

Abstract
Following recent technological advances there has been an increasing interest in genome structural variation, in particular copy-number variants (CNVs) – large-scale duplications and deletions – in the human genome. Although not immediately evident, CNV surveys make a conceptual connection between the fields of population genetics and protein families, in particular with regard to the stability and expandability of families. The mechanisms giving rise to CNVs can be considered as fundamental processes underlying gene duplication and loss; duplicated genes being the results of “successful” copies, fixed and maintained in the population. Conversely, many “unsuccessful” duplicates remain in the genome as pseudogenes. Here, we survey studies on CNVs, highlighting issues related to protein families. In particular, CNVs tend to affect specific gene functional categories, such as those associated with environmental response, and are depleted in genes related to basic cellular processes. Furthermore, CNVs occur more often at the periphery of the protein interaction network. Thereby, functional categories associated with successful duplicates and unsuccessful duplicates are clearly distinguishable. These trends are likely reflective of CNV formation biases and natural selection, both of which differentially influence distinct protein families.
INTRODUCTION
Gene duplication is a major process leading to novel genes and proteins, which may naively be assumed to be a relatively slow process in evolutionary terms. However, recent results from the field of genetics argue that gene duplication has occurred frequently during the recent history of the human population and that gene duplicates occur in humans in variable numbers and may be constantly generated de novo. Studies measuring human genome variation are receiving much attention currently [1], as novel genomics approaches have revealed an unanticipated level of genetic variation in the human population (e.g., [2–7]). A type of variation that was recently found to be abundant in the human genome is genome structural variation [4,5,7–13]. Genome structural variants are generally defined as (e.g., [14,15]), kilobase- to megabase-sized deletions, insertions, duplications, and inversions. Furthermore, structural variants cause a greater amount of sequence divergence between humans than the widely studied single nucleotide polymorphisms (SNPs) [4–7,14,15], if one considers the total number of nucleotides spanned by both forms of variation. Even though genome structural variants can alter the intron/exon-structure of genes by disrupting exons or fusing genes together [5], they frequently span entire genes leading to different gene copy-numbers between individuals. Following the first genome-wide mapping in humans [8,16], genome structural variants have been identified in several mammalian genomes (e.g., [17–22]) and at varying levels of resolution (see Box 1). Here, we use the term copy-number variant (CNV) to refer to a genome structural variant leading to changes in gene copy number (rather than an inversion or a variant not encompassing genes, both of which may also influence protein function, e.g., by influencing gene regulation). While there have been early insights on the origin of small indels (≪ 1kb) [23,24], we are just now beginning to understand mechanisms behind the formation of CNVs. Recent advances have been fuelled by the development of approaches for mapping genome structural variation at the resolution of base-pairs [5,25].
CNVs are of significance in relation to the human proteome in various ways. First, copy-numbers of protein-coding genes can be strikingly different between apparently healthy (“normal”) individuals (e.g., [4,28,29]), with instances of up to 10 additional copies reported for several protein-coding gene loci (e.g., [28–31]). In line with this, CNV de novo-formation is thought to be constantly ongoing in mammalian genomes [21,26,27], affecting recent and current protein evolution; in fact, CNV genesis may occur at rates higher than point mutations with impact on gene function. Finally, results from several studies point to a tight relationship of gene copy-number with messenger RNA and protein expression-level (e.g., [29,32]). Variation at the level of gene expression may represent the underlying basis for several phenotypic traits associated with CNVs, such as dietary preferences across distinct populations [29] or the susceptibility to diseases including HIV [28], breast cancer [33], autism [26], and several auto-immune diseases [30,31,34]. Furthermore, through this “gene-dosage” effect, CNVs are likely to influence protein complex formation and tightly regulated cellular systems. Since some of these require their individual components to be expressed at stoichiometrically precise levels, a CNV may have potential harmful (or beneficial) effects. Many additional phenotypic relationships are likely to be discovered in the near future with the ongoing application and improvement of approaches for ascertaining CNVs (Box 1) and for associating CNVs and phenotypes [35–37]. Thus, CNVs are not only relevant to population genetics, but should to be considered in systems biology and proteomics studies. CNVs may constitute a source of redundancy and thus evolvability or robustness, i.e., provide ‘replacement proteins’. Often, CNVs will behave selectively neutral (similar to most SNPs). Nevertheless, they represent a genomic pool of evolving transcripts, genes, and proteins that in longer evolutionary terms may become fixed in the population as novel genes. Here, we summarize recent findings in the field of genome structural variation and discuss implications for the systems biology and proteomics fields.
An abundance of copy-number variants in the genome
Knowledge on CNVs has dramatically increased following recent technological advances (see Box 1). For instance, a CNV map generated from data of over two hundred individuals has revealed that 12% or more of the human genome is prone to copy-number variation [4]. Recent studies at considerably higher resolution sufficient to map small CNVs (<50 kb) and to identify the precise boundaries (or breakpoints) of CNVs have revealed that the number of genome structural variants (>1 kb) that distinguish genomes of different individuals is at least on the order of 600–900 per individual [5,6]. Of these, approximately ~150 genome structural variants per individual presumably directly affect protein-coding genes by intersecting with them [5]. Moreover, recent surveys have led to a re-estimation of the total amount of sequence divergence between individuals; while it was initially assumed that the genomes of two unrelated individuals differ by ~0.1% (mainly due to SNPs), it has recently been estimated that at least 0.5% of our genomes differ [6], with the majority of variation being due to CNVs.
Considerations for our understanding of protein evolution
Recent findings concerning the abundance of CNVs in the human genome add to current perspectives on gene duplication and loss – essential processes in genome and proteome evolution. For nearly a hundred years, duplication of genetic material has been regarded as an important factor in the evolution of higher organisms (see [38] and references therein) – and protein birth by duplication is widely considered to be more common than formation of proteins ‘from scratch’ [39]. Following gene duplication, one of the newly generated paralogs may escape selective constraints (purifying selection) and become free to acquire a new function (neo-functionalization). Furthermore, both paralogous sequences may experience decreased selective pressure after duplication, which may reflect partitioning between paralogs into different functions which had been combined in the multifunctional ancestral gene [40,41] (sub-functionalization). Gene duplication is also thought to be a major contributor to the evolution of protein networks [42], even though it may not account for the evolution of complex molecular machines [43]. Duplications may evolve in an effectively neutral fashion over extended evolutionary time scales [41]. They further may be advantageous to the cell by increasing the robustness against mutations (e.g., [37]). Moreover, at short evolutionary time scales the potential to modify gene/protein expression levels through gene dosage change may promote gene duplications and losses. In this regard, a genome-wide study [32] has recently reported relationships between CNVs and mRNA levels. Furthermore, Perry et al. found that increased copy-numbers of the amylase gene reflect higher levels of protein expression and are correlated with dietary preferences for starch [29]. Note that a single CNV formation event – a type of mutation that for some genomic loci appears to occur more frequently than nucleotide substitutions (see below) – may be sufficient to specifically promote gene expression modification; thus gene copy-number changes may facilitate evolutionary adaptation involving protein abundance change. Nevertheless, nucleotide substitutions having an effect on the regulation of gene expression are likely to eventually supersede gene-copy number increase (or decrease), i.e., take over in the long run; in particular, maintaining a large number of identical genes per genome during longer evolutionary time scales is likely causing significantly increased ‘costs’ related to genome stability and repair.
De novo CNV formation
The abundance of CNVs in the genome indicates that gene duplication (and loss) probably occurs at a constant and high rate in humans. For a number of loci in the genome involved in commonly recurring genomic disorders – regions in which CNVs may recur frequently – this rate has recently been estimated to be 1e-4 to 1e-6 per generation [44], which is considerably higher than the rate at which point mutations are thought to occur (2e-8; see refs. in [44]). Furthermore, in a recent analysis involving inbred mice, CNV formation rates as high as 1e-2 to 1e-3 have been inferred for loci encoding genes [21]. Note that in order to properly compare these rates we have to take into account the fact that the rate at which CNVs arise has been determined for large loci and large CNVs, e.g., of 100 kb in size, whereas the point mutation rate is given per nucleotide. If we consider that ~1% of the genome comprises coding sequence, then the rate at which protein coding sequence will experience a new point mutation within a given 100 kb locus is approximately 2e-8 * 1e5 * 0.01 = 2e-5. Conversely, any given novel CNV of 100 kb would affect protein coding sequence in the given locus. Thus, for several gene loci, CNVs formed de novo may be significantly more likely to affect coding sequence than point mutations. Frequently, point mutations will remain silent (e.g., if they fall into synonymous sites) and may have little or no effect on protein function. On the other hand, protein duplicates may not always be expressed, and expression differences may sometimes have little or no functional consequence.
CNVs, gene duplicates and formation bias
It is evident from genome-wide surveys that CNVs exhibit a highly non-uniform distribution along chromosomes. This distribution may have different causes: First, it may be due to biases in the ascertainment of CNVs. Second, locus-specific differences in the rate at which CNVs are formed may cause this disparity. Finally, the distribution may be due to natural selection acting differentially throughout the genome, i.e., relative to phenotypic changes caused by different genomic regions that are affected by CNVs.
We believe that the fact that several complementary technologies have detected CNVs at overlapping genomic loci (which becomes quickly apparent when browsing the Database of Genomic Variants (DGV) [16]) indicates that technological biases are unlikely to be responsible for the trend.
However, discerning the remaining two potential causes is not straightforward. Mutation, population-variation and fixation by natural selection or random drift have been studied extensively in relation to SNPs, but much less so for CNVs. The existence of genomic loci undergoing recurrent de novo structural rearrangements in relation to disease [44] suggests that genomic CNV formation biases exist. In this regard, for instance, subtelomeric regions represent hot spots for interchromosomal recombination [45] and segmental duplication sequence [45,46]. In line with this, results from Redon et al. [4] indicate an enrichment of CNVs in subtelomeric regions (within 500 kb of the ends of chromosome arms). Consequently, breakage or fusion of chromosomes during the evolution of mammalian genomes may have influenced the rate of duplication (and loss) of gene families across species.
Natural selection: enrichment and depletion in biological processes
Natural selection can be analyzed by studying the overlap of CNVs with various functional elements. For instance, recent studies have revealed that protein-coding genes, and also other genomic elements including highly conserved non-coding regions, tend to be depleted among CNVs, indicating purifying selection [4,5,47]. In particular, deletions appear to be under stronger selection than duplications [4]. Furthermore, certain functional categories of protein-coding genes are more prone to be affected by CNVs than others. For instance, Table 1 shows a strong enrichment among CNVs for several protein domains. Our survey presented in Figure 1 extends this analysis by assessing which protein functional categories are most strikingly enriched or depleted amongst CNVs: consistent with earlier surveys we find that proteins involved in processes related to environmental response tend to be enriched in CNVs [4,5,8,9,14,48,49] and duplicated genes retained in the genome [50], whereas proteins involved in fundamental cellular functions, such as cellular physiological processes, tend to be depleted. While the latter trend is presumably due to purifying selection owing to constraints, some of the former enrichment may be due to positive selection. Such effects should be observable also in fixed variants. Hence, we extended our survey by comparing “successful duplicates” (i.e., recent segmental duplications) with “unsuccessful duplicates” (nonprocessed pseudogenes, i.e., duplicated genes that were recently inactivated by mutation; e.g., [51,52]). Whereas distributions for successful duplicates reveal trends similar to the ones observed for CNVs (Figure 1b), we note distinct trends for unsuccessful duplicates. Namely, protein-coding genes acting in metabolism and cellular physiological processes, that is dosage-sensitive genes, appear significantly enriched among pseudogenes (Figure 1c) – although also genes putatively involved in environmental response (such as genes mediating locomotion in response to stimuli) were observed to be significantly enriched, consistent with an earlier survey [51]. Overall, the results are consistent with constraint (purifying selection) acting on dosage sensitive genes, leading to the removal of extra gene copies causing dosage imbalance.
Table 1.
Most significantly enriched protein domains in CNVs
| Interpro ID | Enrichment amonst CNVs | P-value (Bonferroni-corrected) | Domain-function |
|---|---|---|---|
| IPR010630 | 2.0 | 1.9 E-10 | Domain of unknown function (DUF1220); recently predicted to be involved in brain functions [62]. |
| IPR012604 | 6.8 | 2.3 E-10 | C-terminal domain found in certain RNA-binding proteins |
| IPR000725 | 1.2 | 2.4 E-10 | Olfactory receptor (GPCR) |
| IPR003597 | 1.3 | 2.4 E-10 | Immunoglobulin C1-set |
| IPR003006 | 1.3 | 5.9 E-10 | Immunoglobulin/major histocompatibility complex motif |
Figure 1.
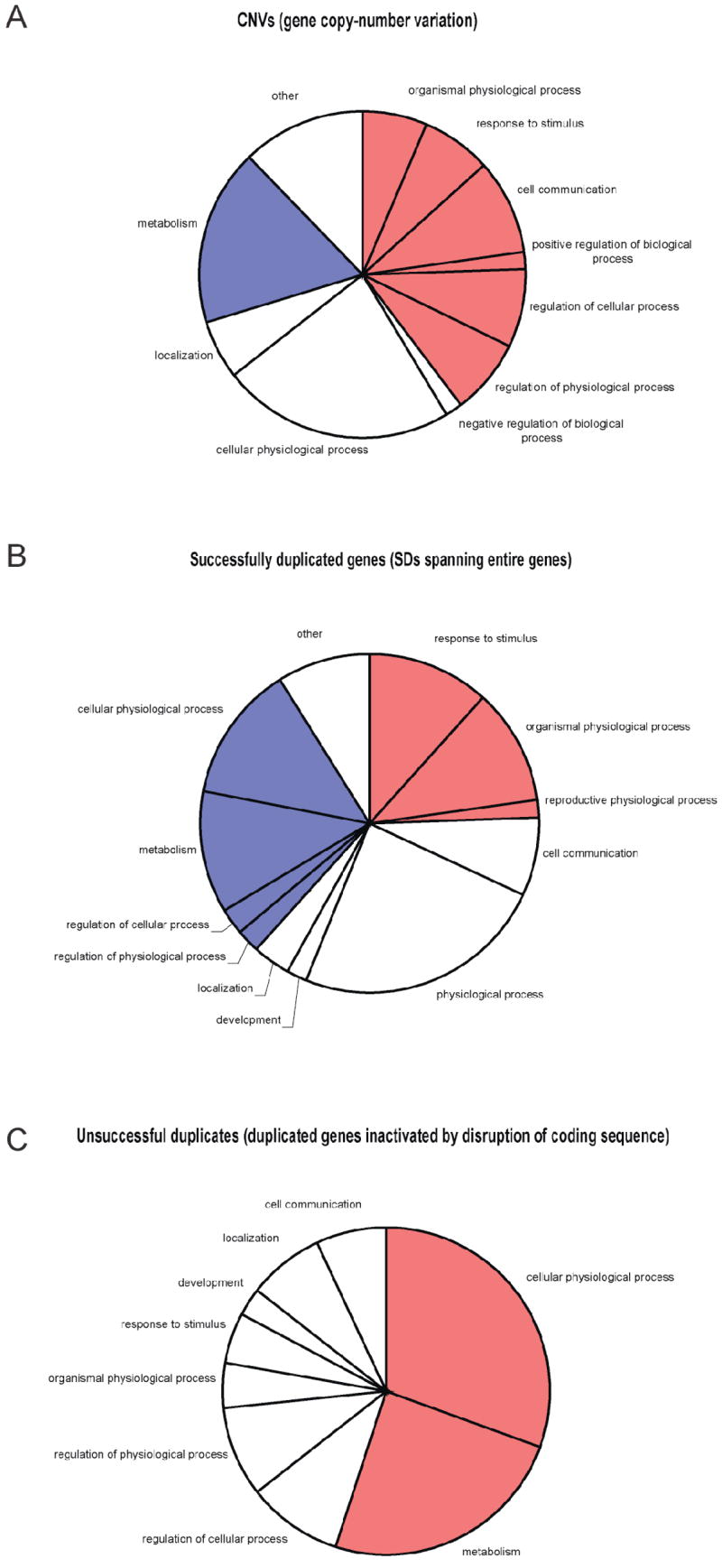
Enrichment and depletion of gene functional categories (Gene Ontology (GO) annotation [67], GO biological process, level 3) among genes affected by CNVs. Significant enrichment (red shading) and depletion (blue shading) of protein-coding genes were determined using software published in [68] (Bonferroni-corrected P-value cutoff of 0.01). Genomic coordinates of CNVs (build hg18) were obtained from DGV [16] on 30th November 2007. A high-confidence list of recently successfully duplicated genes was obtained by collecting RefSeq genes spanned by segmental duplications (SDs) retrieved from http://eichlerlab.gs.washington.edu/database.html. Gene coordinates and GO annotations were obtained from Ensembl (www.ensembl.org/biomart/martview). Functional categories observed in less than 2% of genes were grouped into “other”. (A) Depletion and enrichment of GO categories among CNVs. (B) Depletion and enrichment of GO categories among successful gene duplicates. (C) Depletion and enrichment of GO categories among unsuccessful duplicates (i.e., nonprocessed pseudogenes).
Additionally, our survey shows that unsuccessful duplicates tend to be longer than successful duplicates (Table 2), both at the gene and at the protein level. Although this trend may partially be influenced by the way successful and unsuccessful duplicates have been ascertained, the observations are in line with previous findings that complex genes, such as alternatively spliced ones that are on average longer than non-alternatively spliced genes [53], tend be less prone to duplication than genes with few exons and no or few additional splice forms [54–56].
Table 2.
Influence of the lengths of protein coding genes
| Peptide Length (amino acids) | Gene Length (base pairs) | |
|---|---|---|
| Genome-wide average | 641 | 75,912 |
| Successful duplicates | 398 | 15,638 |
| CNVs | 583 | 38,299 |
| Unsuccessful duplicates | 736 | 52,126 |
Natural selection: relationship of duplications and protein interaction networks
Selection rarely acts on functions carried out by a protein ‘in isolation’. Most proteins, rather than working as a single entity, act in concert as members of a tightly regulated pathway or as a large multi-protein complex. Consequently, the level at which proteins tend to be affected by CNVs is partially reflected in the protein’s role in the protein interaction network, i.e., the entirety of proteins thought to interact in the cell: Recently, it was shown that CNVs are more likely to affect proteins at the periphery of the network (with few interaction partners), whereas proteins at the network center (many interaction partners) are less likely to be variable in copy number [57,58]. These observations are consistent with an over-representation of small protein families (having few or no paralogs) in the center of protein networks [59] and the observation that members of large protein families tend not to be involved in protein complexes [60]. It is plausible that proteins at the network periphery are under less evolutionary constraint and are thus freer to evolve. In contrast, duplicates affecting the network center may be detrimental and thus more likely to be selectively removed. The latter is strongly supported by the fact that unsuccessful gene duplicates are observed at the network center at a significantly higher frequency than successful duplicates (Figure 2).
Figure 2.

Gene duplicates and the human protein interaction network.
(A) Recently successfully duplicated genes are significantly enriched at the periphery of the protein network, as evidenced from a significantly decreased average betweenness centrality with P≪0.01 (the interaction network was constructed and the P-values generated as described in [57]).
(B) Unsuccessful duplicates are significantly enriched at the network center, P<0.01.
Natural selection: other influences on copy number variation
Besides purifying selection, positive or directional selection has been implicated in influencing the distribution of CNVs and successful duplicates in the human genome. For instance, genes frequently affected by CNVs were reported to exhibit elevated rates of amino acid change in evolution [48], which may be an indicator for positive selection. Moreover, a recent case study focusing on the salivary amylase protein Amy1 has concluded that AMY1 gene copy number in human populations likely underlies diet-related positive selection pressures [29]. Furthermore, duplications are, similar to positively selected nucleotide changes, biased to the protein interaction network periphery [57]; this indicates that adaptive evolution – involving SNPs or CNVs – tends to act at the periphery of the network rather than the center. Concerning successful gene duplicates, several groups have reported signs of positive selection (at the level of amino acid replacements) for recently generated gene duplicates in primates (see e.g., [61,62]) and rodents [63]. Finally, a recent computational analysis has presented evidence for substantial positive selection in hotspots of recently formed segmental duplications in humans [64]; these hotspots are presumably subject to recurrent de novo CNV formation.
At least for some genes it appears that gene copy-number may evolve in a neutral fashion: for instance, Nozawa and coworkers [56] reported that no significant difference exists in the amount of CNVs between functional and nonfunctional (i.e., pseudogenic) sensory receptor genes, a gene family particularly prone to structural variation (e.g., [4,65]). On the other hand, the positive effect of gene duplication or loss in the case of CNVs spanning more than one gene may in some instances balance or overshadow the potentially negative impact of protein dosage imbalances and may drive the fixation of CNVs in particular regions of the genome.
Nevertheless, negative effects of commonly occurring CNVs are also visible in current CNV datasets (Figure 3). In particular, a survey in which we linked protein domains present in CNVs to the Online Mendelian Inheritance in Man (OMIM) and the Cancer Gene Census (CGC) [66] databases revealed an enrichment of copy-number variable genes amongst disease-related genes; indeed, positive effects of CNVs need to be balanced against potential harmful influences of genome structural variation.
Figure 3. Disease associations of protein domains in genes affected by copy-number variation.

(A) CNVs are significantly associated with disease genes. Associations between protein domains and diseases were retrieved from OMIM (www.ncbi.nlm.nih.gov/omim). (B) Enrichment of protein domains of cancer-related genes among CNVs. Genes implicated in cancer were obtained from CGC (www.sanger.ac.uk/genetics/CGP/Census).
CONCLUSIONS
CNVs should be considered in systems biology and proteome evolution-related studies due to their effect on protein expression, function and the phenotype, and their likely contribution to protein family evolution. After formation and subsequent fixation following selection or random drift, CNVs may give rise to gene duplicates or losses; thus they represent important genomic intermediates in genome and proteome evolution.
Box 1. Recent technological advances led to an improvement of CNV maps
Our understanding of CNVs was considerably enhanced by novel high-resolution genomics technologies (Figure 4). Genome-wide microarray technologies based on Bacterial Artificial Chromosomes or representational oligonucleotide microarray analysis (ROMA), which uses short oligonucleotides probing genomic loci at a density of one oligonucleotide per 30 kilobases, enabled generation of a first record of CNVs in the human genome [8,16]. Subsequently, mapping-resolutions have considerably increased following the development of computational approaches for mapping fosmid clone-ends to the reference genome [9], the mining and statistical analysis of SNP genotyping data [10,11], and the development of high-resolution oligonucleotide microarray technology [12,25,69,70] For instance, high-resolution comparative genome hybridization (HR-CGH) [70] based on oligonucleotide tiling arrays enables the generation of CNV maps at a resolution below 300 bp. Novel sophisticated computational approaches [65,71,72] have facilitated scoring and interpreting the data, and allowed mapping the actual physical boundaries, or breakpoints, of CNVs systematically [25,65,70]. Other recent surveys provided records of small and medium-sized indels based on comparing raw DNA sequence reads [73] and alternative human genome assemblies [6,74] to the human reference genome. Furthermore, a recent survey based on next-generation DNA sequencing provided a genome-wide account at sub-kilobase resolution of genome structural variants – i.e., deletions, insertions and inversions – in two human genomes by high-resolution and massive paired-end mapping (PEM) [5].
Papers of special interest* and outstanding** interest
**Redon et al., 2006
In this paper two complementary microarray technologies were used to catalogue CNVs genome-wide in over two hundred healthy individuals. Nearly 12% of the human genome was found to be prone to variation in copy-number, and copy-number variable regions mapped encompassed hundreds of genes. The generated data enabled the authors to examine the genomic impact of CNVs.
**Perry et al., 2007
The copy-number of the salivary amylase gene (AMY1) - which shows a strong correlation with protein expression level - is markedly differing across human populations. This study reports that with gene counts being large in populations that use high amounts of starch in their diet, the distribution of AMY1 copy-numbers was likely influenced by positive selection.
*Egan et al., 2007
The authors systematically analyzed the de novo formation of CNVs in the genomes of inbred mice. Surprisingly, the analyzed genomes contained a large extent of recently formed CNVs, distributed in a non-random fashion across the genome and frequently encompassing genes. These findings may have implications on future studies involving model organisms.
*Jiang et al., 2007
The authors devised an algorithmic framework to reconstruct the evolutionary history of recent segmental duplications in the human genome. Many recent duplications occurred in a small subset of genomic hotspots (i.e., core duplicons), which may be centers for human transcript, gene, and protein birth. These centers are enriched for protein-coding genes, many of which show signs of positive selection.
*Kim et al., 2007
This work analyses the relationships of adaptive evolution and genetic variation with proteomic properties, namely topological positioning within the protein interaction network (see also work by Dopman and Hartl, 2007). The network periphery is enriched both for signatures of recent adaptation and genetic variation (SNPs and CNVs). The trends are rationalized in terms of constraints imposed by protein structure, and explained by the approximate mapping of the network to cellular organization.
*Dopman and Hartl, 2007
The authors characterize CNVs across genomic regions in a model organism, Drosophila melanogaster, and report a surprising amount of CNVs in flies. A comprehensive analysis of evolutionary processes revealed various evolutionary trends that are paralleled by findings in relation to CNVs in humans, with negative selection and presumably local biases in mutational mechanisms being main factors shaping genome-wide CNV occurrence patterns. Similar to observations that have been made in the human proteome (by Kim et al., 2007), the authors find that fly CNVs are depleted amongst proteins that are central in the protein interaction network.
*Urban et al., 2006
The authors report the first chromosome-wide tiling microarray experiment enabling the mapping of CNVs at ~300 bp resolution (i.e., below the resolution of most exons), paving the way for future high-resolution analyses of CNVs in the human genome using cost-efficient microarray technology. Using a novel approach, HR-CGH, the authors resolved breakpoints of commonly occurring CNVs as well as large genomic aberrations associated with congenital diseases.
Figure 4.

Efficient high-throughput functional genomics technologies used for identifying CNVs in a genome-wide fashion. Figure adapted from [75].
Acknowledgments
Funding was provided by a Marie Curie Fellowship (J.O.K.) and the NIH (Yale Center of Excellence in Genomic Science grant). The authors thank Pedro Alves and Jeroen Raes for comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pennisi E. Breakthrough of the year. Human genetic variation. Science. 2007;318:1842–1843. doi: 10.1126/science.318.5858.1842. [DOI] [PubMed] [Google Scholar]
- 2.Consortium TIH, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, Eichler EE, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 9.Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 10.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 11.McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 12.Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006;38:82–85. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- 13.Sharp AJ, Locke DP, McGrath SD, Cheng Z, Bailey JA, Vallente RU, Pertz LM, Clark RA, Schwartz S, Segraves R, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 15.Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, et al. Copy number variation: new insights in genome diversity. Genome Res. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- 16.Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Jiang T, Mao JH, Balmain A, Peterson L, Harris C, Rao PH, Havlak P, Gibbs R, Cai WW. Genomic segmental polymorphisms in inbred mouse strains. Nat Genet. 2004;36:952–954. doi: 10.1038/ng1417. [DOI] [PubMed] [Google Scholar]
- 18.Newman TL, Tuzun E, Morrison VA, Hayden KE, Ventura M, McGrath SD, Rocchi M, Eichler EE. A genome-wide survey of structural variation between human and chimpanzee. Genome Res. 2005;15:1344–1356. doi: 10.1101/gr.4338005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feuk L, MacDonald JR, Tang T, Carson AR, Li M, Rao G, Khaja R, Scherer SW. Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies. PLoS Genet. 2005;1:e56. doi: 10.1371/journal.pgen.0010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perry GH, Tchinda J, McGrath SD, Zhang J, Picker SR, Caceres AM, Iafrate AJ, Tyler-Smith C, Scherer SW, Eichler EE, et al. Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci U S A. 2006;103:8006–8011. doi: 10.1073/pnas.0602318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Egan CM, Sridhar S, Wigler M, Hall IM. Recurrent DNA copy number variation in the laboratory mouse. Nat Genet. 2007;39:1384–1389. doi: 10.1038/ng.2007.19. [DOI] [PubMed] [Google Scholar]
- 22.Lee AS, Gutierrez-Arcelus M, Perry GH, Vallender EJ, Johnson WE, Miller GM, Korbel JO, Lee C. Analysis of copy number variation in the rhesus macaque genome identifies candidate loci for evolutionary and human disease studies. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn002. [DOI] [PubMed] [Google Scholar]
- 23.Levinson G, Gutman GA. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol. 1987;4:203–221. doi: 10.1093/oxfordjournals.molbev.a040442. [DOI] [PubMed] [Google Scholar]
- 24.Messer PW, Arndt PF. The majority of recent short DNA insertions in the human genome are tandem duplications. Mol Biol Evol. 2007;24:1190–1197. doi: 10.1093/molbev/msm035. [DOI] [PubMed] [Google Scholar]
- 25.Perry GH, Ben-Dor A, Tsalenko A, Sampras N, Rodriguez-Revenga L, Tran CW, Scheffer A, Steinfeld I, Tsang P, Yamada NA, et al. The Fine-Scale and Complex Architecture of Human Copy-Number Variation. Am J Hum Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watkins-Chow DE, Pavan WJ. Genomic copy number and expression variation within the C57BL/6J inbred mouse strain. Genome Res. 2008;18:60–66. doi: 10.1101/gr.6927808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, Catano G, Nibbs RJ, Freedman BI, Quinones MP, Bamshad MJ, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 29.Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, Werner J, Villanea FA, Mountain JL, Misra R, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39:1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, Heward JM, Gough SC, de Smith A, Blakemore AI, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D, van de Kerkhof PC, Traupe H, de Jongh G, den Heijer M, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank B, Bermejo JL, Hemminki K, Sutter C, Wappenschmidt B, Meindl A, Kiechle-Bahat M, Bugert P, Schmutzler RK, Bartram CR, et al. Copy number variant in the candidate tumor suppressor gene MTUS1 and familial breast cancer risk. Carcinogenesis. 2007;28:1442–1445. doi: 10.1093/carcin/bgm033. [DOI] [PubMed] [Google Scholar]
- 34.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 35.McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39:S37–42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 36.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 37.Conrad B, Antonarakis SE. Gene duplication: a drive for phenotypic diversity and cause of human disease. Annu Rev Genomics Hum Genet. 2007;8:17–35. doi: 10.1146/annurev.genom.8.021307.110233. [DOI] [PubMed] [Google Scholar]
- 38.Taylor JS, Raes J. Duplication and divergence: the evolution of new genes and old ideas. Annu Rev Genet. 2004;38:615–643. doi: 10.1146/annurev.genet.38.072902.092831. [DOI] [PubMed] [Google Scholar]
- 39.Ohno S. Evolution by Gene Duplication. New York: Springer-Verlag; 1970. [Google Scholar]
- 40.Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 42.Barabasi AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 43.Kim PM, Lu LJ, Xia Y, Gerstein MB. Relating three-dimensional structures to protein networks provides evolutionary insights. Science. 2006;314:1938–1941. doi: 10.1126/science.1136174. [DOI] [PubMed] [Google Scholar]
- 44.Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 45.Linardopoulou EV, Williams EM, Fan Y, Friedman C, Young JM, Trask BJ. Human subtelomeres are hot spots of interchromosomal recombination and segmental duplication. Nature. 2005;437:94–100. doi: 10.1038/nature04029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 47.Derti A, Roth FP, Church GM, Wu CT. Mammalian ultraconserved elements are strongly depleted among segmental duplications and copy number variants. Nat Genet. 2006;38:1216–1220. doi: 10.1038/ng1888. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen DQ, Webber C, Ponting CP. Bias of selection on human copy-number variants. PLoS Genet. 2006;2:e20. doi: 10.1371/journal.pgen.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cooper GM, Nickerson DA, Eichler EE. Mutational and selective effects on copy-number variants in the human genome. Nat Genet. 2007;39:S22–29. doi: 10.1038/ng2054. [DOI] [PubMed] [Google Scholar]
- 50.Li WH, Gu Z, Cavalcanti AR, Nekrutenko A. Detection of gene duplications and block duplications in eukaryotic genomes. J Struct Funct Genomics. 2003;3:27–34. [PubMed] [Google Scholar]
- 51.Harrison PM, Gerstein M. Studying genomes through the aeons: protein families, pseudogenes and proteome evolution. J Mol Biol. 2002;318:1155–1174. doi: 10.1016/s0022-2836(02)00109-2. [DOI] [PubMed] [Google Scholar]
- 52.Torrents D, Suyama M, Zdobnov E, Bork P. A genome-wide survey of human pseudogenes. Genome Res. 2003;13:2559–2567. doi: 10.1101/gr.1455503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhuang Y, Ma F, Li-Ling J, Xu X, Li Y. Comparative analysis of amino acid usage and protein length distribution between alternatively and non-alternatively spliced genes across six eukaryotic genomes. Mol Biol Evol. 2003;20:1978–1985. doi: 10.1093/molbev/msg203. [DOI] [PubMed] [Google Scholar]
- 54.Kopelman NM, Lancet D, Yanai I. Alternative splicing and gene duplication are inversely correlated evolutionary mechanisms. Nat Genet. 2005;37:588–589. doi: 10.1038/ng1575. [DOI] [PubMed] [Google Scholar]
- 55.Su Z, Wang J, Yu J, Huang X, Gu X. Evolution of alternative splicing after gene duplication. Genome Res. 2006;16:182–189. doi: 10.1101/gr.4197006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nozawa M, Kawahara Y, Nei M. Genomic drift and copy number variation of sensory receptor genes in humans. Proc Natl Acad Sci U S A. 2007;104:20421–20426. doi: 10.1073/pnas.0709956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim PM, Korbel JO, Gerstein MB. Positive selection at the protein network periphery: evaluation in terms of structural constraints and cellular context. Proc Natl Acad Sci U S A. 2007;104:20274–20279. doi: 10.1073/pnas.0710183104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dopman EB, Hartl DL. A portrait of copy-number polymorphism in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2007;104:19920–19925. doi: 10.1073/pnas.0709888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hughes AL, Friedman R. Gene duplication and the properties of biological networks. J Mol Evol. 2005;61:758–764. doi: 10.1007/s00239-005-0037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- 61.Ciccarelli FD, von Mering C, Suyama M, Harrington ED, Izaurralde E, Bork P. Complex genomic rearrangements lead to novel primate gene function. Genome Res. 2005;15:343–351. doi: 10.1101/gr.3266405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Popesco MC, Maclaren EJ, Hopkins J, Dumas L, Cox M, Meltesen L, McGavran L, Wyckoff GJ, Sikela JM. Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science. 2006;313:1304–1307. doi: 10.1126/science.1127980. [DOI] [PubMed] [Google Scholar]
- 63.Shiu SH, Byrnes JK, Pan R, Zhang P, Li WH. Role of positive selection in the retention of duplicate genes in mammalian genomes. Proc Natl Acad Sci U S A. 2006;103:2232–2236. doi: 10.1073/pnas.0510388103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang Z, Tang H, Ventura M, Cardone MF, Marques-Bonet T, She X, Pevzner PA, Eichler EE. Ancestral reconstruction of segmental duplications reveals punctuated cores of human genome evolution. Nat Genet. 2007;39:1361–1368. doi: 10.1038/ng.2007.9. [DOI] [PubMed] [Google Scholar]
- 65.Korbel JO, Urban AE, Grubert F, Du J, Royce TE, Starr P, Zhong G, Emanuel BS, Weissman SM, Snyder M, et al. Systematic prediction and validation of breakpoints associated with copy-number variants in the human genome. Proc Natl Acad Sci U S A. 2007;104:10110–10115. doi: 10.1073/pnas.0703834104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martin D, Brun C, Remy E, Mouren P, Thieffry D, Jacq B. GOToolBox: functional analysis of gene datasets based on Gene Ontology. Genome Biol. 2004;5:R101. doi: 10.1186/gb-2004-5-12-r101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selzer RR, Richmond TA, Pofahl NJ, Green RD, Eis PS, Nair P, Brothman AR, Stallings RL. Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH. Genes Chromosomes Cancer. 2005;44:305–319. doi: 10.1002/gcc.20243. [DOI] [PubMed] [Google Scholar]
- 70.Urban AE, Korbel JO, Selzer R, Richmond T, Hacker A, Popescu GV, Cubells JF, Green R, Emanuel BS, Gerstein MB, et al. High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc Natl Acad Sci U S A. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Colella S, Yau C, Taylor JM, Mirza G, Butler H, Clouston P, Bassett AS, Seller A, Holmes CC, Ragoussis J. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marioni JC, Thorne NP, Valsesia A, Fitzgerald T, Redon R, Fiegler H, Andrews TD, Stranger BE, Lynch AG, Dermitzakis ET, et al. Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biol. 2007;8:R228. doi: 10.1186/gb-2007-8-10-r228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16:1182–1190. doi: 10.1101/gr.4565806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Khaja R, Zhang J, MacDonald JR, He Y, Joseph-George AM, Wei J, Rafiq MA, Qian C, Shago M, Pantano L, et al. Genome assembly comparison identifies structural variants in the human genome. Nat Genet. 2006;38:1413–1418. doi: 10.1038/ng1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scherer SW, Lee C, Birney E, Altshuler DM, Eichler EE, Carter NP, Hurles ME, Feuk L. Challenges and standards in integrating surveys of structural variation. Nat Genet. 2007;39:S7–15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]