and indels at different coverage, we chose several subsets of reads and called variants from those subsets. We compared the indels and filtered substitution calls to the true variants we added to the diploid genome in the simulation and measured the accuracy by false-positive rate (FP) and false-negative rate (FN) (Fig. 2B). MAQ consistently generates very few false positives but does miss true substitutions. Most of these missing substitutions fall in “filtered regions,” which tend to consist of repetitive sequences. In the human genome as represented by the X chromosome, we can call variants on ∼85% of the sequence using single end reads and 93% using paired-end reads.
Figure 2.

Accuracy of variant calling. In the figure, “filtered regions” are regions covered by three or fewer reads or by no reads with mapping quality higher than 60. For substitutions, FP equals the number of positions called as different from homozygous reference that in fact should be identical to the reference according to the simulation, divided by the total number of MAQ substitution calls; FN equals the number of positions that are different from the reference according to the simulation but are missed by MAQ, divided by the total number of mutations added in the simulation. For indels, FP equals the number of indel calls within 5-bp flanking regions of a true indel, divided by the total number of MAQ indel calls; FN equals the number of true indel calls missed by MAQ, divided by the total number of indels in simulation. (A) Variants are called based on single-end alignment. (B) Variants are called based on paired-end alignment. (C) Theoretical accuracy of k-allele method, where we call an allele as long as at least k reads are supporting the allele, assuming all reads are correctly aligned (see also Supplemental material).
The difference between the blue and the red curves indicates the fraction of missing substitutions in the regions trusted by MAQ. This difference decreases from ∼15% at 8× down to 1% at 30×. Note that we apply more filters on SNPs than on filtered regions, which leads to the 1% difference between the two curves at high depth. Most of difference at low depth is accounted for by sampling variation. At, say, 10× coverage there is 5× coverage on average of each haplotype. However, the actual number of reads at a site will be distributed around the average at best according to a Poisson distribution. Given that we may need to see a variant several times to be confident enough to call it, there is a significant probability that not enough reads will be aligned and the variant will be missed.
A simple model to this issue is to assume we require k reads to call an allele. We call this strategy the k-allele method. If we assume all read bases have an error rate 0.003, or phred quality 25, the theoretical FN and FP are shown in Figure 2C. If we require low FP rate, the FN rate of MAQ’s model largely agrees with that of the k-allele methods, allowing for the fact that some of the data have Q value lower than 25 or low mapping quality.
A uniquely aligned read tends to be wrongly mapped if it has many good alternative hits. Mapping quality helps to downweight such a read in SNP calling and to reduce the false SNPs caused by wrong alignments. To see the effect of mapping quality, we altered our method to ignore the mapping quality and to use only uniquely mapped reads in calling SNPs. We filtered the resulting SNPs using the same five rules as previously, except the third one, as we assumed no mapping quality is available in this case. In comparison, without mapping quality, MAQ discovered 217 false SNPs out of 127,910 predictions, and with mapping quality, MAQ gave 186 out of 126,228, yielding a 14% reduction in FP. This reduction amounts to 31 false SNPs, which is small in comparison to the 136,012 true substitutions in the simulation. However, in real data the FP is higher and in some applications, such as in the study of somatic mutations in cancer, the number of true SNPs will be much lower and the rate of false SNPs more critical.
This simulation only gives a rough evaluation of MAQ’s performance. On one hand, in the simulation process, reads are evenly distributed along the genome, no contamination exists, base qualities are accurate and sequencing errors are entirely independent. All these factors make SNP calling simpler. The true accuracy on real data will almost always be lower than the simulation. On the other hand, although errors are independent, we use a dependent model to infer the consensus. Using an independent model would achieve higher accuracy for simulated data. Moreover, we were using the same set of filters across all depths. Adjusting the threshold in filters might help to reach a better balance point between FN and FP at different depths.
SNP calling for bacterial genomes
To evaluate MAQ on real data, we obtained one lane of 2.9 million 36-bp Illumina read sequences of S. paratyphi A AKU12601 strain collected by the pathogen group at the Sanger Institute. The short reads are purity filtered. To calibrate the quality values, we put PhiX sample on the fifth lane of the same run, calculate a quality calibration table from the alignment against the known PhiX genome, and then apply the table on reads from other lanes to infer base qualities. S. paratyphi is a 4.8-Mbp bacterium, including plasmid (Holt et al. 2007), and so we had ∼20× coverage. An initial reference genome sequence of the same strain (AC: FM200053) was also produced by the pathogen group with capillary sequencing. Read sequences have been submitted to European Read Archive (AC: ERA000012).
After mapping and consensus base calling, we filtered the SNPs based on the same five rules as for the human X simulation, but in comparison to SNP calling on simulated human X chromosome, we did not filter SNPs around indels as we only had single-end reads; we decreased the threshold on mapping quality (rule 3) to 40 because single end reads usually have lower mapping quality than using mate-pair reads; and we increased the threshold on consensus quality (rule 5) to 40 because for haploid genome where there are no true heterozygotes, it is easier to get higher consensus quality.
After these filters, MAQ predicted two homozygous differences. Checking the capillary reads used in reference assembly confirms that the current reference is wrong at one of the homozygous sites. The other homozygous site is covered by 19 reads, with all of them identical to each other but different from the reference. This site is possibly a true mutation between the reference sequence and the Illumina-sequenced sample.
As well as these two homozygous differences, MAQ also predicted four heterozygotes. All four cases look confident from read alignment and show excessively high read depth in comparison to the average depth. Three are clustered together, and it appears likely that there is an additional copy of this region that was not identified in the reference. The fourth position may also be in a duplicated region (see below).
Alignment against the same reference strain only evaluates the FP of MAQ SNP calling. To assess the FN, we aligned the reads to a previously published sequence from another reference strain ATCC9150 (McClelland et al. 2004).
We downloaded the sequence of strain ATCC9150 (AC: NC_006511) from NCBI and aligned it, using cross_match (P. Green, unpubl.; http://www.phrap.org/phredphrapconsed. html), against the AKU12601 stain with the two homozygous SNPs discovered previously masked as N. Cross_match gave seven alignments, spanning the complete ATCC9150 and 99.97% of AKU12601 genome. 211 substitutions and 39 indels (five of them longer than 20 bp) are contained in the alignment. MAQ did not give any false positives and predicted 173 true substitutions. Of the missing 38 substitutions, 35 were covered by no uniquely aligned reads, and one site was covered by only one uniquely aligned read. Discovering SNPs at these 36 sites is almost impossible with single end short reads. Of the remaining two (38 − 36) sites, one site was called as a SNP initially but was filtered out due to low read coverage (two reads), and the other was dropped because it was covered by no read with mapping quality higher than 24 and so did not pass the filter, either. In regions passing the SNP calling filters (96.9% of ATCC9150 genome), no SNPs were missed. Interestingly, the four heterozygotes in the AKU12601 read mapping were not called as SNPs any more. One site became a repeat in ATCC9150, and the other three were called confident monomorphic sites with about the average read depth. This observation possibly revealed that around these three sites, there are no copy number changes between ATCC9150 strain and the sample resequenced by Illumina.
It is worth noting that AKU12601 and ATCC9150 are highly similar strains. Aligning short reads against a reference genome that is more distant to the sample being resequenced would be harder, especially when there are highly variable regions. In these regions, doing de novo assembly (Zerbino and Birney 2008) first and then aligning the contigs may greatly help.
Discussion
MAQ is capable of human whole-genome alignments and supports SNP calling on a diploid sample. It has been used to map short sequencing reads for structural variant calling in cancer samples (Campbell et al. 2008) and for whole-genome methylation analyses (Down et al. 2008). It is able to accurately estimate the error probability of each alignment and of each consensus genotype as well. MAQ can also simulate reads from a diploid genome based on a haploid reference. Simulation suggests that 20- to 30-fold coverage is needed for achieving FNs below 1% in the nonrepeat regions of a diploid sample.
The reliability of short read alignments
The reliability of read alignments can substantially affect the accuracy of the detection of variations. Knowing which alignment is reliable is key to the subsequent analyses. The most convenient way to measure the reliability is to define uniqueness: A read is said to be uniquely mapped if its second best hit contains more mismatches than its best hit. Generally this simple criterion works well, but potential difficulties are illustrated by the following scenarios: (1) a read has two one-mismatch hits, one with a Q30 mismatch and the other with a Q3 mismatch; (2) a read has one perfect hit and 100 one-mismatch hits; and (3) a read has a perfect hit and a Q3-mismatch hit. In the first case, although the read is not unique, the hit with a Q30 mismatch may still be reliable. In the remaining two cases, although the read can be uniquely aligned, the alignments are not reliable. For the human genome, these types of scenarios may happen at times due to the large fraction of repetitive sequences.
In our view, it is better to regard the position a read is mapped to as a random variable, and the reliability of an alignment can be naturally interpreted as the likelihood of the read being mapped to the correct position. At this point, mapping quality directly measures the reliability. It considers the repeat structure of the reference and the base quality of read sequences, which is implied in Equation 1 (see Methods), and can easily handle the three cases shown above.
Time complexity
If we map N reads to an L long reference and use k bits in indexing, the time complexity of MAQ alignment algorithm is O(c1NlogN + c2L + c32−kNL). The first term NlogN corresponds to the time spent on sorting the indexes; the second, on scanning the whole reference sequence; and the third term, on processing the alignment when there is a seed hit. In MAQ alignment, k is 24 and N is typically 2 million and therefore 2−kN ≈ 0.1, but as constant c3 is usually much larger than c2 and the human genome has many repeats, the time spent on the last two terms is approximately equal.
By default, MAQ scans the reference three times against six hash tables. It would be possible to save time by stopping the scan for a read once a perfect or one-mismatch hit was found. The perfect and one-mismatch hits, which exist for the majority of reads, are found in the first scan. However, stopping after the first scan for these reads would greatly reduce the resolution of mapping qualities. Reads that can be mapped confidently may not be effectively distinguished from those poorly aligned when the suboptimal hits were not available.
Evaluating the accuracy
Short reads tend to be wrongly aligned because one or two mutations or sequencing errors may make the best position wrong. When evaluating the accuracy of alignments, we have to look at the fraction of discarded reads (FD) and the fraction of wrongly aligned reads (FW) at the same time. Only counting one type of the errors might be misleading.
While on simulated data it is possible to estimate both FD and FW of alignments, on real data we cannot calculate FW as we do not know what the correct alignment is. As a consequence, we cannot directly measure the accuracy of the alignment using real data. To see what alignment strategy works best, we must evaluate a measurable outcome from the alignment, such as the accuracy of SNP calls, structural variations, or the agreement between expression profiling and microarray results. The criteria may vary with different applications.
In resequencing, accuracy can be measured by the SNP accuracy, which, again, should be measured by the fraction of missing polymorphic sites (FN) and the fraction of wrong calls (FP) at the same time. We can always trade one type of error for the other and therefore once again counting one type of error is misleading.
Unlike in an alignment, both FP and FN of SNPs on real data can be estimated from other sources of data. FP can be evaluated by capillary resequencing or genotyping a small subset of SNP calls. FN can be estimated by comparing SNP calls to the whole-genome chip-genotyping results. The fraction of chip-genotyping polymorphic sites that are not found is the FN. It should be noted that such a fraction is only the FN on the sites where probes can be designed for the genotyping microarray. These sites tend to be unique in the reference genome and are usually easier to find by short-read resequencing. The overall FN across the whole genome is higher.
In resequencing, it is also a good idea to explicitly define the resequenceable regions (or the regions where SNPs can be confidently called). We want to distinguish low SNP-density regions from hard-to-resequence regions. Using MAQ, the fraction of the human genome that is resequenceable with 35-bp reads is ∼85%, and with read pairs separated by 170 bp it is ∼93%. Achieving higher coverage would require a mixture of varying insert sizes and longer reads.
Methods
Single end read mapping
To map reads efficiently, MAQ first indexes read sequences and scans the reference genome sequence to identify hits that are extended and scored. With the Eland-like (A.J. Cox, unpubl.) hashing technique, MAQ, by default, guarantees to find alignments with up to two mismatches in the first 28 bp of the reads. MAQ maps a read to a position that minimizes the sum of quality values of mismatched bases. If there are multiple equally best positions, then one of them is chosen at random.
In this article, we will call a potential read alignment position a hit. The algorithm MAQ uses to find the best hit is quite similar to the one used in Eland. It builds multiple hash tables to index the reads and scans the reference sequence against the hash tables to find the hits. By default, six hash tables are used, ensuring that a sequence with two mismatches or fewer will be hit. The six hash tables correspond to six noncontiguous seed templates (Buhler 2001; Ma et al. 2002). Given 8-bp reads, for example, the six templates are 11110000, 00001111, 11000011, 00111100, 11001100, and 00110011, where nucleotides at 1 will be indexed while those at 0 are not. By default, MAQ indexes the first 28 bp of the reads, which are typically the most accurate part of the read.
In alignment, MAQ loads all reads into memory and then applies the first template as follows. For each read, MAQ takes the nucleotides at the 1 positions of the template, hashes them into a 24-bit integer, and puts the integer together with the read identifier into a list. When all the reads are processed, MAQ orders the list based on the 24-bit integers, such that reads with the same hashing integer are grouped together in memory. Each integer and its corresponding region are then recorded in a hash table with the integer as the key. We call this process indexing.
At the same time that MAQ indexes the reads with the first template, it also indexes the reads with the second template that is complementary to the first one. Taking two templates at a time helps the mate-pair mapping, which will be explained in the section below.
After the read indexing with the two templates, the reference will be scanned base by base on both forward and reverse strands. Each 28-bp subsequence of the reference will be hashed through the two templates used in indexing and will be looked up in the two hash tables, respectively. If a hit is found to a read, MAQ will calculate the sum of qualities of mismatched bases q over the whole length of the read, extending out from the 28-bp seed without gaps (the current implementation has a read length limit of 63 bp). MAQ then hashes the coordinate of the hit and the read identifier into another 24-bit integer h and scores the hit as q⋅224 + h. In this score, h can be considered as a pseudorandom number, which differentiates hits with identical q: If there are multiple hits with the same q, the hit with the smallest h will be identified as the best, effectively selecting randomly from the candidates. For each read, MAQ only holds in memory the position and score of its two best scored hits and the number of 0-, 1-, and 2-mismatch hits in the seed region.
When the scan of the reference is complete, the next two templates are applied and the reference will be scanned once again until no more templates are left.
Using six templates guarantees to find seed hits with no more than two mismatches, and it also finds 57% of hits with three mismatches. In addition, MAQ can use 20 templates to guarantee finding all seed hits with three mismatches at the cost of speed. In this configuration, 64% of seed hits with four mismatches are also found, though our experience is that these hits are not useful in practice.
Single end mapping qualities
MAQ assigns each individual alignment a mapping quality. The mapping quality Qs is the phred-scaled probability (Ewing and Green 1998) that a read alignment may be wrong:
For example, Qs = 30 implies there is a 1 in 1000 probability that the read is incorrectly mapped. In this section, we only consider a simplistic case where all reads are known to come from the reference and an ungapped exhaustive alignment is performed. A practical model for alignment with heuristic algorithms will be presented in the Supplemental material.
Suppose we have a reference sequence x and a read sequence z. On the assumption that sequencing errors are independent at different sites of the read, the probability p(z|x,u) of z coming from the position u equals the product of the error probabilities of the mismatched bases at the aligned position. For example, if read z mapped to position u has two mismatches: one with phred base quality 20 and the other with 10, then p(z|x,u) = 10−(20 + 10)/10 = 0.001.
To calculate the posterior probability ps(u|x,z), we assume a uniform prior distribution p(u|x), and applying the Bayesian formula gives
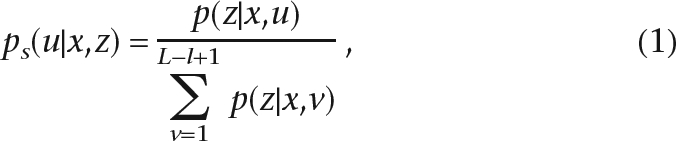 |
where L = |x| is the length of x and l = |z|. Scaling ps in the phred way, we get the mapping quality of the alignment:
The calculation of Equation 1 requires summing over all positions on the reference. It is impractical to calculate the sum given a human-sized genome. In practice, we approximate Qs as:
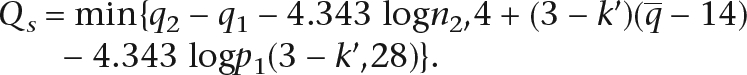 |
Where q1 is the sum of quality values of mismatches of the best hit, q2 is the corresponding sum for the second best hit, n2 is the number of hits having the same number of mismatches as the second best hit, k′ is the minimum number of mismatches in the 28-bp seed, q is the average base quality in the 28-bp seed, 4.343 is 10/log10, and p1(k,28) is the probability that a perfect hit and a k-mismatch hit coexists given a 28-bp sequence that can be estimated during alignment. Detailed deduction of this equation is given in the Supplemental material.
It is also worth noting that in minimizing the sum of quality values of mismatched bases, MAQ is effectively maximizing the posterior probability ps(u|x,z). This is the statistical interpretation of MAQ alignments.
On sequencing real samples, reads may also be different from the reference sequence due to the existence of sequence variants in different samples or strains. These variants behave in a similar manner to sequencing errors for mapping purposes, and therefore at the alignment stage, we should set the minimum base error probability as the rate of differences between the reference and the reads. However, this strategy is an approximation. When there are differences between the reference and reads, the best position might consistently give wrong alignments even if there are no sequencing errors, which can invalidate the calculation of mapping qualities. It would be possible in an iterative scheme to update the reference with an estimate of the new sample sequence from the first mapping and then remap to the updated reference.
Paired-end read alignment
MAQ jointly aligns the two reads in a read pair and fully utilizes the mate-pair information in the alignment.
In the paired-end alignment mode, MAQ will by default build six hash tables for each end (12 tables in total). In one round of indexing, MAQ indexes the first end with two templates and the second end also with two templates. Four hash tables, two for each end, will be put in memory at a time. In the scan of the reference, when a hit of a read is found on the forward strand of the reference sequence, MAQ appends its position to a queue that always keeps the last two hits of this read on the forward strand. When a hit of a read is found on the reverse strand, MAQ checks the queue of its mate and tests whether its mate has a hit on the forward strand within a maximum allowed distance ahead of the current read. If there is one, MAQ will mark the two ends as a pair. In this way, MAQ jointly maps the reads without independently storing all the potential hits of each end.
For each end, MAQ will only hold in memory two hash tables corresponding to two complementary templates (e.g., 11110000 and 00001111 for 8-bp reads). This strategy guarantees that any hit with no more than one mismatch can be always found in each round of the scan. Holding more hash tables in memory would help to find pairs containing more mismatches, but doing this would also increase memory footprint.
Paired-end mapping qualities are derived from single end mapping qualities. There are two different cases when a pair can be wrongly mapped. In the first case, one of the two ends is wrongly aligned and the other is correct. This scenario may happen if a repetitive sequence appear twice or more in a short region. In the second instance, a pair is wrong because both ends are wrong at the same time.
In MAQ, if there is a unique pair mapping in which both ends hit consistently (i.e., in the right orientation within the proper distance), we give the mapping quality Qp = Qs1+Qs2 to both reads, assuming independent errors. If there are multiple consistent hit pairs, we take their single end mapping qualities as the final mapping qualities.
Detecting short indels
MAQ first aligns reads with the ungapped alignment algorithm described above and then finds short indels by utilizing mate-pair information. Given a pair of reads, if one end can be mapped with confidence but the other end is unmapped, a possible scenario is that a potential indel interrupts the alignment of the unmapped read. For this unmapped read, we can apply a standard Smith-Waterman gapped alignment (Smith and Waterman 1981) in a region determined by the aligned read. The coordinate and the size of the region is estimated from the distribution of all the aligned reads by taking the mean separation of read pairs plus or minus twice the standard deviation. As Smith-Waterman will only be applied to a small fraction of reads in short regions, efficiency is not a serious issue.
Consensus genotype calling
By default, MAQ assumes the sample is diploid. It calculates the posterior distribution of genotypes and calls the genotype that maximizes the posterior probability.
Before consensus calling, MAQ first combines mapping quality and base quality. If a read is incorrectly mapped, any sequence differences inferred from the read cannot be reliable. Therefore, the base quality used in SNP calling cannot exceed the mapping quality of the read. MAQ reassigns the quality of each base as the smaller value between the read mapping quality and the raw sequencing base quality.
We first calculate the probability of data given each possible genotype. In consensus calling, if there are no sequencing errors, at most two different nucleotides can be legitimately seen. Therefore, we can consider only the two most frequent nucleotides at any position and ignore others as errors. Assume we are observing data D which consist of k nucleotides b and n−k nucleotides b′ with b,b′∈{A,C,G,T} and b ≠ b′. Then the three possible genotypes are 〈b,b〉, 〈b,b′〉, and 〈b′,b′〉. If the true genotype is 〈b,b〉, we have n−k errors from n bases. Let the probability of observing these errors be αn,n-k, and therefore P(D|〈b,b〉) = αn,n-k. Similarly we have P(D|〈b′,b′〉) = αnk. If the true genotype is 〈b,b′〉, the probability can be approximated with a binomial distribution: P(D|〈b,b′〉) = (nk)/2n.
If we further assume the prior of genotypes is P(〈b,b〉) = P(〈b′,b′〉) = (1 − r)/2 and P(〈b,b′〉) = r, we can calculate the posterior probability P(g|D) of genotype g given the observation D. Then the estimated genotype is ĝ = argmaxgP(g|D) with a quality Qg = −10log10[1 − P(ĝ|D)]. Here r is the probability of observing a heterozygote. We usually use r = 0.001 for the discovery of new SNPs and r = 0.2 for inferring genotypes at known SNP sites. In principle, a site-specific r can be used given known allele frequencies.
The real difficulty is to calculate αnk, the probability of k errors observed from n nucleotides. If errors arise independently and error rates are identical for all bases, αnk can be calculated with a binomial distribution. When errors are correlated and not identical, MAQ approximates αnk by
 |
Where εi is the ith smallest base error probability and c′nk is a function of εi but varies little with εi. The only unknown parameter is θ , which controls the dependency of errors. The deduction of this equation and the calculation of c′nk will be presented in the Supplemental material.
Taking a form like Equation 2 is inspired by CAP3 (Huang and Madan, 1999), where θ is arbitrarily set to 0.5. In principle, θ can be estimated from real data. In practice, however, the estimate is complicated by the requirement of large data set where SNPs are known, by the inaccuracy of sequencing qualities, by the dependencies of mapping qualities, and also by the approximation made to derive the equation. To estimate θ, we just tried different values and selected the one that was giving the best final genotype calls. We found θ = 0.85 is a reasonable value for Illumina Genetic Analyzer data.
Simulating diploid genomes and short reads
MAQ also generates in silico mutated diploid sequences by adding random mutations to the known reference sequence. The human reference genome does not contain heterozygotes, but when we resequence a human sample and map reads to the reference genome, we will see both homozygous and heterozygous variants in comparison to the reference. If the sample and the reference come from the same population and at a potential polymorphic site the allele frequency is f, the probability of observing a heterozygote is 2f(1 − f) and of observing a homozygous variant is f(1 − f) (= f2(1 − f) + f(1 − f)2). Consequently, on the condition that a site is different from the reference, the probability of a heterozygote is always 2/3, regardless of the allele frequency f, assuming the sample comes from the same population as the reference. Based on this observation, we can simulate a diploid genome as follows. We first used the reference genome as the two preprocessed haplotypes. We then generated a set of polymorphic sites, randomly selected two thirds of them as heterozygotes, and took the rest as homozygotes. At a heterozygous site, we randomly selected one haplotype and mutated the base into another one; on a homozygous site, we mutated both haplotypes. Both substitutions and indels can be simulated in this way. This simulation ignores linkage disequilibrium between variants. Although coalescent-based simulation (Hudson 2002) gives a more accurate long-range picture, the procedure described here is sufficient for the evaluation of the variant calling method for a single individual.
From a known sequence, paired-end reads can be simulated with insert sizes drawn from a normal distribution and with base qualities drawn from the empirical distribution estimated from real sequence data. Sequencing errors are introduced based on the base quality. With sufficiently large data, we are able to estimate the position-specific distributions of base qualities and the correlation between adjacent qualities as well. An order-one Markov chain is constructed, based on these statistics, to capture the fact that low-quality bases tend to appear at the 3′-end of a read and to appear successively along a read.
Alignment for Applied Biosystems SOLiD reads
SOLiD reads are presented in the color space, which comprises four colors with each color representing four types of combinations of two adjacent nucleotides. The SOLiD sequencing machine gives the last primer nucleotide base and the color read sequence. This information makes it possible to write down the nucleotide read sequence based on the meaning of colors. However, a single color error will completely change the nucleotide sequencing following that error. Mapping reads in the color space is preferable to mapping in the nucleotide space.
To map reads in the color space, we need to convert the reference sequences into color sequences and to perform the alignment in the color space. Between the color alignment and nucleotide alignment, the main difference is that the complement of a color is identical to itself, and therefore in the color space, reads coming from the reverse strand of the reference only need to be reversed without complementation. Most alignment programs can be adapted to perform such an alignment with little effort. Another difference is for paired-end reads. In SOLiD sequencing, the two ends of a read pair should always come from the same strand, instead from two different strands like Illumina sequencing.
MAQ is able to map SOLiD mate-pair reads to the reference, but it has to trim off the primer nucleotide base and the following color because currently MAQ cannot work with color sequences and nucleotide sequences at the same time. Trimming the first color is equivalent to using reads 1 bp shorter, which should not greatly affect the alignment results.
Acknowledgments
We thank Tony Cox, Keira Cheetham, Richard Carter, and David Bentley from Illumina for beneficial discussions on consensus genotype calling. We thank Julian Parkhill, Kathryn Holt, and the Sanger Institute pathogen sequencing unit for providing the S. paratyphi sequence, and the sequencing team and the sequencing informatics group for generating and processing the data. We also thank Ken Chen, David Spencer, LaDeana Hillier, and all the MAQ users for their valuable feedback as MAQ has matured; Klaudia Walter and the members of the Durbin research group for their helpful comments; and the anonymous referees for their helpful suggestions. This work was funded by the Wellcome Trust.
Footnotes
[Supplemental material is available online at www.genome.org. Short-read sequences have been deposited in the European Read Archive (ERA) under accession no. ERA000012 (ftp://ftp.era.ebi.ac.uk/ERA000012/).]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.078212.108.
References
- Altschul S.F., Madden T.L., Schaffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D., Pollara V.J., Cowles C.R., Van Etten W.J., Baldwin J., Linton L., Lander E.S. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- Barski A., Cuddapah S., Cui K., Roh T.-Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bentley D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Buhler J. Efficient large-scale sequence comparison by locality-sensitive hashing. Bioinformatics. 2001;17:419–428. doi: 10.1093/bioinformatics/17.5.419. [DOI] [PubMed] [Google Scholar]
- Campbell P.J., Stephens P.J., Pleasance E.D., O’Meara S., Li H., Santarius T., Stebbings L.A., Leroy C., Edkins S., Hardy C., et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down T.A., Rakyan V.K., Turner D.J., Flicek P., Li H., Thorne N.P., Kulesha E., Graf S., Tomazou E.M., Backdahl L., et al. A Bayesian deconvolution strategy for immunoprecipitation-based DNA methylation analysis. Nat. Biotechnol. 2008;26:779–785. doi: 10.1038/nbt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R., Dear S. Base qualities help sequencing software. Genome Res. 1998;8:161–162. doi: 10.1101/gr.8.3.161. [DOI] [PubMed] [Google Scholar]
- Ewing B., Green P. Base-calling of automated sequencer traces using phred. ii. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B., Hillier L., Wendl M.C., Green P. Base-calling of automated sequencer traces using phred. i. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Hillier L.W., Marth G.T., Quinlan A.R., Dooling D., Fewell G., Barnett D., Fox P., Glasscock J.I., Hickenbotham M., Huang W. Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- Holt K.E., Thomson N.R., Wain J., Phan M.D., Nair S., Hasan R., Bhutta Z.A., Quail M.A., Norbertczak H., Walker D., et al. Multidrug-resistant Salmonella enterica serovar paratyphi A harbors IncHI1 plasmids similar to those found in serovar typhi. J. Bacteriol. 2007;189:4257–4264. doi: 10.1128/JB.00232-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Madan A. Cap3: A DNA sequence assembly program. Genome Res. 1999;9:868–877. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson R.R. Generating samples under a wright-fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–338. doi: 10.1093/bioinformatics/18.2.337. [DOI] [PubMed] [Google Scholar]
- Johnson D.S., Mortazavi A., Myers R.M., Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Kent W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B., Tromp J., Li M. Patternhunter: Faster and more sensitive homology search. Bioinformatics. 2002;18:440–445. doi: 10.1093/bioinformatics/18.3.440. [DOI] [PubMed] [Google Scholar]
- Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.-J., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth G.T., Korf I., Yandell M.D., Yeh R.T., Gu Z., Zakeri H., Stitziel N.O., Hillier L., Kwok P.Y., Gish W.R. A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 1999;23:452–456. doi: 10.1038/70570. [DOI] [PubMed] [Google Scholar]
- McClelland M., Sanderson K.E., Clifton S.W., Latreille P., Porwollik S., Sabo A., Meyer R., Bieri T., Ozersky P., Mclellan M., et al. Comparison of genome degradation in Paratyphi and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat. Genet. 2004;36:1268–1274. doi: 10.1038/ng1470. [DOI] [PubMed] [Google Scholar]
- Ning Z., Cox A.J., Mullikin J.C. SSAHA: A fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G., Hirst M., Bainbridge M., Bilenky M., Zhao Y., Zeng T., Euskirchen G., Bernier B., Varhol R., Delaney A., et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Schwartz S., Kent W.J., Smit A., Zhang Z., Baertsch R., Hardison R.C., Haussler D., Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T.F., Waterman M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Stephens M., Sloan J.S., Robertson P.D., Scheet P., Nickerson D.A. Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat. Genet. 2006;38:375–381. doi: 10.1038/ng1746. [DOI] [PubMed] [Google Scholar]
- Weckx S., Del-Favero J., Rademakers R., Claes L., Cruts M., De Jonghe P., Van Broeckhoven C., De Rijk P. novoSNP, a novel computational tool for sequence variation discovery. Genome Res. 2005;15:436–442. doi: 10.1101/gr.2754005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.D., Watanabe C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- Zerbino D.R., Birney E. Velvet: Algorithms for de novo short read assembly using de Brujin graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Wheeler D.A., Yakub I., Wei S., Sood R., Rowe W., Liu P.P., Gibbs R.A., Buetow K.H. SNPdetector: A software tool for sensitive and accurate SNP detection. PLoS Comput. Biol. 2005;1:e53. doi: 10.1371/journal.pcbi.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]