

Abstract
Cystathionine β-synthase (CBS) domains are found in myriad proteins from organisms across the tree of life, and have been hypothesized to function as regulatory modules that sense the energy charge of the cell. Here we characterize the structure and stability of PAE2072, a dimeric, tandem CBS domain protein from the hyperthermophilic crenarchaeon Pyrobaculum aerophilum. Crystal structures of the protein in unliganded and adenosine monophosphate (AMP)-bound forms, determined at resolutions of 2.10 Å and 2.35 Å respectively, reveal a remarkable conservation of key functional features seen in the γ subunit of the eukaryotic AMP-activated protein kinase (AMPK). The structures also confirm the presence of a suspected intermolecular disulfide bond between the two subunits that is shown to stabilize the protein. Our AMP-bound structure represents a first step in investigating the function of a large class of uncharacterized prokaryotic proteins. In addition, this work extends previous studies that have suggested that, in certain thermophilic microbes, disulfide bonds play a key role in stabilizing intracellular proteins and protein-protein complexes.
Keywords: Cystathionine β-synthase, AMP-activated protein kinase, disulfide bond, protein stabilization, hyperthermophile
INTRODUCTION
It is now clear that disulfide bonding is a common, naturally occurring effector of stability in proteins from certain thermophilic microbes. For many years, observations on the reductive nature of the cytoplasm in well-studied organisms supported the idea that disulfide bonds could not play a significant role in stabilizing intracellular proteins1; 2; 3; 4. However, an accumulation of crystal structures revealing intracellular disulfide bonds in proteins from various thermophilic organisms challenged the universal validity of this dogma5; 6; 7; 8; 9; 10. Subsequent structural, biochemical, genomic, and proteomic investigations by our group and others have now established that disulfide bonding is in fact a common feature in proteins from a certain subset of thermophilic archaea and bacteria10; 11; 12; 13; 14; 15; 16. Several of these proteins have been characterized, and in each case removal of the disulfide(s) by chemical reduction or mutagenesis results in a significant decrease in thermostability10; 12; 13; 14. These findings support the notion that the widespread disulfide bonding observed represents a stabilizing structural adaptation to life at high temperature. Although the underlying cellular redox systems of these organisms have only begun to be explored, several lines of evidence suggest that an enzyme referred to as PDO (Protein Disulfide Oxidoreductase) is a key player in facilitating intracellular disulfide bonding11; 15; 17; 18.
Recently, a proteomics study in Pyrobaculum aerophilum identified a number of oligomeric protein complexes containing intermolecular disulfide bonds12, one of which was PAE2072, a protein of unknown function containing two tandem cystathionine β-synthase (CBS) domains19. CBS domains are ubiquitous in proteins from all three kingdoms of life, in many cases occurring in conjunction with enzymatic or transmembrane domains. Their importance is underscored by the range of hereditary diseases associated with mutations in the CBS domains from various proteins20. The best-studied of these is the γ subunit of AMP-activated protein kinase (AMPK), an energy-sensing kinase with homologs in all eukaryotes that is central to the control of cellular metabolism21. The γ subunit, composed of four tandem CBS domains, senses the energy charge of the cell by competitively binding AMP and ATP and regulates the kinase activity of the catalytic α subunit accordingly. Recent structures of core αβγ AMPK heterotrimers from three species have revealed the architecture of the inter-subunit interactions in the complex22; 23; 24. Many other CBS domains have also been shown to specifically bind adenosine derivatives such as AMP, ADP, ATP, or S-adenosyl methionine, leading to the idea that CBS domains may generally function as regulatory modules that regulate the activity of effector domains in response to the energy charge of the cell20. If this hypothesis is correct, intriguing questions are posed by the existence of a large, uncharacterized class of prokaryotic “standalone” CBS domain proteins such as PAE2072, which consist of only the sensor modules without any effector domain: What are the activities being regulated? Which CBS domain proteins affect which activities? What are the mechanisms of regulation?
Here we report crystal structures of the homodimeric, tandem CBS domain protein PAE2072 unliganded and in complex with AMP. The structures reveal strong conservation of the functional features seen in the γ subunit of AMPK, providing a starting point for investigating the function of standalone CBS domain proteins in prokaryotes. We also characterize the contribution of an intermolecular disulfide bond to the stability of the protein, extending previous studies on disulfide bonding as a stabilizing adaptation in certain prokaryotes.
RESULTS
Structure Determination
PAE2072 was recombinantly expressed in Escherichia coli, purified to homogeneity, and crystallized (see Materials and Methods). No special care was taken concerning the disulfide bond during either purification or crystallization other than the omission of reducing agents from all buffers. Non-reducing SDS-PAGE showed that the disulfide bond in PAE2072 was present in nearly quantitative yield at the end of the purification procedure. Although the oxidation state of the cysteines during expression in the E. coli cytoplasm was not investigated, we surmise based on the chemically reducing nature of the E. coli cytosol that the disulfide bond is probably formed during purification at some point after lysis of the cells. The structure of unliganded PAE2072 was determined to 2.10 Å resolution by molecular replacement (see Table 1 for scaling and refinement statistics). The crystallographic asymmetric unit contained two molecules arranged in a parallel or head-to-head fashion (Figure 1). These two molecules in the crystal were interpreted as the biological dimer due to the large interfacial surface area and the presence of the expected intermolecular disulfide bond between cysteines 120 and 120′. Crystals of PAE2072 in complex with AMP were obtained by crystallization of the protein in the presence of 2.5 mM nucleotide. The structure of the PAE2072-AMP complex was determined to 2.35 Å resolution by molecular replacement, using the unliganded structure as a search model (Table 1). Two of the dimers seen in the unliganded structure were contained in the asymmetric unit of this crystal, and this interpretation of the biological unit was further reinforced by the observation that the nucleotide binding sites are formed by constellations of residues from both subunits of the dimer (see below). The final models of the protein chains begin at residue 2 in all cases and end between residues 129 and 132 (out of 138 in the full-length protein).
Table 1.
Summary of data collection and refinement statistics
| Data collection | Unliganded PAE2072 | AMP-bound PAE2072 |
|---|---|---|
| Space group | P21212 | P41212 |
| Cell dimensions | ||
| a (Å), b (Å), c (Å) | 57.8, 98.5, 51.6 | 55.7, 55.7, 336.4 |
| Resolution (Å) | 2.10 | 2.35 |
| Rmerge# (%) | 9.4 (45.2) | 6.4 (37.3) |
| I/σI | 17.7 (4.18) | 9.79 (2.67) |
| Completeness (%) | 96.8 (95.7) | 93.7 (99.6) |
| Redundancy | 6.3 (6.0) | 2.6 (2.7) |
| Radiation wavelength (Å) | 1.5418 | 1.00 |
| Refinement | ||
| Resolution (Å) | 51.50–2.10 (2.16–2.10) | 18.91–2.35 (2.41–2.35) |
| Total no. working reflections | 16361 | 19638 |
| Total no. test reflections | 873 | 1118 |
| Rwork†/Rfree‡ (%) | 19.7/24.3 | 23.9/29.4 |
| No. atoms | ||
| Protein | 2089 | 3984 |
| Ligand/ion | 39 | 195 |
| Water | 161 | 66 |
| Average B-factors (Å2) | ||
| Protein | 27.4 | 60.2 |
| Ligand/ion | 33.9 | 46.9 |
| Water | 33.6 | 45.5 |
| R.M.S. deviations | ||
| Bond lengths (Å) | 0.008 | 0.005 |
| Bond angles | 1.2º | 1.1º |
| Ramachandran plot statistics (%) | ||
| Residues in most favored regions | 95.7 | 96.7 |
| Residues in additional allowed regions | 4.3 | 3.3 |
| Residues in generously allowed regions | 0 | 0 |
| Residues in disallowed regions | 0 | 0 |
| PDB ID | 2RIH | 2RIF |
Values in parentheses are for the last shell.
Rwork = Σ|Fobs − Fcalc |/ΣFobs
Rfree = Σ|Fobs − Fcalc|/ΣFobs, calculated using a random set containing 5% of reflections that were not included throughout refinement
Figure 1. Overall structure of PAE2072.
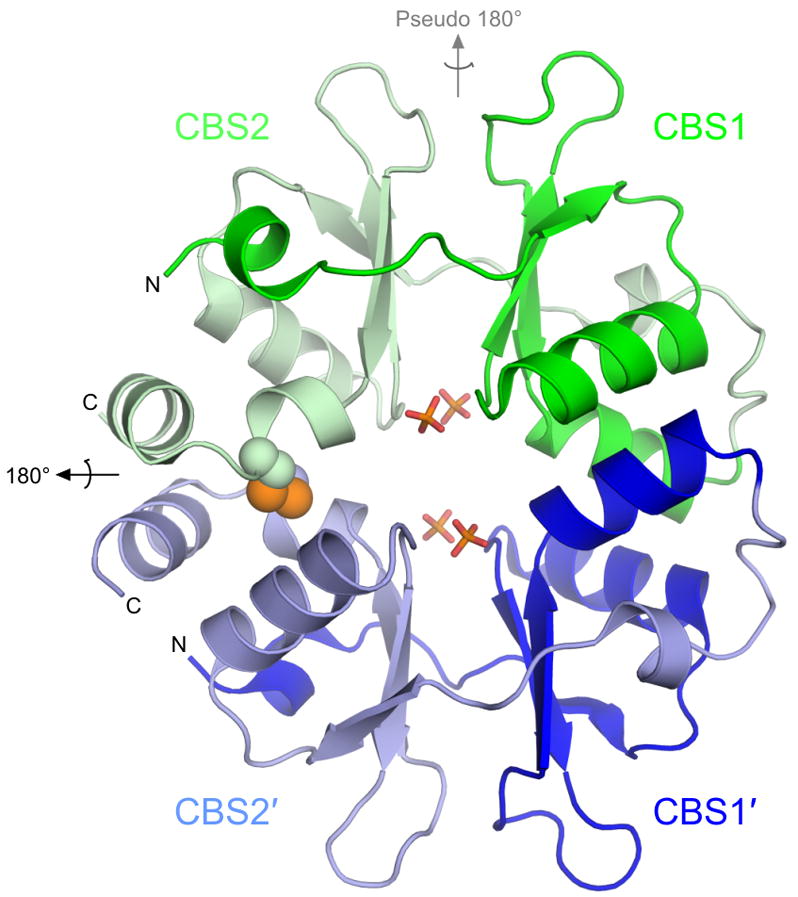
The unliganded PAE2072 dimer is shown in cartoon representation, with each CBS domain colored individually. The intermolecular disulfide bond between cysteines 120 and 120′ is shown as spheres, and the sulfate ions that occupy the same positions as the phosphate groups of AMP in the AMP-bound structure are shown in stick representation. The dimeric two-fold axis is indicated in black, and the pseudo two-fold axis in gray. The core β-α-β-β-α fold conserved in CBS domain proteins is easily identifiable; an additional short helix punctuates the extended N-terminus of each CBS domain.
General structural features
The overall structural features of the dimer are highly similar between the unliganded and the AMP-bound structures. Comparison of the protein backbones in the two structures yields an RMSD of 0.71 Å over 254 Cα atoms. Therefore, the following observations concerning the general structural features of PAE2072 apply to both forms of the protein. A similar lack of conformational change has been observed in structures of the γ subunit of AMPK complexed with various adenine nucleotides23; 24; 25, and is potentially significant for interpreting the mechanism of regulation by this class of proteins. The PAE2072 dimer forms a disk-like structure approximately 55 Å tall, 45 Å wide, and 25 Å thick (Figure 1). As expected based upon examination of other CBS domain protein structures, the two tandem CBS domains in each protein chain are related by pseudo two-fold symmetry to form a single “Bateman domain”26. In the case of PAE2072, the two CBS domains account for the entire polypeptide. We therefore refer to the protein as a “standalone” CBS domain protein, indicating the lack of other, non-CBS domains. Within each CBS domain, the β-α-β-β-α fold typical of this family is conserved. However, an additional small helix is present at the N-terminus of each CBS domain, and the last helix of the second domain (CBS2) is interrupted by an approximately 85 degree kink directly after the cysteine residue involved in the disulfide. The major interface between the two tandem CBS domains is formed by a conserved hydrophobic core between two beta sheets that surround the pseudo-dyad running through the center of the subunit. Another inter-domain interaction involves packing of the small helix at the N-terminus of each CBS domain against the other domain to form an additional pair of hydrophobic cores. This helix is followed by a well-ordered, extended segment without secondary structure that leads into the first strand of the three-stranded beta sheet.
The dimeric interface
The dimeric interface of PAE2072 is formed by the four main alpha helices from each of the two subunits, two from each of the CBS domains. In most dimeric CBS domain proteins the pseudo two-fold axis within each subunit is roughly orthogonal to the dimeric interface, which presents the peculiar possibility that dimerization could result in either parallel or anti-parallel assemblies. A review of the available structures of dimeric tandem CBS domain proteins revealed that examples of each type of dimer can be found (parallel: PDB codes 1Y5H, 1PVM, 1VR9, and 1YAV; anti-parallel: PDB codes 1O50, 1PBJ). The structures of PAE2072 reported here show that it is a parallel dimer, such that CBS1 interacts with CBS1′ and CBS2 interacts with CBS2′ (Figure 1). The existence of the stabilizing disulfide bond linking CBS2 and CBS2′strongly implies that this is the biologically relevant arrangement of the subunits. Unambiguous density is observed for the disulfides in both structures in simulated annealing omit maps (Figure 2A), and the geometries of the disulfides are compatible with previously determined consensus geometries27, although the disulfides in the AMP-bound structure are slightly extended compared to that in the unliganded structure. The dimeric interface buries 1884 Å2 of surface area per subunit (23% of the total surface area of each subunit) and is dominated by two large hydrophobic patches, one in each CBS domain (Figure 2B). In addition, there is a large network of interacting charged and polar side chains from residues of both subunits at the center of the disk-like dimer, but these interactions appear to be more important for ligand binding than for dimerization (see below).
Figure 2. The dimeric interface.
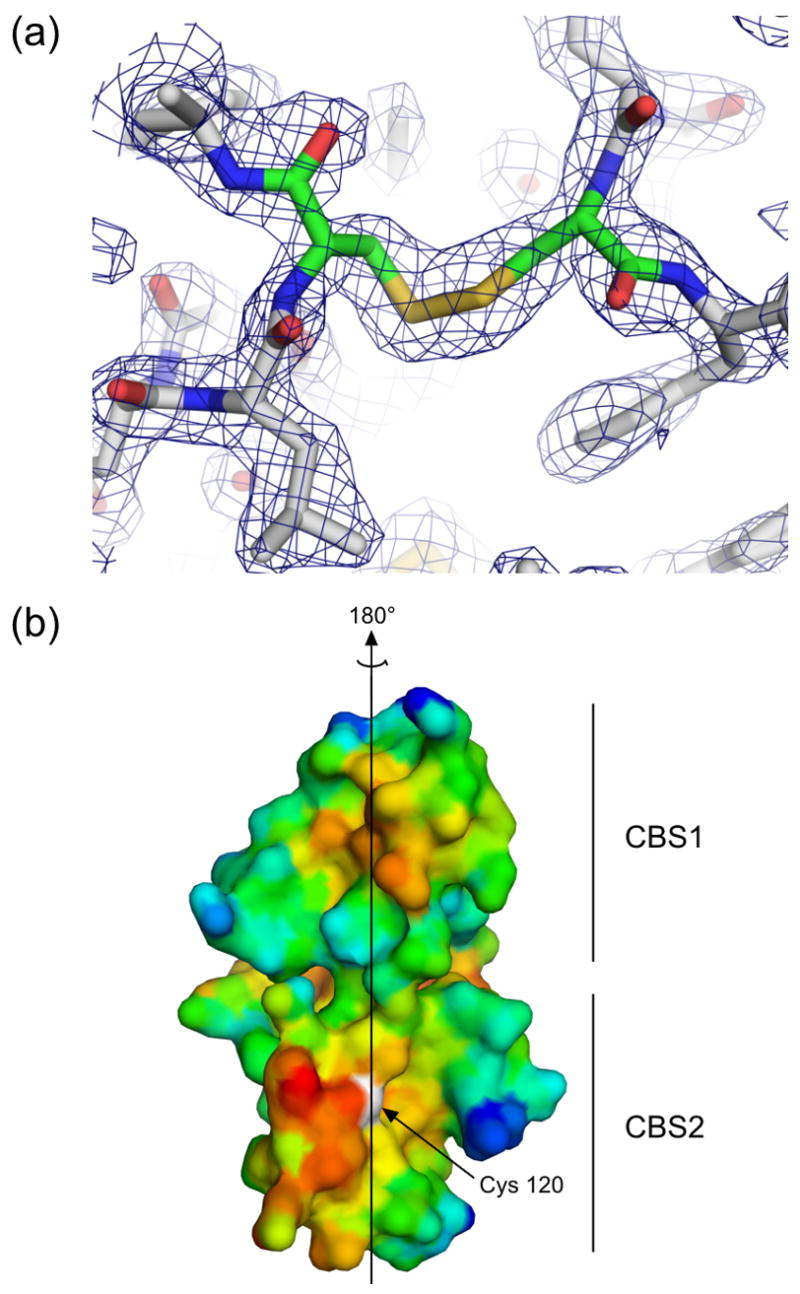
(a) Residues in the area of the disulfide bond in the unliganded structure are shown in stick representation, and electron density from a simulated annealing 2Fo-Fc omit map in which residues Cys120 and Cys120′ were omitted is contoured at 1.2 σ, demonstrating clear density for the disulfide bond. Similar density was seen for the disulfides in an omit map made using the coordinates and structure factors from the AMP-bound structure. (b) A surface representation of the dimeric interface of one subunit of PAE2072 is shown, colored according to hydrophobicity. Red indicates regions of higher hydrophobicity, while blue indicates more polar areas. The dimeric twofold axis is shown, and a prominent hydrophobic patch can be seen in each CBS domain. The sulfur atom of cysteine 120, representing the site of the intermolecular disulfide bond, is colored white.
Ligand binding by PAE2072
Analysis of crystals of PAE2072 grown in the presence of AMP resulted in a structure with four molecules of AMP bound per dimer, or one AMP per CBS domain (Figure 3A). Simulated annealing omit maps show clearly interpretable density for the nucleotide in each of the eight binding sites in the asymmetric unit (Figure 3B). Overall, the structural basis of nucleotide binding by PAE2072 is nearly identical to that observed in the cytoplasmic domain of the chloride transporter ClC-528 and the γ subunit of AMPK23; 24; 25 (Figure 3D), except that both potential binding sites per subunit are occupied in PAE2072. This is reflected in a strong conservation of the AMP-binding amino acid residues between the two pseudo-symmetry related binding sites per subunit, despite the low overall sequence identity (18%) between the CBS1 and CBS2 domains. The binding sites are located at the interface between the two CBS domains in each subunit, such that residues from both CBS domains contribute to nucleotide coordination. The adenine base in each binding site is sandwiched in a hydrophobic pocket, with aliphatic side chains (Val33/Val113 and Val50/Ile98) packing against both sides of the purine ring. Protein backbone groups appear to provide both positive and negative determinants of base specificity by hydrogen bonding to nitrogens 1 and 6 of adenine (Val13/Leu35 and Ile78/His100), while sterically precluding accommodation of the 2-amino group of guanine (Pro11 and Ser76). The side chain of an aspartate residue in each binding site (Asp55 and Asp118) forms an interaction with the 2′ and 3′ hydroxyls of the ribose ring that has been shown to be essential for ligand binding in the CBS domains of ClC-528. Finally, the phosphate of each AMP makes several contacts with polar and basic side chains concentrated in the center of the disc-like dimer. The locations of the four phosphates in each dimer can be described by a rhombus with sides of slightly less than 9 Å, and the network of interactions connecting them suggests that ligand binding may be cooperative (Figure 3C). Notably, the imidazole ring of His100 makes symmetric interactions with the phosphates of both AMP molecules bound in each subunit, while Arg99 makes direct contacts with one intra-subunit phosphate and water-mediated contacts to the phosphates of both of the AMP molecules bound in the opposite subunit. Overall, six arginines (three from each subunit) are involved in coordinating the four phosphate groups of the bound nucleotides, and several more basic residues are located nearby. Only two negatively charged side chains (Glu53 and Glu53′) participate in this network of interactions surrounding the AMP phosphates. In a PAE2072 dimer lacking bound nucleotides, such a concentration of positively charged side chains would be expected to create considerably unfavorable electrostatic interactions. Consistent with this idea is the observation that in the unliganded structure, four well-ordered sulfate ions from the crystallization solution occupy essentially identical positions to those of the phosphate groups in the AMP-bound structure (Figure 1).
Figure 3. Ligand binding by PAE2072.

(a) A view of one of the two crystallographically independent dimers in the AMP-bound structure shows the positions of the four bound AMP molecules. Note the lack of any significant conformational change from the unliganded structure in Figure 1. (b) Difference density from a simulated annealing Fo-Fc omit map, contoured at 3.0 σ, reveals readily interpretable density for AMP. The coordinates from the final model are shown in stick representation. (c) Three residues from each subunit (Arg99, His100, and Arg117) are involved in coordinating multiple AMP molecules, which may suggest cooperative binding. Several well-ordered water molecules form bridging interactions, and may indicate space where additional phosphate groups, such as those on ATP, could be accommodated. The dimeric two-fold axis is indicated in the center of the figure and runs perpendicular to the page; the pseudo two-fold is shown in gray. (d) The mechanism of adenine nucleotide binding is strongly conserved in the CBS domains of PAE2072 (both binding sites are shown), mammalian AMPK (PDB code 2V92), and human ClC-5 (PDB code 2J9L), but not in bacterial MgtE (PDB code 2YVY). The four central beta strands of one subunit of PAE2072 were aligned with those of the tandem CBS domains of MgtE (RMSD = 0.81 Å over 20 Cα atoms) to model the potential position of a bound AMP molecule. Strong steric clashes between the protein and the modeled AMP can be seen, and no basic residues are in position to coordinate the phosphate residues. It therefore seems unlikely that MgtE binds adenine nucleotides in a manner similar to that observed in the other CBS domains shown here.
Sequence-structure analysis of Pyrobaculum CBS domain proteins
A search of the Pfam database (release 22.0)29 revealed sixteen different proteins in the P. aerophilum genome that contain recognizable CBS domains. Remarkably, fourteen of these are standalone CBS domain proteins, more than were found in any other organism in the Pfam database. Two of the fourteen CBS domain proteins in P. aerophilum contain a large number of CBS domains (four in PAE2364 and seven in PAE3169), whereas the other twelve proteins are composed of two tandem CBS domains. These latter twelve formed the basis for a multiple sequence alignment (Figure 4A). The residues involved in binding AMP in PAE2072 were nearly universally conserved across the twelve proteins in the alignment; out of 132 total putative ligand-binding residues (twelve residues in each of the eleven other proteins), only two substitutions likely to affect contacts to adenine nucleotides were found. This degree of conservation stands out in view of the much lower similarity between the proteins over their full length; pairwise alignments yielded average values of 36% identity and 60% amino acid similarity. Aside from the termini, the segments of the multiple sequence alignment containing the most variation map to 1) the prominent loop connecting strands 2 and 3 of CBS1, and 2) the N-terminal, extended portion of CBS2, including the small N-terminal helix. In the structures of fission yeast, budding yeast, and mammalian AMPK, the surface defined by precisely these regions of the γ subunit is responsible for the interactions with the other subunits of the heterotrimeric complex (Figure 4B)22; 23; 24.
Figure 4. Sequence-structure analysis of Pyrobaculum CBS domain proteins.

(a) A multiple sequence alignment of 12 standalone CBS domain proteins from P. aerophilum, generated using the alignment program T-COFFEE, revealed nearly universal conservation of the residues involved in adenylate binding in PAE2072. Residues with side chains (*) or backbone atoms (^) that contact AMP are indicated. Two substitutions likely to result in the loss of a contact are boxed. The sequences of two regions other than the termini were found to be highly variable in this family of proteins (residues 40–46 and 71–84 in PAE2072). The first 10 residues of PAE2961 and 27 residues of PAE2866.a are represented by ellipses for clarity. (b) The regions of high sequence variability in Pyrobaculum CBS domain proteins map to a single surface patch on PAE2072 which coincides precisely with the site of interaction between the γ subunit of AMPK and the α and β subunits. The four central beta strands of PAE2072 were aligned with those of the γ subunit of S. pombe AMPK (PDB ID 2OOX; RMSD = 0.88 over 20 Cα atoms) to model PAE2072 in the place of the γ subunit. The surface of PAE2072 is shown and colored according to sequence conservation; red indicates highly conserved positions, while blue indicates highly variable positions. The α and β subunits of AMPK are shown in cartoon representation and can be seen to dock onto the surface patch defined by the highly variable segments (blue and cyan). The AMP molecules bound to PAE2072 are shown in sticks to emphasize their surface accessibility, which is presumably important for the mechanism of regulation by PAE2072.
Structural adaptations to high temperature
Although several structures of prokaryotic standalone CBS domain proteins were available, the lack of data on the stability of these proteins precluded a meaningful analysis of possibly stabilizing structural adaptations in PAE2072. However, it is noted that the main features likely to contribute to the stability of the protein are several large hydrophobic cores (including the dimeric interface) and the intermolecular disulfide bond.
The role of the disulfide bond in protein stability was tested by thermal and chemical denaturation of PAE2072 proteins with and without the disulfide. Because the melting temperature of PAE2072 is above the boiling point of water even in the absence of the disulfide (data not shown), thermal denaturation experiments were conducted in the presence of moderate amounts of the chemical denaturant guanidinium hydrochloride (GdmHCl). PAE2072 exhibited a large decrease in circular dichroism at 86 °C in 3.5 M GdmHCl, indicative of an unfolding transition (Figure 5A). Addition of the disulfide reducing agent dithiothreitol under otherwise identical conditions lowered the apparent melting temperature to 58 °C, a change of nearly 30 degrees, strongly suggesting that the intermolecular disulfide bond contributes significantly to the stability of the protein at high temperatures. Chemical denaturation of wild-type PAE2072 and a C120S mutant lacking the disulfide provided additional support for this interpretation. The midpoint of GdmHCl denaturation of the wild-type protein was approximately 4.6 M, while the midpoint for the C120S mutant was approximately 3.8 M (Figure 5B). Although chemical denaturation of both wild-type PAE2072 and the C120S mutant was for the most part reversible, a small amount of aggregation upon refolding (data not shown) prohibited calculation of equilibrium thermodynamic parameters.
Figure 5. Contribution of the disulfide bond to protein stability.
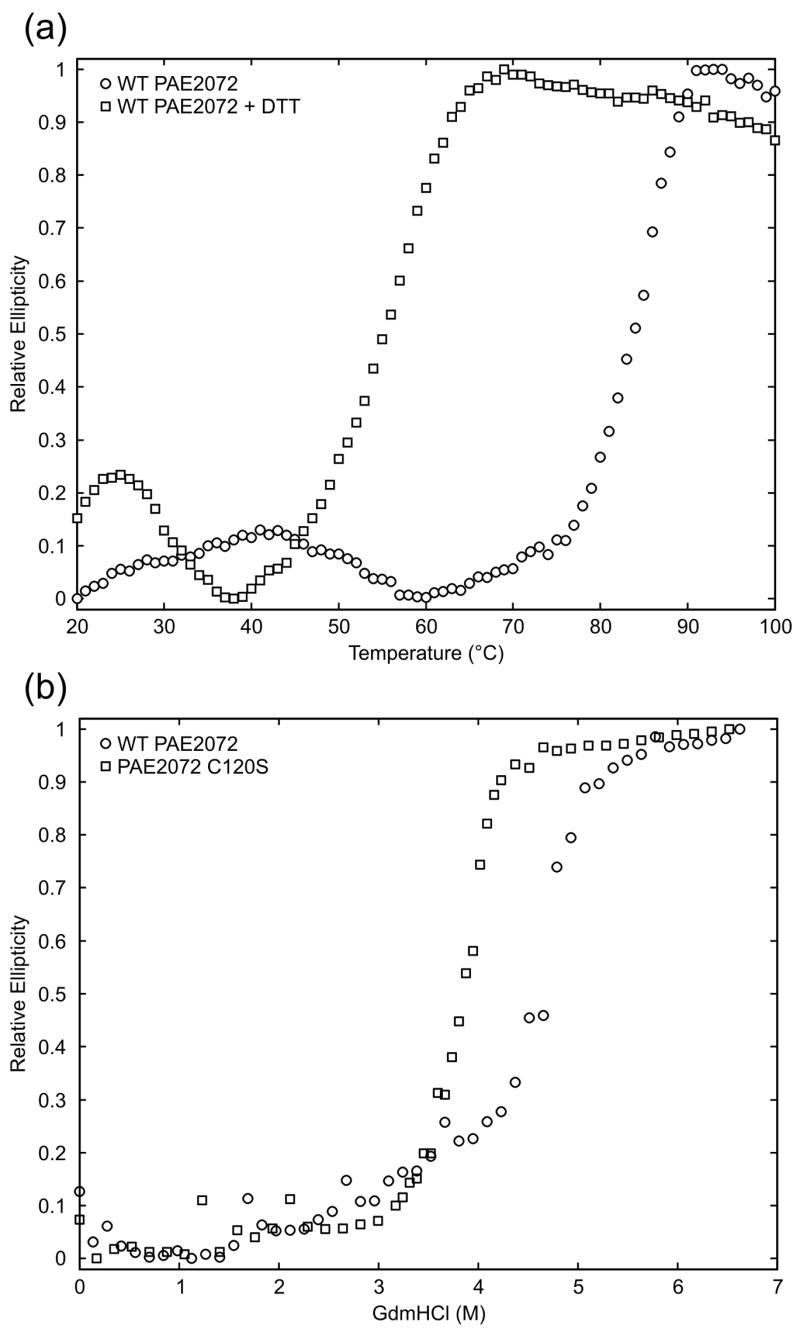
(a) Thermal melts of PAE2072 in the presence and absence of DTT indicate a dramatic loss of protein stability upon reduction of the disulfide bond. (b) Chemical denaturation of wild-type PAE2072 and a C120S mutant also reveal a stabilizing role for the disulfide bond as evidenced by the lower midpoint of denaturation for the mutant lacking the disulfide.
DISCUSSION
Several recently published studies have revealed a first glimpse of the structural basis of nucleotide binding by eukaryotic CBS domains, a subject currently attracting considerable interest22; 23; 24; 25; 28. We have demonstrated that a prokaryotic, standalone CBS domain protein binds AMP, and that the structural basis for nucleotide coordination is essentially identical to that seen in the CBS domains of AMPK and human ClC-5. This strong conservation of the ligand binding mechanism across proteins from such phylogenetically distant organisms supports the hypothesis of Hardie and coworkers that CBS domains generally function as energy-sensing modules that bind adenosine derivatives20. However, a pair of studies on transmembrane transporters has presented evidence for alternative mechanisms of regulation by CBS domains. In the first study, the tandem CBS domain pair in the cytoplasmic domain of the osmoregulatory ABC transporter OpuA was shown to regulate transporter activity as a function of intracellular ionic strength30. To our knowledge, the ability of the CBS domains in this system to bind nucleotides has not been tested. Another study provided crystallographic evidence that the dimer of tandem CBS domain pairs in the cytoplasmic domain of the bacterial magnesium transporter MgtE changes conformation upon binding magnesium ions at specific sites, suggesting a possible homeostatic regulation mechanism31. Due to significant differences in the regions of the MgtE CBS domains that correspond to the binding sites in PAE2072, it appears unlikely that MgtE is capable of binding adenine nucleotides in a similar manner (Figure 3D). Thus, it appears that some CBS domains sense signals in the cell other than energy charge. Nevertheless, the strong conservation of the mechanism of nucleotide binding observed in AMPK, ClC-5, and PAE2072 supports the idea that many CBS domains, and perhaps most of those in standalone CBS domain proteins, sense cellular energy charge by binding adenosine derivatives. Given the critical nature of this function to the metabolism of the eukaryotic cell, as evidenced by the many inherited diseases associated with mutations that affect nucleotide binding in CBS domains20, it is likely that standalone CBS domain proteins in prokaryotes also play important roles in cellular metabolism.
The lack of conformational change upon ligand binding observed in our structures also mirrors results recently obtained in the AMPK system, and supports the model of a regulatory mechanism based mainly upon electrostatic differences at the nucleotide binding site. These differences would result from the contribution of charge from the phosphate group(s) of bound nucleotides23; 24. Previous studies of ligand binding by eukaryotic CBS domains have provided comparisons of structures with various nucleotides bound (i.e. AMP, ADP, ATP, and ZMP), demonstrating that the structural rearrangements involved in accommodating additional phosphate groups are limited to changes in the conformations of a few side chains, whereas large differences in electrostatic potential are observed23; 24; 25; 28. Although we did not obtain crystals of PAE2072 in complex with ATP, based upon our AMP-bound structure it appears that PAE2072 could readily accommodate ATP in one or more of the four binding sites in an identical manner. A major contribution of the present study is that we reveal for the first time structures of a CBS domain protein in both the unliganded and nucleotide-bound states. Our structures show that binding of the nucleoside portion of the ligand induces essentially no change in the protein structure. Therefore, a combination of several structural studies has now demonstrated that the most significant property of adenosine nucleotide-binding CBS domain proteins affected by the presence and identity of bound ligand is the electrostatic potential at the binding site. Presumably, interactions with effector proteins will be different in the AMP- and ATP-bound states as a result of the additional negative charge from ATP, leading to regulation of effector protein activity. The conservation of this principle across such distantly related proteins both strengthens the proposal that it is important for the regulation of AMPK activity by the γ subunit23; 24 and suggests that this mechanism of regulation is conserved in prokaryotic CBS domains.
We have also shown that eleven other standalone CBS domain proteins in P. aerophilum exhibit essentially universal conservation of the residues responsible for nucleotide binding and, intriguingly, that the most variable sequence segments of these proteins are localized to a single surface patch that corresponds precisely with the region responsible for intersubunit interactions in fungal and mammalian AMPK γ subunits22; 23; 24. These observations are compatible with a model in which the standalone CBS domain proteins in P. aerophilum specifically interact with different effector proteins whose activity they regulate, with the variable surface patch contributing to interaction specificity. We submit this as a hypothesis that requires further testing. Although our results do not yield any clues concerning the identities of the interacting effector proteins, they would presumably be involved in processes in which regulation by energy charge would be advantageous. It is of note that AMPK appears to be strictly eukaryotic; no prokaryotic homolog of AMPK has been described. It is possible that non-homologous (or undetectably homologous) kinase domains interact with and are regulated by standalone CBS domain proteins in prokaryotes, thereby forming prokaryotic energy-sensing kinase complexes that serve physiological roles analogous to that of AMPK. Alternatively, standalone CBS domain proteins may interact directly with and regulate the activity of metabolic enzymes in a more allosteric fashion, without making use of a central, intermediary kinase. The large number of standalone CBS domain proteins in the P. aerophilum genome would seem to support the latter possibility; however, this is an unusual feature of this particular organism and should not be taken as evidence for the allosteric regulatory model in general.
The intermolecular disulfide bond in PAE2072 was shown to contribute to the stability of the protein, and provides another example of the use of intracellular disulfide bonding as a stabilizing structural adaptation in the proteins of certain thermophilic microbes11; 15. This initially controversial idea10; 16 is now established among those studying proteins from thermophilic organisms15. Furthermore, recent studies have revealed unusual geometrical mechanisms by which disulfide bonds stabilize some thermophilic proteins. For instance, the two subunits of the dimeric citrate synthase from P. aerophilum were found to be topologically interlinked by an intramolecular disulfide bond in each subunit, such that the polypeptide chains are catenated and cannot untangle from each other while the disulfides are intact12. A similar situation was noted earlier by Guss and coworkers32. Those surprising results raised the possibility that disulfide bonding and complex chain topology could combine to provide novel stabilization strategies in proteins from thermophiles. A subsequent study supported this hypothesis by using engineered disulfide bonds to investigate the effects of disulfide bonding and topology on the stability of the naturally slipknotted protein alkaline phosphatase33. In the structure of PAE2072 reported here, the disulfide bond does not appear to introduce any particularly unusual features into the chain topology of the protein dimer. Further studies on disulfide-bonded protein complexes from P. aerophilum and other disulfide-rich organisms will be required in order to determine how often and in what variety disulfide bonding might be used to generate stabilizing topological features in these systems.
In summary, we provide evidence for strong conservation of the key functional features seen in the γ subunit of AMPK in a CBS domain protein from a phylogenetically distant organism. Our results confirm and extend previous studies on eukaryotic CBS domains to provide support for a model in which prokaryotic, standalone CBS domain proteins interact with and regulate the activities of effector proteins by binding adenine nucleotides in a manner that depends upon the energy charge of the cell. At present there are not sufficient data to confidently predict the identities of the effector proteins, but given the central roles of CBS domains in eukaryotic metabolism it is likely that they also play important roles in the cellular physiology of prokaryotes. We have initiated functional studies on this large class of prokaryotic proteins, and provide a hypothesis that should guide future experiments. Finally, we add another example to the growing list of intracellular proteins from disulfide-rich thermophiles that are stabilized by disulfide bonds.
MATERIALS AND METHODS
Protein expression, purification, and crystallization
The PAE2072 gene (GenBank accession number AE009855.1) was cloned from P. aerophilum genomic DNA into the pETM11 expression vector. The resulting construct contained an N-terminal, TEV-cleavable (His)6 tag, which, after cleavage by TEV protease, left a Gly-Met-Ala tripeptide at the N-terminus in place of the initial methionine. The C120S mutant was made using the Quikchange II site-directed mutagenesis kit (Stratagene). Wild-type and C120S PAE2072 were expressed and purified in an identical fashion. The expression vectors carrying the wild-type and C120S PAE2072 genes were transformed into Rosetta™ 2(DE3) E. coli cells (Novagen). Cells were grown in LB in the presence of 30 mg L−1 kanomycin at 37 °C until the OD600 reached 0.8. Protein expression was induced by addition of 1 mM isopropyl-thio-β-D-galactopyranoside and allowed to proceed for 4.5 hours before cells were harvested. Cells were lysed by sonication in 12.5 mM Tris/12.5 mM MOPS pH 7.5, 0.1% Tween20. Lysates were treated with DNase and Protease Inhibitor Cocktail (Sigma) before being cleared by centrifugation. Lysates were then subjected to a heat purification step at 75 °C for 15 minutes, and the precipitate was pelleted by centrifugation. PAE2072 was purified from the resulting supernatant by nickel affinity chromatography on a HisTrap HP column (Pharmacia) and eluted in the presence of 125–200 mM imidazole. Purified protein was dialyzed into 50 mM Tris pH 8.0, 75 mM NaCl, 1 mM DTT, 0.5 mM EDTA, diluted to 1 mg mL−1, and cleaved by incubation with TEV protease (1 mg TEV per 10 mg PAE2072) overnight at room temperature. Pure PAE2072 lacking the N-terminal (His)6 tag was collected in the flowthrough of another nickel affinity chromatography step, dialyzed into 30 mM Tris pH 7.2, 30 mM NaCl, concentrated to 9.3 mg mL−1 (wild-type) or 6.9 mg mL−1 (C120S), and stored at 4 °C. Protein concentrations were determined by the Bio-Rad version of Bradford’s dye-binding assay 34. Crystals of unliganded PAE2072 were obtained by hanging drop vapor diffusion at room temperature. The drop contained 1 μL of the reservoir solution (0.1 M HEPES pH 6.0, 0.17 M LiSO4, 16.5% PEG3350) mixed with 1 μL of the concentrated protein. Crystals were transferred to paraffin oil for a few seconds before flash freezing at 100 K. Co-crystallization of PAE2072 with AMP was performed by hanging drop vapor diffusion, following pre-incubation of the protein with AMP at a concentration of 5 mM. Plate-like crystals were routinely obtained using a reservoir solution of 0.2 M lithium acetate pH 7.8–8.0, 13.5% PEG3350; however, these crystals were deemed unsuitable for data collection due to anisotropic diffraction that did not exceed a resolution of 3.0 Å. A single, large, three-dimensional crystal was obtained in condition 24 (1.0 M CsCl) of the Additive Screen HR2-428 (Hampton Research) using a reservoir solution of 0.2 M lithium acetate pH 7.8, 13.5% PEG3350. The ratio in the drop of protein/AMP to reservoir solution to additive was 5:4:1, for a final AMP concentration of 2.5 mM and a final CsCl concentration of 0.1 M. The crystal was transferred to reservoir solution supplemented with 30% glycerol for a few seconds before flash freezing at 100 K.
Data collection, structure determination, and refinement
Diffraction data from the unliganded crystals were collected using a Rigaku FR-D rotating anode X-ray generator equipped with an R-axis IV++ imaging plate detector (Rigaku). Data extending to 2.10 Å were processed using DENZO/SCALEPACK35. Phases were obtained by molecular replacement using PHASER36 and the coordinates from a single subunit of the M. tuberculosis Hypoxic Response Protein I (PDB code 1Y5H; M.L. Sharpe et al., unpublished) as a search model, which shares less than 20% sequence identity with PAE2072. An initial model was built using the graphics program Coot37 and refined using CNS simulated annealing and conjugate gradient least squares refinement38. Subsequent rounds of model building and refinement were carried out in Coot and REFMAC39 to introduce TLS anisotropic displacement parameters into the refinement. The final model had R and Rfree values of 20.0% and 24.3%, respectively. A dataset from the AMP-bound crystal was collected at the Advanced Light Source, beamline 5.0.2, using an ADSC Quantum 315 CCD detector to record the data. Diffraction data extending to 2.35 Å were processed using DENZO/SCALEPACK. Phases were obtained by molecular replacement using PHASER and the coordinates from the unliganded PAE2072 dimer as a search model. An initial round of refinement using CNS simulated annealing and conjugate gradient least squares refinement was followed by iterative rounds of model building and refinement carried out in Coot and REFMAC. TLS parameters and strong non-crystallographic symmetry constraints were utilized throughout, although the NCS constraints were released during the last round of refinement. The modeling of cesium ions was aided by the appearance of peaks in an anomalous difference Fourier map, attributed to the anomalous scattering of cesium (3.8 anomalous electrons) at 1 Å wavelength. The final model had R and Rfree values of 23.9% and 29.4%, respectively. The refinement statistics for both models are given in Table 1. The geometric quality of the models was assessed with the structure validation tools ERRAT40, PROCHECK41, and VERIFY3D42. ERRAT reported overall quality factors of 96.7% for the unliganded structure and 93.2% for the AMP-bound structure.
Structural analyses
Surface area buried upon dimerization was calculated using the program AREAIMOL43, which is distributed with the CCP4 suite of crystallographic programs44. Omit maps of the disulfide bonds and AMP molecules were made by deleting all atoms of Cys120 residues or all AMP molecules, respectively, followed by simulated annealing and conjugate gradient least squares refinement in CNS to reduce model bias. All figures of protein structures were made using PyMOL45.
Sequence analyses
For the purpose of counting the number of standalone CBS domain proteins per organism in Pfam, these were defined as proteins that: 1) contained one or more copies of Pfam Family PF00571 (CBS domain pair), 2) contained no other domains recognized by Pfam, and 3) had at least 70% of the length of their amino acid sequence covered by the PF00571 domain(s). The multiple sequence alignment of the P. aerophilum standalone CBS domain proteins was generated using T-COFFEE46. The per-residue conservation scores for PAE2072 output by T-COFFEE were used to color the protein according to sequence conservation in Figure 4. Pairwise identities and similarities were calculated using the BLASTall program47.
Circular dichroism
Circular dichroism measurements were recorded using a Jasco J-715 spectrometer with a Peltier temperature-controlled cell holder (Jasco). Samples for thermal denaturation analysis were prepared by dilution of concentrated protein to a final concentration of 4.9 μM (0.75 mg mL−1) in 30 mM Tris pH 7.2, 30 mM NaCl, 3.5 M GdmHCl, with or without 1 mM DTT. Samples were equilibrated for an hour at room temperature before CD measurements were taken. The far-UV CD spectra of the protein in buffer and after incubation in 3.5 M GdmHCl, 1 mM DTT, or both were checked to ensure that these conditions did not induce structural changes at 20 °C. Thermal denaturations were performed by monitoring the CD signal at 218 nm with a bandwidth of 2 nm while increasing the temperature linearly from 20–100 °C at a rate of 1 °C min−1. Samples for chemical denaturation analysis were prepared by diluting stocks of wild-type or C120S PAE2072 to a final concentration of 2.5 μM in 30 mM Tris pH 7.2, 30 mM NaCl, with varying concentrations of GdmHCl. Samples were allowed to equilibrate overnight at 25 °C before CD measurements were taken. Each data point represents the CD signal at 220 nm averaged over 45 seconds, with the temperature held constant at 25 °C. At least two curves were collected for each denaturation experiment, and the raw CD data were normalized for the purpose of comparison to the minimum (CDmin) and maximum (CDmax) CD signal on the temperature or GdmHCl range according to the equation:
Acknowledgments
The authors would like to thank Danny Boutz for work on the initial stages of this project, Julie Elbogen for cloning, Martin Phillips for assistance with CD spectrometry, and Luki Goldschmidt, Chris Miller, and Morgan Beeby for helpful and informative discussions. This work was funded by award R01GM081652 from the National Institutes of Health.
Abbreviations
- CBS
Cystathionine β-synthase
- AMPK
AMP-activated protein kinase
Footnotes
ACCESSION CODES: The coordinates and structure factors for the unliganded and AMP-bound forms of PAE2072 have been deposited in the Protein Data Bank with accession numbers 2RIH and 2RIF, respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gilbert HF. Molecular and cellular aspects of thiol-disulfide exchange. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 2.Derman AI, Beckwith J. Escherichia coli alkaline phosphatase fails to acquire disulfide bonds when retained in the cytoplasm. J Bacteriol. 1991;173:7719–22. doi: 10.1128/jb.173.23.7719-7722.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prinz WA, Aslund F, Holmgren A, Beckwith J. The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem. 1997;272:15661–7. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 4.Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes. Annu Rev Biochem. 2003;72:111–35. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 5.Chiu HJ, Johnson E, Schroder I, Rees DC. Crystal structures of a novel ferric reductase from the hyperthermophilic archaeon Archaeoglobus fulgidus and its complex with NADP+ Structure. 2001;9:311–9. doi: 10.1016/s0969-2126(01)00589-5. [DOI] [PubMed] [Google Scholar]
- 6.DeDecker BS, O’Brien R, Fleming PJ, Geiger JH, Jackson SP, Sigler PB. The crystal structure of a hyperthermophilic archaeal TATA-box binding protein. J Mol Biol. 1996;264:1072–84. doi: 10.1006/jmbi.1996.0697. [DOI] [PubMed] [Google Scholar]
- 7.Jiang Y, Nock S, Nesper M, Sprinzl M, Sigler PB. Structure and importance of the dimerization domain in elongation factor Ts from Thermus thermophilus. Biochemistry. 1996;35:10269–78. doi: 10.1021/bi960918w. [DOI] [PubMed] [Google Scholar]
- 8.Maes D, Zeelen JP, Thanki N, Beaucamp N, Alvarez M, Thi MH, Backmann J, Martial JA, Wyns L, Jaenicke R, Wierenga RK. The crystal structure of triosephosphate isomerase (TIM) from Thermotoga maritima: a comparative thermostability structural analysis of ten different TIM structures. Proteins. 1999;37:441–53. [PubMed] [Google Scholar]
- 9.Meyer J, Clay MD, Johnson MK, Stubna A, Munck E, Higgins C, Wittung-Stafshede P. A hyperthermophilic plant-type [2Fe-2S] ferredoxin from Aquifex aeolicus is stabilized by a disulfide bond. Biochemistry. 2002;41:3096–108. doi: 10.1021/bi015981m. [DOI] [PubMed] [Google Scholar]
- 10.Toth EA, Worby C, Dixon JE, Goedken ER, Marqusee S, Yeates TO. The crystal structure of adenylosuccinate lyase from Pyrobaculum aerophilum reveals an intracellular protein with three disulfide bonds. J Mol Biol. 2000;301:433–50. doi: 10.1006/jmbi.2000.3970. [DOI] [PubMed] [Google Scholar]
- 11.Beeby M, O’Connor BD, Ryttersgaard C, Boutz DR, Perry LJ, Yeates TO. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS Biol. 2005;3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boutz DR, Cascio D, Whitelegge J, Perry LJ, Yeates TO. Discovery of a thermophilic protein complex stabilized by topologically interlinked chains. J Mol Biol. 2007;368:1332–44. doi: 10.1016/j.jmb.2007.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cacciapuoti G, Moretti MA, Forte S, Brio A, Camardella L, Zappia V, Porcelli M. Methylthioadenosine phosphorylase from the archaeon Pyrococcus furiosus. Mechanism of the reaction and assignment of disulfide bonds. Eur J Biochem. 2004;271:4834–44. doi: 10.1111/j.1432-1033.2004.04449.x. [DOI] [PubMed] [Google Scholar]
- 14.Karlstrom M, Stokke R, Steen IH, Birkeland NK, Ladenstein R. Isocitrate dehydrogenase from the hyperthermophile Aeropyrum pernix: X-ray structure analysis of a ternary enzyme-substrate complex and thermal stability. J Mol Biol. 2005;345:559–77. doi: 10.1016/j.jmb.2004.10.025. [DOI] [PubMed] [Google Scholar]
- 15.Ladenstein R, Ren B. Reconsideration of an early dogma, saying “there is no evidence for disulfide bonds in proteins from archaea”. Extremophiles. 2008;12:29–38. doi: 10.1007/s00792-007-0076-z. [DOI] [PubMed] [Google Scholar]
- 16.Mallick P, Boutz DR, Eisenberg D, Yeates TO. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc Natl Acad Sci U S A. 2002;99:9679–84. doi: 10.1073/pnas.142310499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pedone E, Limauro D, D’Alterio R, Rossi M, Bartolucci S. Characterization of a multifunctional protein disulfide oxidoreductase from Sulfolobus solfataricus. Febs J. 2006;273:5407–20. doi: 10.1111/j.1742-4658.2006.05533.x. [DOI] [PubMed] [Google Scholar]
- 18.Ren B, Tibbelin G, de Pascale D, Rossi M, Bartolucci S, Ladenstein R. A protein disulfide oxidoreductase from the archaeon Pyrococcus furiosus contains two thioredoxin fold units. Nat Struct Biol. 1998;5:602–11. doi: 10.1038/862. [DOI] [PubMed] [Google Scholar]
- 19.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–3. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 20.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–84. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–85. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 22.Amodeo GA, Rudolph MJ, Tong L. Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature. 2007;449:492–5. doi: 10.1038/nature06127. [DOI] [PubMed] [Google Scholar]
- 23.Townley R, Shapiro L. Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science. 2007;315:1726–9. doi: 10.1126/science.1137503. [DOI] [PubMed] [Google Scholar]
- 24.Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, Martin SR, Carling D, Gamblin SJ. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- 25.Jin X, Townley R, Shapiro L. Structural insight into AMPK regulation: ADP comes into play. Structure. 2007;15:1285–95. doi: 10.1016/j.str.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 26.Kemp BE. Bateman domains and adenosine derivatives form a binding contract. J Clin Invest. 2004;113:182–4. doi: 10.1172/JCI20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pellequer JL, Chen SW. Multi-template approach to modeling engineered disulfide bonds. Proteins. 2006;65:192–202. doi: 10.1002/prot.21059. [DOI] [PubMed] [Google Scholar]
- 28.Meyer S, Savaresi S, Forster IC, Dutzler R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat Struct Mol Biol. 2007;14:60–7. doi: 10.1038/nsmb1188. [DOI] [PubMed] [Google Scholar]
- 29.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–8. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biemans-Oldehinkel E, Mahmood NA, Poolman B. A sensor for intracellular ionic strength. Proc Natl Acad Sci U S A. 2006;103:10624–9. doi: 10.1073/pnas.0603871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hattori M, Tanaka Y, Fukai S, Ishitani R, Nureki O. Crystal structure of the MgtE Mg2+ transporter. Nature. 2007;448:1072–5. doi: 10.1038/nature06093. [DOI] [PubMed] [Google Scholar]
- 32.Duff AP, Cohen AE, Ellis PJ, Kuchar JA, Langley DB, Shepard EM, Dooley DM, Freeman HC, Guss JM. The crystal structure of Pichia pastoris lysyl oxidase. Biochemistry. 2003;42:15148–57. doi: 10.1021/bi035338v. [DOI] [PubMed] [Google Scholar]
- 33.King NP, Yeates EO, Yeates TO. Identification of rare slipknots in proteins and their implications for stability and folding. J Mol Biol. 2007;373:153–66. doi: 10.1016/j.jmb.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 34.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–8. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–9. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laskowski R, Macarthur M, Moss D, Thornton J. PROCHECK - A program to check the stereochemical quality of protein structures. J Applied Crystallogr. 1993;26:283–291. [Google Scholar]
- 42.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–5. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 43.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 44.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; 2002. [Google Scholar]
- 46.Notredame C, Higgins DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 47.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]