

Abstract
Genetic and chemically induced neuronopathic mouse models of Gaucher disease were developed to facilitate understanding of the reversibility and/or progression of CNS involvement. The lethality of the skin permeability barrier defect of the complete gene knock out [gba, (glucocerebrosidase) GCase] was avoided by conditional reactivation of a low activity allele (D409H) in keratinocytes (kn-9H). In kn-9H mice, progressive CNS disease and massive glucosylceramide storage in tissues led to death from CNS involvement by the age of 14 days. Conduritol B epoxide (CBE, a covalent inhibitor of GCase) treatment (for 8–12 days) of wild type, D409H, D409V or V394L homozygotes recapitulated the CNS phenotype of the kn-9H mice with seizures, tail arching, shaking, tremor, quadriparesis, extensive neuronal degeneration loss and apoptosis, and death by the age of 14 days. Minor CNS abnormalities occurred after daily CBE injections of 100 mg/kg/day for 6 doses, but neuronal degeneration was progressive and glucosylceramide storage persisted in D409V homozygotes in the 2 to 5 months after CBE cessation; wild type and D409H mice had persistent neurological damage without progression. The persistent CNS deterioration, histologic abnormalities, and glucosylceramide storage in the CBE-treated D409V mice revealed a threshold level of GCase activity necessary for the prevention of progression of CNS involvement.
Keywords: Gaucher mice, keratin 14, Cre, conduritol B epoxide, CNS degeneration
INTRODUCTION
In Western countries neuronopathic Gaucher disease occurs in ~6% of Gaucher patients [1]. The major CNS pathology in types 2 and 3 disease includes brainstem dysfunction, neuronal degeneration and neuronal loss [2–6]. Some of these patients develop a progressive encephalopathy that is neocortical in origin [6, 7]. The visceral phenotype in neuronopathic variants can range from mild to severe, and occasionally with little storage material in organs outside the brain [8]. The molecular and cellular bases for the phenotypic variation and the progression of the neuronopathic features in Gaucher disease are poorly understood.
The most severe neuronopathic variants are fatal in humans and often are caused by GBA mutations that express no acid β-glucosidase (glucocerebrosidase, GCase; EC3.2.1.45) protein [9, 10]. Few studies are available from human tissues that correlate neuronopathic progression, pathologic involvement, and the clinical manifestations [11, 12]. To fill this void, several mouse models with GCase null or point mutations or chemical inhibition were developed [13–16]. The GCase-null mutations exhibited a defective skin permeability barrier, similar to that in collodion babies [17–19]. Skin permeability barrier defects lead to death shortly after birth and have limited their utility in understanding the pathogenesis in CNS of type 2 and 3 Gaucher disease [13, 14]. In our previous studies, the presence of a neo selection marker in intron 8 of gba produced a null allele for GCase, irrespective of the introduced missense point mutation in exon 9. All these neo-containing gba-mutant mice died within 24 hours of birth with skin permeability barrier defects [16] similar to that in the full knock-out [13]. This observation led to the potential approach of producing a GCase null mouse, involving all tissues, except skin, by excising the floxed loxP-neo cassette in skin with a keratin promoter-driven Cre recombinase. Such a mouse is used here to evaluate the visceral and CNS effects of GCase null, except in specific skin cells.
The GCase irreversible inhibitor, conduritol B epoxide (CBE), has been used to chemically induce a Gaucher-like phenotype in WT mice [15]. CBE is structurally related to inositol, crosses the blood brain barrier [15], has a high degree of specificity for GCase [15, 20, 21], and can induce the biochemical [9, 22], clinical [9, 15] and histologic characteristics of Gaucher disease in neurons [3]. The CNS signs in these mice included tail arching and high amplitude action tremor suggesting gray matter lesions [15]. This chemical model could provide a conditional approach to evaluating the neuronopathic mechanisms, the relationships of the pathophysiological progress of the CNS phenotypes, and the therapeutic approaches for neuronopathic Gaucher disease.
Here, CBE was used in combination with WT or low activity (D409V, D409H and V394L) GCases to evaluate the initiation and propagation of CNS Gaucher-like lesions during GCase inhibition as well as during “recovery” to the respective endogenous GCase activity levels. Such a conditional system allows for “reconstitution” of GCase activity in all cells to the respective steady state levels derived from the mutant gba allele. This system would facilitate the evaluation of the critical threshold levels of GCase that are necessary and potentially sufficient, to reverse and/or prevent progression of CNS lesions. The defined threshold level could be used as a benchmark for enzyme reconstitution approaches to treat such disorders. Such studies show that initial CNS lesions are not reversible, even by reestablishment of the endogenous GCase activity to WT levels upon withdrawal of CBE inhibition. Many of the CBE-induced phenotypic, histological and biochemical abnormalities progress during recovery (i.e., absence of CBE) to mutant or WT level, if the accumulated substrate glucosylceramide (GC) could not be degraded by the endogenous GCases.
MATERIALS AND METHODS
Materials
The following were from commercial sources: Conduritol B epoxide (CBE, Calbiochem, San Diego, La Jolla, CA). 4-methyl-umbelliferyl-β-D-glucopyranoside (4MU-Glc; Biosynth AG, Switzerland). Sodium taurocholate and Protease Inhibitor Cocktail (Calbiochem, La Jolla, CA). Triton X-100 and primulin (Sigma, St. Louis, MO). PVDF membrane (Millipore, Bedford, MA). Alkaline phosphates (AP)-conjugated goat anti-rabbit IgG and AP-developing reagents A & B (Bio-Rad, Hercules, CA). Antibody sources were as follows: Anti-INFγ, CCL3, CD68, and IL-4 (Serotec, Raleigh, NC); Anti-Arg2 (Santa Cruz Biotechnology, Santa Cruz, CA); Goat anti-rabbit or -rat-biotinylated and streptavidin-Alexa Frou-610 (Molecular Probes, Irvine, CA); Mouse anti-neuronal nuclei (NeuN) monoclonal antibody (Chemicon, Temecula, CA); Goat anti-rabbit-FITC (ICN/CAPPEL, Aurona, OH); Rabbit anti-Cre (Novagen, Madison, WI); Goat anti-Keratin 14 (Covance, Berkeley, CA); TUNEL labeling kit (R&D system, Minneapolis, MN); ABC Vectastain and DAB peroxidase (Vector, Burlingame, CA). Rabbit anti-mGCase (mouse GCase) antiserum was raised to purified mature mGCase protein that had been expressed in pET21/BL21 system and purified by absorbance against fibroblast lysates from GCase null mice in our laboratory.
Kn-9H mice and genotyping
The kn-9H mice were generated by crossing keratin14-Cre mice [23] with GCase loxP-neo-D409H (neo-9h) mice [16]. The strain background of D409H mice was a mix of 129/SvEvBrd and C57BL/6 [16]. The keratin 14-Cre mice were in the 129Ola/C57BL/6 background. Kn-9H was obtained from keratin 14-Cre and D409H mice crosses. For CBE studies, wild type FVB mice were from Jackson Laboratory (FVB/NJ, Stock No 001800). Other point mutant mice V394L and D409V were in the same protein background as the D409H mice. The mice were housed under pathogen-free conditions at the animal facility of Cincinnati Children's Hospital Research Foundation. Mice were genotyped using PCR systems for Cre [23], or neo and 9H as described [16]. The identity of the PCR products from the 9H allele, K14-Cre transgene and neo cassette were verified by DNA sequencing.
Mice treated with CBE
GCase point-mutated mice D409V (9V), D409H (9H), V394L (4L) and wild type (WT) mice were injected intraperitoneally with 100 mg CBE/kg body weight/day [15]. Injections were initiated at postnatal day 5 or 15 and continued for 6 to 11 doses. The mice were sacrificed either 3–4 hours or 24 hours after last CBE injection. Mice were perfused with PBS prior to organ harvesting for enzyme activity assays, lipid analyses, and histological preparation.
GCase enzyme activity assay
For GCase activities, tissues (~50 mg) were homogenized 1:10 [w(mg):v(µl)] in 1% sodium taurocholate/Triton-X100, sonicated (4°C, 30s × 3), and clarified (10,000×g; 5 min, 4°C) [24]. The assay mixtures were pre-incubated in the presence or absence of CBE (1 mM; 30 min, 37°C), then substrate (4MU-Glc) was added, and the reactions were stopped after incubation (1 hr, 37°C). Protein concentrations were determined using the BCA kit according to manufacturer’s instructions.
Tissue lipid analyses
Lipid analyses were as described [16]. Tissue samples (20 mg wet weight) were analyzed in triplicates. Relative proportions of lipids were determined in tissue aliquots (100 µg wet weight) after resolution by [chloroform/methanol/water (65:25:4, v/v/v)] thin-layer chromatography using borate impregnated plates (TLC, 10 cm2 Merck HPTLC silica gel 60, 200µm) [25]. Lipids were visualized with primulin (100 mg/l; 80% acetone) and quantified with blue fluorescence scanning (Storm 860, Amersham Pharmacia Biotech).
Histological studies
For histological studies, 3 to 4 mice from each group and minimum of two sections were examined from each tissue. For light microscopic studies organs (brain, liver, lung, thymus, spinal cord, spleen, and skin) were dissected after the mice had been perfused with PBS and the tissues fixed in 10% buffered formalin and embedded in paraffin for H&E staining. For immunohistochemistry or immunofluorescence studies, tissues were fixed with 4% paraformaldehyde in PBS, pH 7.4. Sections were incubated (overnight, 4°C) with the desired antibodies in PBS containing 5% goat serum. Second antibody-conjugated FITC or biotin-streptavidin were used as the reporters. Detection was with the ABC Vectastain and DAB peroxidase substrate for immunohistochemistry or fluorescein for immunofluorescence studies.
RESULTS
Phenotype of Kn-9H mice
The neo-9h construct contained the neo selection marker inserted in intron 8 of gba in all tissues and produced a null phenotype (Fig. 1 [16]). The expressed Cre driven by the K14 promoter excised loxP-neo cassette from the gba locus (Fig. 1) in selected cells of the skin, but not in other tissues. This led to the D409H allele being expressed only in skin and producing a GCase enzyme with low activity [16], whereas the presence of the neo in the D409H allele in all other tissues prevented the expression of an active enzyme. In the absence of the K14-Cre, all 9H-neo homozygotes were similar to the gba complete knock-outs and died in the first few hours of life [13]. The neo cassette (~1,850 bp) in gba produced a null allele without gba mRNA production (data not shown). Genotyping of kn-9H homozygotes showed Cre (~300 bp) was present in all tissues. The fragments corresponding to the 9H mutation (485 bp) were present in tail DNA, but not in that from internal organs (Fig. 1B and 1C). In skin, excision of the neo-loxP sequence by K14-Cre left a 94 bp loxP junction sequence in intron 8 of gba alleles together with the 9H point mutation in exon 9. PCR generated a 391 bp fragment from wild type (WT) gba DNA (Fig. 1C, bottom band), and a 485 bp fragment (391 + 94 bp lox-P junction sequence) from 9H point mutated gba DNA (Fig. 1C, top band).
Figure 1. Schematic of mice containing k14-Cre/neo-9h gene constructs and PCR genotyping.
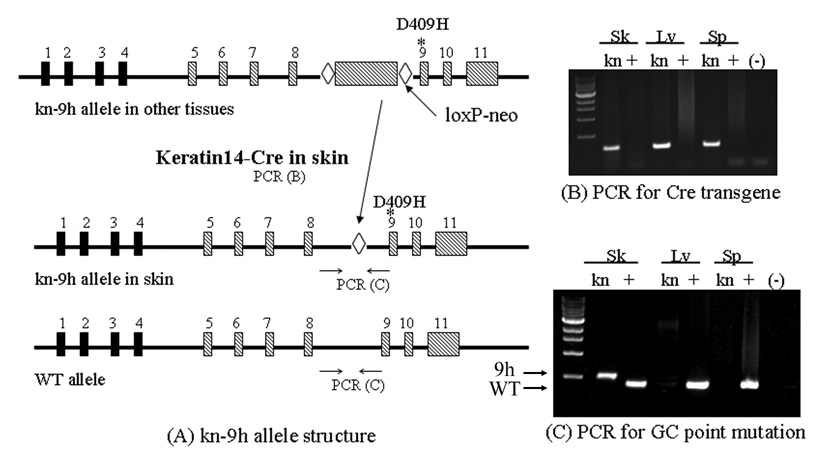
(A) The gba structure of k14-Cre/neo-D409H (kn-9H) mice containing the loxP-neo in intron 8 gba with a D409H encoding point mutation in exon 9(*) (top line). The neo was excised in skin by k14-Cre (second line). The resultant recombinant D409H allele (*) contained one loxP (◊) site only in skin; the other tissues retain neo. WT gba is shown in the lower diagram. The B and C panels show the PCR genotyping for Cre transgenes. (B) A 400 bp PCR Cre containing fragment was in tissues of kn-9H (kn) mice, but not in WT (+) mice. (C) PCR genotyping shows that the mutant D409H allele (9H) was only re-activated in skin, but not in other tissues. The 391 bp PCR fragment (bottom band) is from WT (+) and the 485 bp PCR fragment (upper band) is from the skin of kn-9H (391+loxP, D409H) gba mice. Sk: skin; Lv: liver; and Sp: spleen. The (−) lanes are no template DNA controls.
Kn-9H newborn pups (Fig. 2B) had normal skin compared with the red wrinkled, friable skin in neo-9h (Fig. 2A) or complete knock-out newborn pups. From 236 newborn pups, 40 (17%) were kn-9H homozygotes and ~20% of these survived until the age of 14 days. All kn-9H pups were small (Fig. 2C) with a mean body weight of ~52% (n=5) of WT littermates (n=8). The weights of the brain, liver, lung, or spleen in kn-9H (n=4) were 68, 45, 45, or 20% of the respective WT weights (n=7).
Figure 2. kn–9H mouse phenotype.
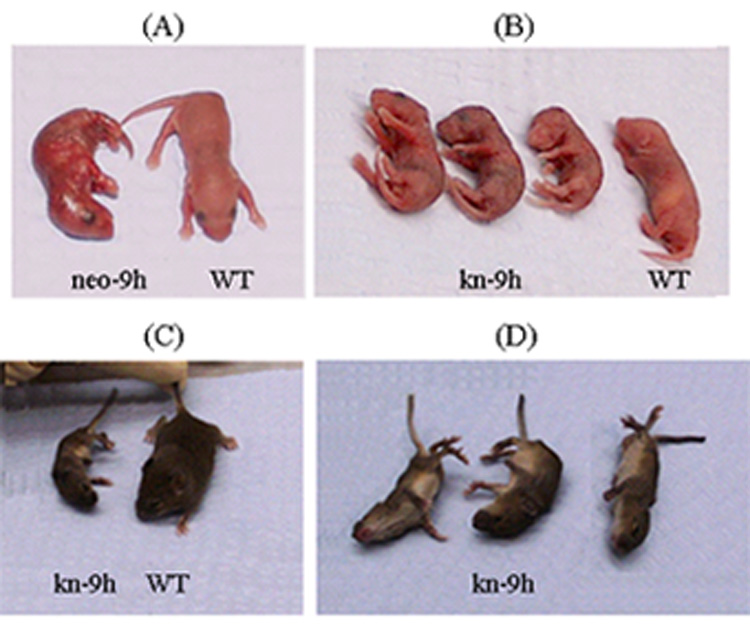
(A) Homozygote that had the neogene in intron 8 (neo-9h/neo-9h) (Fig. 1) pups was similar to the gba null mouse [13] with red-wrinkled dry skin and death after birth. (B) kn-9H pups (i.e., neo in intron 8 was present in all tissues but skin) were smaller than WT mice and had nearly normal skin. (C and D) These mice survived to the age of 14 days, but were small and severely neurologically impaired. They often flipped on their side and displayed tail arching, tremor, myoclonus and tonic/clonic seizures.
The surviving kn-9H homozygotes were generally normal until ~10 days of age. They then developed progressive slowing of movements, a wobbly gait, tremor, tail arching, myoclonus, spastic paraparesis, and seizures (Fig. 2C and D) with death by 14 days. Thus, the K14 driven Cre “reactivated” the 9H allele in selected skin cells allowed expression of the mutant enzyme (see below), and re-established the skin permeability barrier. The observed ratio of kn-9h homozygotes was 17% that is close to the expected Mendelian ratios of 25%. These mice were small and sick, and often not taken care of or eaten by mother even on the days 2 or 3 post birth. To increase the survival rate, the most normal appearing pups (kn-9h-neo/WT and WT) were provided foster mothers to reduce the mother-care competition. Although the survival rate was low and the life span was short, a fully developed phenotype of GCase was present in null in visceral organs and the CNS. The low neonatal survivability and shortened life span (14 days) of kn-9H homozygotes likely relates to the absolute deficiency of GCase function in all other tissues.
Expression of GCase in skin of Kn-9H mice
Expression of Cre and mGCase in the skin of kn-9H homozygotes was detected by immunofluorescence staining with anti-Cre, anti–keratin14 or anti-mGCase antibodies. The results showed that Cre was co-expressed with mGCase and k14 in selected skin cells of the kn-9H homozygotes (Fig. 3, top and middle panels). In control experiments, Cre was not detected in skin of non-transgenic WT mice (Fig. 3, bottom). Using immunofluorescence, mGCase signals were not present in other tissues from kn-9H homozygotes, but were clearly evident in the corresponding WT littermate tissues (Fig. 4). The kn-9H homozygotes were used as genetic controls for the studies with CBE.
Figure 3. Immunofluorescence analyses of skin from kn-9H or WT mice.
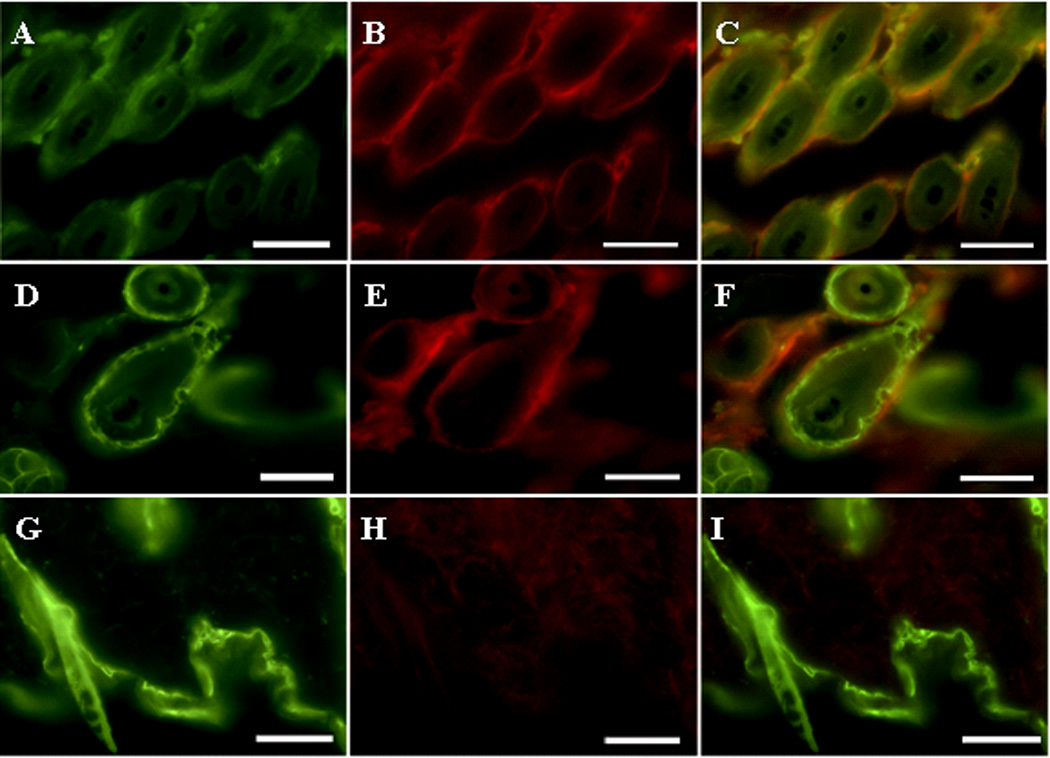
Biopsies were analyzed using anti-mouse GCase (mGCase) or anti-Cre, or -K14, antibodies. (A–C) mGCase signals (A, FITC) and Cre (B, Alexa-610) were present in skin cells of kn-9H mice. The merged images (C) show co-localization of Cre and mGCase (i.e., the D409H protein). K14 (D, FITC) and Cre signals (E, Alexa-610) were detected in skin of K14-Cre mice. Panel F merges D & E. In WT mice, only K14 (G), but no Cre (H) signals were detected. The magnifications are 400X ; scale bar, 50 µm.
Figure 4. Expression of mGCase in kn-9H and WT mice.
Lung, liver, cerebellum and cerebrum sections from kn-9H (A–D) or WT (E–H) were treated with rabbit anti-mGCase antiserum for immunofluorescence, respectively. The mGCase positive signals (red) were detected in tissues from WT (E–H), but not from kn-9H (A–D). Cell nuclei were counterstained with DAPI (blue). The magnifications are 400X; scale bar, 50 µm.
Phenotypes of mice treated with CBE
Two age groups (5- and 15-day old) of 9V, 9H, 4L, and WT mice were injected intraperitoneally with CBE (100 mg/kg/day). The 5-day old 9V, 9H or WT mice injected with CBE for 10–11 days displayed a neuronopathic phenotype similar to that of kn-9H homozygotes, and died by day 11–12 of neurological disease. In this age group, the 9V, 9H and WT mice that received only 6 daily CBE injections had different phenotypes than those obtained with 10–11 daily injections. Following the 6th CBE injection, without additional injections, these three mouse genotypes experienced a 3–4 day latency of little or no CNS signs. By day 4–5 post-CBE obvious CNS signs developed and included myoclonus, tremor, and incoordination (Fig. 5). These initial signs persisted in the 9V mice for an additional 2–3 days. Increasing shakiness of the head and limbs, wobbling gait, paresis and dragging of the rear legs and incoordination of the forelimbs followed this period. These signs progressed slowly so that by 2 months following discontinuation of CBE, the tremor was much greater, and complete paralysis of the hind limbs was apparent. These signs persisted in the 9V mice with increasing disability until sacrifice for ethical reasons at 2 to 5 months. In comparison, similarly treated WT and 9H mice experienced gradual reduction in the initial signs, but the general shakiness/tremor persisted until sacrifice at 5 mos for control purposes.
Figure 5. CNS phenotypes of CBE-injected mice.
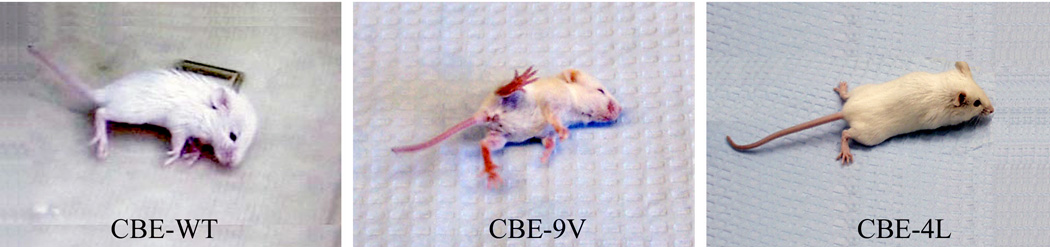
After 6 daily injections (CBE; 100 mg/kg/day) beginning at post-partum day 5, WT and 9V mice displayed similar CNS signs to those of kn-9H mice. The CBE-4L mouse only showed paresis of the hind limbs after 24 injections.
A separate group of 15 day old 9V or WT mice received 8 or 9 daily injections of CBE. These mice rapidly became very ill with phenotypes that included difficulty moving, paralysis of the hind- and fore- limbs, and frequent generalized tonic/clonic seizures. The 9V or WT mice died within 2 or 7–12 days, respectively, after last CBE injection, i.e., the CBE treated 15-day old WT mice survived longer after CBE was stopped, but they had the same general phenotype.
Additional experiments were conducted in 5- and 15-day old 4L mice. The 5-day old mice, who received 9 CBE injections, showed minor tremor. The 15-day old mice did not develop a CNS phenotype after 9 injections (100 mg/kg/day). However, after 24–36 injections of CBE (100 mg/kg/day), the 4L mice manifested a wide-based gait and rear limb paresis (Fig. 5). In an additional group of 15-day old 4L mice, doses were increased to 1000 mg CBE/kg/day. The severe CNS signs that appeared after 8 doses were nearly identical to those in the 5-day old 9V, 9H or WT mice that had received CBE (100mg/kg/day) for 10–11 doses. Importantly, no other gross phenotypic or histological evidence for CBE toxicity was found, i.e., CBE even at high doses was quite specific to GCase. In vitro analyses showed that the inhibition (IC50) of purified 4L GCase required 3 to 4 fold higher concentrations than the WT, 9H or 9V GCase [26]. This probably explained the higher doses needed to induce the CNS signs in the 4L mice. These studies show that the CBE effect was genotype and somewhat age related, and that the induced GCase deficiency caused CNS manifestations that were very similar in young mice to the GCase null in all organs, except skin, of the kn-9H homozygotes. In addition, these findings show that the CNS phenotypes once established were irreversible even if endogenous WT or mutant GCase is fully reconstituted by endogenous synthesis in a natural genomic setting.
GCase protein and enzyme activities in kn-9H and CBE mice
GCase activities in kn-9H mice, neo-9h and gba null mice were similar in all internal tissues (Table 1). In comparison, tissue GCase activities in 9H homozygotes were somewhat higher [16]. The presence of variable amounts of other β-glucosidase (i.e., non-CBE inhibitable) activities in all tissues obscured perfect correlations of genotype and GCase activity even in the presence of CBE [27].
Table 1.
GCase Residual Activity in Tissues from kn-9H and neo-9h Mice
| Genotype | Brain | Liver | Lung | |||
|---|---|---|---|---|---|---|
| nmoles/h/mg 1 | %2 | nmoles/h/mg | % | nmoles/h/mg | % | |
| WT (n=10–13) |
28.83 (±1.37) | 100 | 28.72 (±1.43) | 100 | 43.13 (±4.5) | 100 |
| kn-9H/kn-9H (n=5) |
2.78 (±0.14) | 9.6 | 0.89 (±0.09) | 3.1 | 0.73 (±0.12) | 1.7 |
|
neo-9h/neo-9h (n=5) |
2.50 (±0.16) | 8.7 | 0.92 (±0.09) | 3.2 | 0.70 (±0.22) | 1.6 |
| GCase null (n=5) |
2.77 (±0.53) | 9.6 | 0.88 (±0.119) | 3.1 | 0.87 (±0.23) | 2.0 |
| D409H/D409H (n=9) |
7.38 (±0.29) | 25.6 | 3.29 (±0.16) | 11.4 | 3.26 (±0.36) | 7.6 |
GCase activity (±SEM)
% WT activity.
Tissue GCase activity levels in CBE injected mice were similar to those from kn-9H mice (Table 2). Tissues from CBE injected mice were collected 3 or 24 hrs after a CBE injection. By 24 hr the CBE-9V mice (n=7) had GCase recoveries to ~37% (brain), ~64% liver and ~100% lung of pretreatment 9V GCase levels. In comparison, by 24 hr WT GCase recovered to ~10% (brain), ~20% (liver), or ~15% (lung), whereas 9H GCase recovered to ~51% (brain), ~83% (liver), or ~83% (lung) of pretreatment 9H GCase activity levels by 24 hr (Table 2). Thus, the recovery time to baseline endogenous levels were more rapid or similar to WT in the 9V and 9H mice, but the total activities at 24 hr were significantly greater in WT organs. To estimate the recovery rate of GCase in these tissues, the GCase activities in CBE-WT or CBE-9V treated mice also were determined at 3 hours (n=5) after CBE injection (Table 2). Since the clearance of CBE in mouse tissues is ~5 hr [28], the tissue GCase activities at 3 hours after injection reflected the free GCase; i.e., not in the GCase-CBE covalent complex. Because the inhibition of GCase by CBE is irreversible, the difference of GCase activity between 3 hours and next day results from newly synthesized GCase over a 24 hour period. The data showed that the recovery rate of GCase varied from tissue to tissue in the sequence of high to low in the following order liver>lung>brain in CBE injected 9V and WT mice (Table 2). Furthermore, the attainment of full reconstitution to pretreatment levels was slower in WT than in 9V, due to the more rapid turnover of the D409V enzyme. However, the exposure of WT tissues to GCase activity levels was greater than those in 9V within the first 24 hr after CBE was stopped.
Table 2.
Recovery of GCase Activity after CBE Injections
| Genotype | Injections/ | Brain | Liver | Lung | |||
|---|---|---|---|---|---|---|---|
| Harvest 1 | nmoles/h/mg 2 | %3 | nmoles/h/mg | % | nmoles/h/mg | % | |
| WT-CBE (n=5) |
3/3 hr | 1.30(±0.35), | 4.5 | 0.79(±0.13), | 2.8 | 0.99 (±0.09), | 2.3 |
| WT-CBE (n=10) |
6/24 hr | 2.95 (±0.25), | 10.2 | 5.82 (±0.52), | 20.3 | 6.52 (±1.10), | 15.1 |
| 9V-CBE (n=5) |
3/3 hr | 0.63 (±0.04), | 2.2 | 0.17 (±0.04), | 0.6 | 0.14 (±0.06), | 0.3 |
| 9V-CBE (n=7) |
6/24 hr | 2.09 (±0.16), | 7.2 | 1.21 (±0.20), | 4.2 | 1.48 (±0.10), | 3.4 |
| 9H-CBE (n=5) |
3/3 hr | 1.44 (±0.07), | 5.0 | 0.39 (±0.05), | 1.4 | 0.84 (±0.0.10), | 1.9 |
| 9H-CBE (n=8) |
6/24 hr | 3.78 (±0.22), | 13.1 | 2.73 (±0.15), | 9.5 | 2.70 (±0.0.22), | 6.3 |
| 9V/9V (n=9) |
No-CBE | 5.56 (±0.52), | 19.1 | 1.86 (±0.25), | 6.5 | 1.67 (±0.24), | 3.9 |
Injections=number of 100 mg/kg/day single dose injections; Harvest=time (hours) of tissue harvest following the injection of CBE.
GCase activity (±SEM).
% of non-CBE treated WT mouse GCase activity (see in Table 1).
Histology of kn-9H and CBE mice
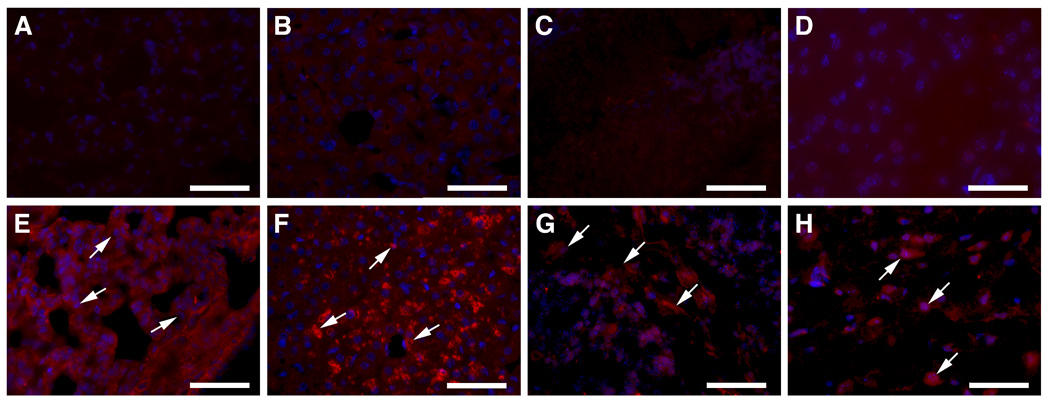
Storage cells were present in all tissues from four kn-9H homozygotes by the age of 14 days (Fig. 6). These cells were positive for the macrophage cell surface antigen, CD68 (Fig. 6, I to L). The data shown in Figure 6 is representative for all examined mice. Such cells are not present in WT (Fig. 6), 9H or 9V tissues at this age [16]. In CBE-treated 9V or WT mice, storage cells were not observed in visceral organs. However, increased numbers of CD68 positive cells were detected in liver, lung, thymus and BM after 6 injections in CBE-9V (Fig. 7) or CBE–WT mice (data not shown), compared to non-treated mice. Moderate numbers of CD68 cells were present in these tissues 2 mos after the 6 injections were discontinued in CBE-9V mice (Fig. 7I–L) and to a much lesser degree in the corresponding CBE–WT mice (data not shown). The response of tissue macrophages to CBE-induced GCase deficiency varied in different tissues. The CD68 signals were significant and the cell size was larger in liver and thymus compared with that in lung and bone marrow after treatment by 6 CBE injections (Fig. 7E–H). During the recovery phase the CD68 signals were weak and the cell size was smaller in liver and lung at 2 month’s after CBE withdrawal (Fig. 7I–J). But, the morphology of CD68 positive macrophages in thymus and bone marrow was maintained and unchanged for 2 months after CBE withdrawal (Fig. 7K–L). The differential response of macrophages to CBE-induced GCase defect may be tissue type related, due to the differential functions of these tissue-specific macrophages. Likely, liver and lung macrophages have mostly phagocytosis and cleavage functions, whereas BM monocytes/macrophages could function mainly to supply precursors to support macrophage needs.
Figure 6. Histological and immunochemical analyses of kn-9H mouse visceral tissues.
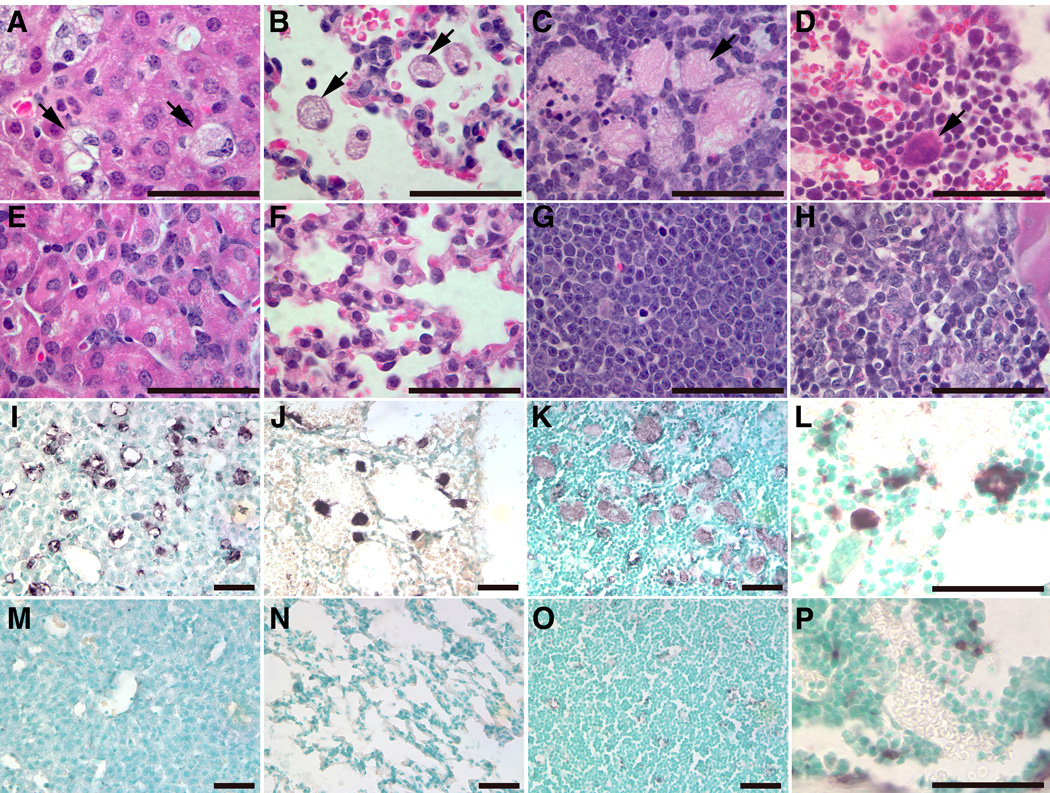
Storage cells (arrows) were in liver (A), lung (B), thymus (C) and (D) bone marrow (BM) from 13 day old kn-9H mice, but not in littermate WT control tissues (E–H). The magnifications of H&E stained sections (A–H) are 1000X (Scale bar, 50 µm). CD68 positive macrophages (stained as dark brown) were detected in liver (I), lung (J), thymus (K), or BM (L) of kn-9H mice, but not in matched littermate WT control tissues (M–P). The magnifications of CD68 stained sections are 400X for liver, lung and thymus, and 1000X for BM (Scale bar, 50 µm).
Figure 7. Macrophage presence in visceral tissues of CBE-injected D409V mice.
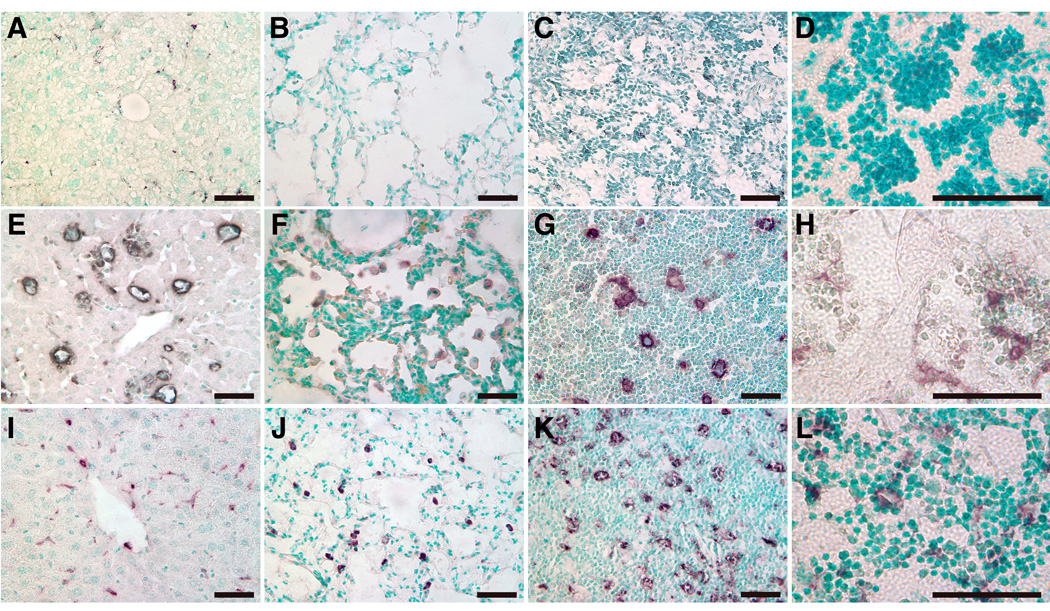
Liver, lung, thymus, or BM from CBE-injected 9V mice with 6 doses (middle row, E–H) or 2-month after the 6 doses (bottom row, I–L) were analyzed with anti-CD68 antibody. The CD68 positive cells were stained as dark brown. No-CBE controls showed absence of CD68 positivity (top row, A–D). The magnifications are 400X for liver, lung and thymus, and 1000X for BM. Scale bar, 50 µm.
CNS lesions and neuronal loss
In kn-9H homozygotes, degenerate neuronal cells were observed in cerebrum and spinal cord (Fig. 8A and 8G). Lesser numbers were in cerebellum (not shown). No abnormalities were observed in WT brains or spinal cords (Fig. 8D and 8J). In kn-9H, but not WT (Fig. 8E and 8K), many CD68 positive microglial cells were present in cerebrum and spinal cord (Fig. 8B and 8H), and lesser numbers in cerebellum. TUNEL staining showed numerous apoptotic cells in spinal cord (Fig. 8I), and fewer in the cerebrum (Fig. 8C). These CNS abnormalities were not observed in 9H homozygotes (not shown). TUNEL positivity was not observed in lung and liver, suggesting differential sensitivity of CNS cells, particularly spinal cord neuronal cells, to the GCase deficiency and lipid storage. Similarly, CBE-injected mice showed neuronal degeneration and death. In CBE-9V mice, many degenerate neurons were detected in cerebrum and spinal cord (Fig. 9), and a few in cerebellum (data not shown). The neuronal cells show progressive deterioration by 2 months after the 6 initial injections (Fig. 9). Similar CNS histology, but much less extensive lesions were observed in CBE-9H or CBE-WT mice (data not shown). These observations indicate a progressive CNS lesions and neuronal loss caused by a complete GCase deficiency. These findings also indicate a progressive neuronal loss was related to the recovery level of GCase, but that the final return to even WT levels did not completely alleviate the phenotype or histologic abnormalities.
Figure 8. Histological and immunochemical analyses of kn-9H mouse CNS.
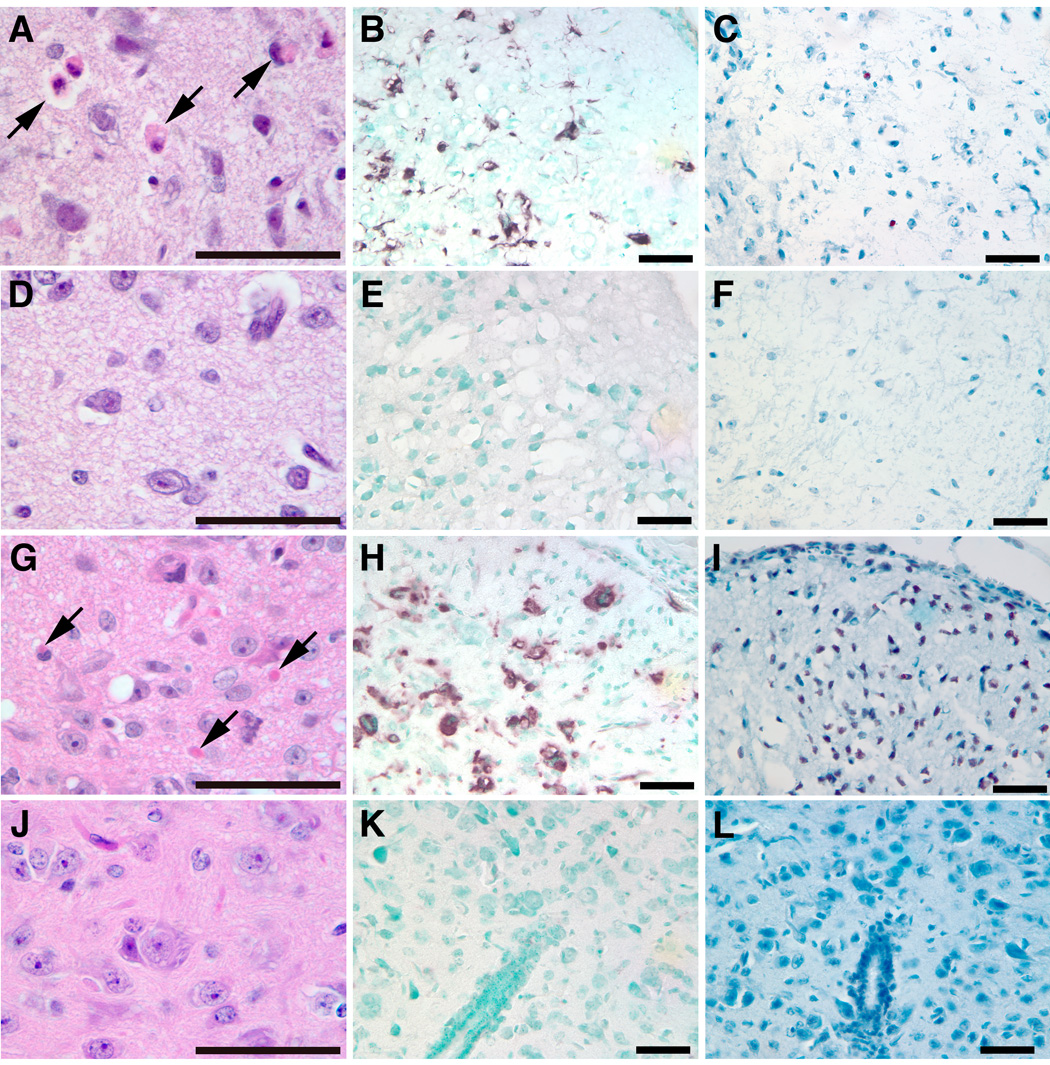
Degenerated neuron cells (arrows) were observed in cerebrum (A) or spinal cord (G) of day-13 kn-9H mice, but not in WT mice (D or J). CD68 positive microglial cells were present in cerebrum (B) and spinal cord (H) of kn-9H mice (dark brown cells), but not in WT control tissues (E or K). Many TUNEL positive cells (dark brown cells) were in the spinal cord (I), but only a few in cerebrum (C) of kn-9H mice. In WT (F or L) control mice, no neuronal degeneration, microglial response or apoptosis was observed in the cerebrum and spinal cord.
Figure 9. Neuronal lesions in CBE-9V mouse CNS.
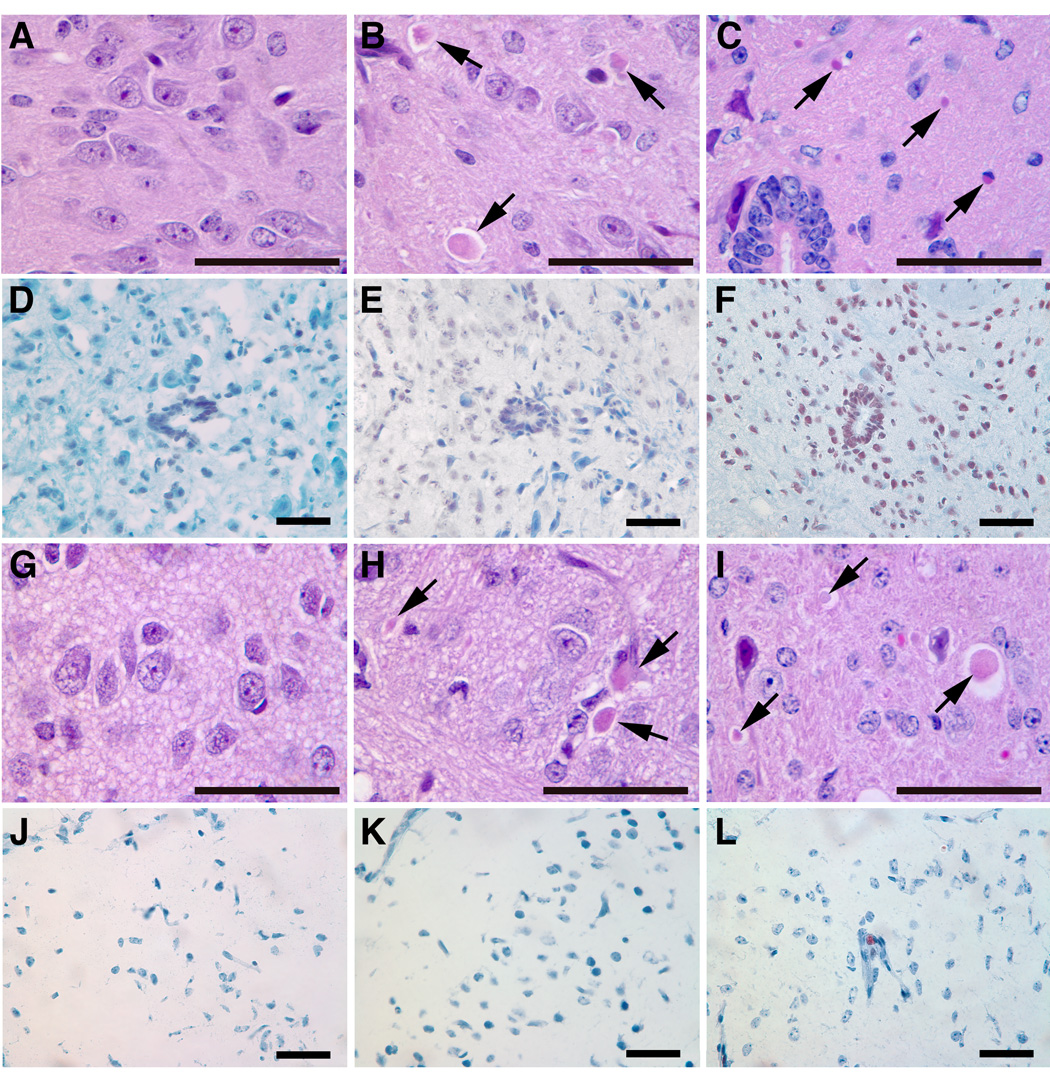
Spinal cord and cerebral sections from CBE-9V mice after 6 injections (middle column, B, E, H and K) or 2 month after the initial 6 injections of CBE (right column, C, F, I and L). Degenerated neuronal cells are shown by arrows and TUNEL positive cells are shown as dark brown. The corresponding no-CBE controls are in left column (A, D, G and J). The magnifications are 400X; scale bar, 50 µm.
The degenerating cells stained with anti-NeuN antibody, a neuron-specific nuclear protein [29, 30]. This antibody was used to quantify neuronal cell populations in the spinal cord of kn-9H homozygotes. The NeuN positive cells in kn-9H spinal cord (thoracic spine) were [900, 832 and 768/~5100 µm2 (n=3)] per total coronal section of spinal cord or ~65% of age matched WT littermates [1312, 1170 and 1250 (n=3)]. Similar levels of neuronal loss were observed in thoracic spinal cord of CBE injected mice. With longer CBE treatment courses (8~10 injections), more severe neuron loss was observed in 9V or WT mice (data not shown). These studies indicate an irreversible and progressive neuronal loss after initial lesions induced by the GCase deficiency (null or CBE induced) and even after return of the endogenous steady state 9V GCase levels for 2 to 5 months.
Pro-inflammatory and anti-inflammatory cytokines in CNS
To investigate the microglial activation in the CNS, the expression of selected pro-inflammatory and anti-inflammatory cytokines were evaluated by immunofluorescence. Moderate levels of the pro-inflammatory cytokine, CCL3, were detected in spinal cord of kn-9H homozygotes at the age of 13 days (Fig. 10). Weak signals for the pro-inflammatory cytokine, INFγ, or the anti-inflammatory cytokines (IL-4 and arginase 2) were in spinal cord, cerebrum and cerebellum of kn-9H homozygotes (data not shown). The pro-and anti-inflammatory cytokines co-localized with the CD68 signals (Fig. 10) indicating the microglial expression of these cytokines in the CNS and the involvement of pro-and anti-inflammatory factors in the pathophysiology of CNS lesions.
Figure 10. Expression of CCL3 and CD68 in CNS of kn-9H mice.
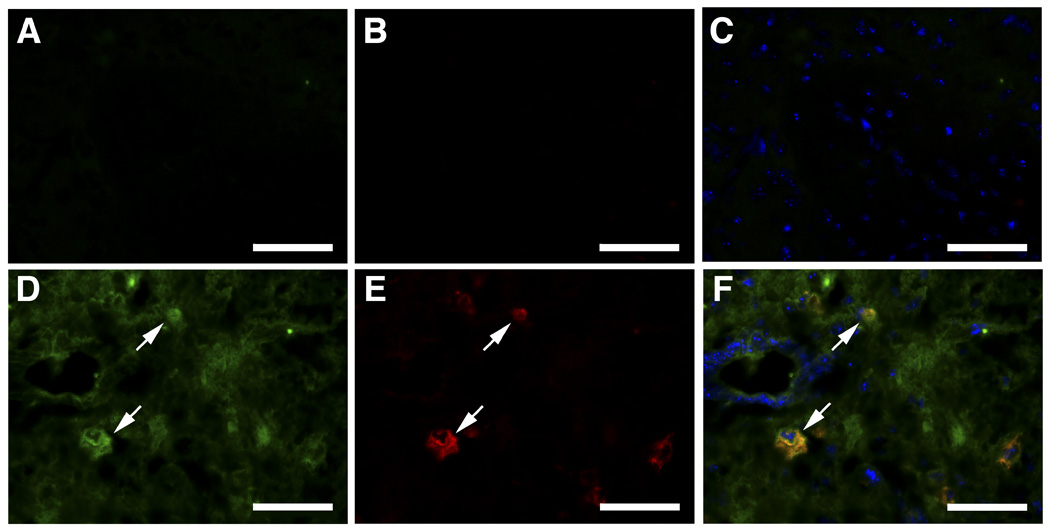
Spinal cord sections from kn-9H (bottom, D–F) and WT (top, A–C) mice were stained for CCL3 (FITC green, D, arrows) or anti-CD68 (Alexa-610 red, E, arrows). The co-localization of CD68 and CCL3 is shown in merged image (yellow, F, arrows). The magnifications are 400X; scale bar, 50 µm.
Tissue lipid analysis
Glucosylceramide accumulated in tissues of kn-9H and CBE treated mice. The 13-day old kn-9H mice had large glucosylceramide accumulations in brain, lung and kidney (Fig. 11A). In 9H homozygotes, little glucosylceramide accumulation was observed in brains or lungs at this age [16]. CBE treatment (6 injections) of WT, 9H or 9V mice caused accumulation of glucosylceramide in brain (Fig. 11B). In the absence of CBE, glucosylceramide was nearly undetectable. Significant levels of glucosylceramide persisted for 2 to 5 months after the initial 6 injections were stopped in CBE-9V mice, but not in CBE-WT (Fig. 11B) or CBE-9H (not shown) brains. Thus, the CNS phenotype of kn-9H homozygotes and CBE treated mice were related to the levels of glucosylceramide accumulation.
Figure 11. Glycosphingolipid (GSL) TLC profiles from kn-9H and CBE-treated mouse tissues.
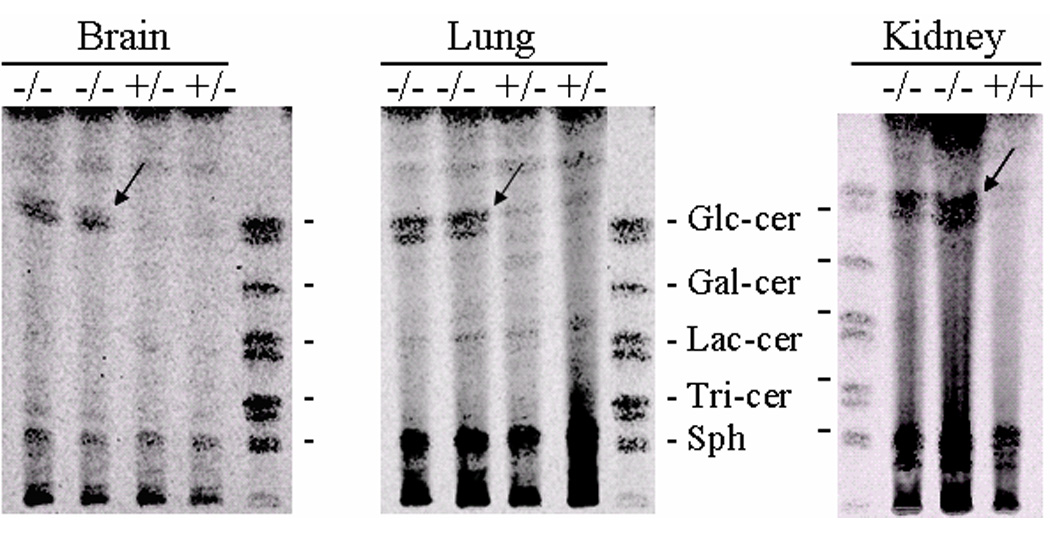
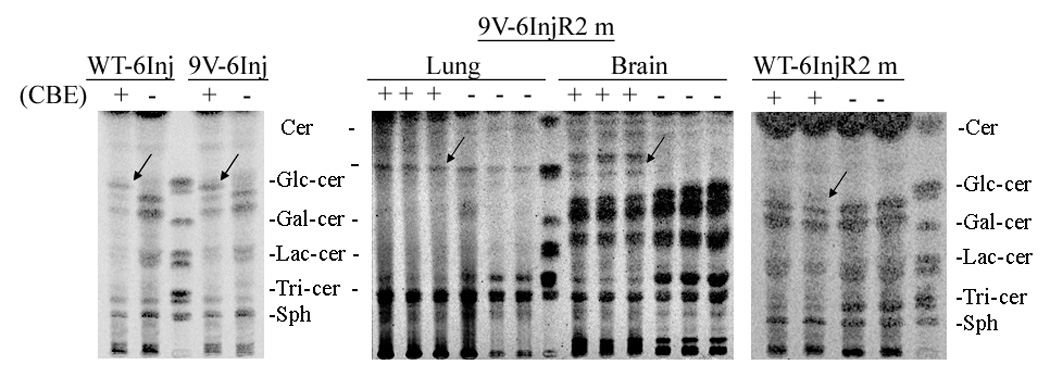
(A) GSLs of brain, lung, and kidney tissues from 13 day kn-9H (−/−) or littermates (+/+ or +/−) mice. Glucosylceramide was in many fold excess in kn-9H tissues compared to the unaffected counterpart (almost undetectable). (B) GSLs in brains from CBE-9V or CBE-WT mice. Mice were sacrificed after 6 injections (6Inj) of CBE or sacrificed 2 months after CBE injections (6InjR2m). CBE+ or − indicates injected or un-injected age and genotype matched mice. CBE-WT and CBE-9V show greatly and similarly increased GC after 6Inj (left panel). Large amounts of GC persisted in the brain of three 9V-6InjR2m mice (middle panel), whereas, little, if any, GC was present in matched WT-6InjR2m brains (right panel). GC in lung of 9V-6InjR2m was increased compared the untreated 9V mutant mice (middle panel). The 9V-6InjR2m mice that did not receive CBE show no GC accumulation in brain (middle panel). Arrows indicate the glucosylceramide in tissues. Migration standards are made from visceral organ spleen. These are GlcCer, glucosylceramide; GalCer, galactosylceramide; LacCer, lactosylceramide; Tri-cer, ceramide trihexoside; Sph, sphingomyelin.
DISCUSSION
Essential to the treatment of lysosomal storage diseases is an understanding of the differential involvement and reversibility of the induced lesions in various tissues. This is a pan-therapeutic issue, including enzyme therapy, gene transfer approaches, substrate reduction techniques and pharmacological chaperone methods [31–38]. Furthermore, the timing of such interventions compared to the onset of disease manifestations may have substantial impact on patient outcome. These issues are also important for newborn screening programs directed at the detection of affected individuals with lysosomal storage diseases [39]. Clearly, over the past decade, the use of enzyme therapy for the treatment of Gaucher disease type 1 has led to a remarkable change in the outcomes for affected individuals [31, 35]. This dramatic improvement in hematologic, hepatic and splenic manifestations has been tempered by the less than robust response of established architectural bone disease in affected adults. In comparison, the use of enzyme therapy in similar cohorts of children has indicated that early intervention and thereby, potential treatment of early lesions, can prevent or at least substantially diminish or delay the development of irreversible bony lesions. These lessons are clearly important for the development of new and/or adjunctive therapies that maybe less expensive than or provide added benefit to enzyme therapy for the treatments of Gaucher disease, Fabry disease, Pompe disease, selected mucopolysaccharidoses and other lysosomal storage diseases [40–46]. These concerns have the most importance for the relatively inaccessible CNS compartment.
For the CNS, there has been debate and concern over the use of varying enzyme doses for treatment of the neuropathic variants of Gaucher disease [47–50]. This led to anecdotal observations relating to the response of the CNS to high doses of enzyme or other approaches [51]. However, no substantive studies are available to access the potential reversibility and/or impact of initial therapy at an early age in either humans or mouse models of this disease. In the current studies, a complete knock out of GCase function in all tissues, except skin, was developed to provide a mouse model that has short term survival (14 days), and dies of overwhelming Gaucher disease in the visceral and CNS organs. By using the K14 promoter to drive Cre and restore some residual activity to the skin, this model showed substantive accumulations of glucosylceramide in the brain and visceral organs by 14 days of age. This genetic model allowed for evaluation of the effects of complete GCase deficiency in internal organs that were encumbered by the lethal defective skin permeability barrier [13]. It should be noted that a complete knock out of the GCase activity in macrophages only, leads to a late onset (7–10 months) visceral variant of Gaucher disease [52]. The fact that such a prolonged period is required for accumulation of glucosylceramide in the macrophage, compared to just 14 days for massive accumulation of glucosylceramide in the kn-9H model, indicates the contribution of all cell types to the storage material in Gaucher disease. In this study, 9H-neo mice were used to develop a live GCase null mouse model, although the survival times were short. As shown in our previous studies, 9H-neo is null because the neo cassette interfered with GCase RNA splicing to mature RNA [16]. Cre expression driven by K14 promoter in skin re-activated null GCase 9H-neo to a low activity 9H GCase that maintains the skin permeability function as well as the WT GCase. The low neonatal survivability and shortened life span (14 days) of kn-9H homozygotes are similar to that in severe neuronopathic Gaucher disease of humans.
The kn-9H homozygote model also provided a genetic control for the chemically induced abnormalities that were obtained with CBE. Indeed, the kn-9H homozygote was mimicked by CBE by treatment for 10–14 days with complete recapitulation of the CNS abnormalities. Although CBE can potentially act on other glycosidases here it was highly specific and provides a reliable and informative phenotype. Moreover, the 4L homozygote required 10 times the dose of CBE to induce similar CNS abnormalities. This is due to the mutant V394L enzyme having a decreased interaction with CBE [26]. As such, the 4L experiments provide additional confidence that the majority, if not all, of the abnormalities within the CBE induced models were due to selective GCase inhibition and glucosylceramide accumulation, rather than non-specific effects.
The treatment of 9V, 9H or WT mice for 6 days with CBE followed by a “recovery,” provided analyses of the CNS abnormalities that could be induced acutely by glucosylceramide accumulation and inhibition of GCase. This early treatment of 9V, 9H or WT with CBE mice showed that the CNS abnormalities, phenotypically, histologically, biochemically and GC accumulation, were similar to those in the genetically deficient kn-9H model. The differences between each of these CBE-induced models occurred in the “recovery phases.” The WT and 9H CBE treated mice [6-days] never showed progressive CNS disease and did not require sacrifice due to ethical considerations. However, they did retain the initial induced CNS abnormalities that consisted mostly of shakiness and mild ataxia. The lack of progression correlated with the disappearance of glucosylceramide from CNS tissues, and a diminution in microglial cell activation and TUNEL positivity. In comparison, the CBE-treated 9V mice experienced progressive CNS disease during the initial period following CBE withdrawal, and then a slowly progressive CNS disorder over a period of 2 to 5 months that required humane sacrifice. These mice, in comparison to WT and 9H, showed significant persistence of glucosylceramide storage for two to five months after withdrawal of CBE and continued significant microglial cell activation.
These experiments provide some insight into the level of residual activity that may be required for return to biochemical normalcy in the CNS of these mice. Clearly, the level obtained by 9V, was insufficient to clear glucosylceramide from the brain, whereas, wild type and 9H did clear the stored material. Thus, the level of 9H enzyme in the CNS suggests a threshold for sufficient amounts of activity present continuously needed to prevent progression of CNS disease. Thus, a level of total exposure at or above that attained by the 9H enzyme would be needed globally at least on an intermittent basis, to prevent progression. The WT or 9H levels of enzyme would be equivalent to either low dose or high dose enzyme or gene therapy studies in the presence of a continuous production of glucosylceramide from endogenous sources in the steady state environment. Clearly, these studies establish an initial threshold above that obtainable by 9V, for at least biochemical normalization of the CNS. What is equally clear is the irreversibility of existing lesions within the CNS, the stabilization of these with WT or 9H enzyme levels, and the progression of histologic and phenotypic lesions in the CNS at 9V levels. Clearly, attempts to establish therapeutic efficacy will require direct attention to pre-existing disease and the level of residual enzyme obtainable within the CNS.
The general applicability of these findings to other lysosomal storage diseases or other neurodegenerative diseases will clearly be based on the nature of the insult to the CNS and the integrity of neurons. For the above mouse as well as the human neuronopathic variants of Gaucher disease the primary pathological process is neuronal cell death [6, 12]. These are clearly not recoverable without additional influx of new neurons from stem cells that have adequate enzyme levels to prevent disease initiation. The achievability of such “therapy” remains under active investigation. For other lysosomal storage diseases that involve the CNS, the pathological process might relate primarily to substrate accumulation without major degeneration of neurons, i.e., they are primarily neuronal functional abnormalities. These disorders, e.g., murine MPS VII, may be amenable to reversibility of the disease manifestations if neuronal function can be recaptured/rescued. Thus, the continuation of progress toward the treatment of the CNS disorders caused by the lysosomal storage diseases will require greater understanding of the basic pathophysiology, nature of the progression, age at initial insults, and mechanisms of propagation, including proinflammatory contributions, to the overall course of individual diseases.
ACKNOWLEDGEMENTS
The authors thank Amy Hirsch for her clerical expertise and to Venette Johnson and Matt Zamzow for their excellent technical assistance; Dr. Keith Stringer for scientific input on the immunohistochemistry study, Chris Wood for photography procession, and Lisa McMillin, Meredith Farmer for skilled tissue preparation. This work was supported by a grant from the NIH to GAG (DK 36729).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Rosenbloom BE, Scott CR, Wappner RS, Weinreb NJ, Zimran A. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch. Intern. Med. 2000;160:2835–2843. doi: 10.1001/archinte.160.18.2835. [DOI] [PubMed] [Google Scholar]
- 2.Volk BW, Wallace BJ, Adachi M. Infantile Gaucher's disease: electron microscopic and histochemical studies of a cerebral biopsy. J Neuropathol Exp Neurol. 1967;26:176–177. [PubMed] [Google Scholar]
- 3.Adachi M, Wallace BJ, Schneck L, Volk BW. Fine structure of central nervous system in early infantile Gaucher’s disease. Arch Pathol. 1967;83:513–526. [PubMed] [Google Scholar]
- 4.Goker-Alpan O, Schiffmann R, Park JK, Stubblefield BK, Tayebi N, Sidransky E. Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr. 2003;143:273–276. doi: 10.1067/S0022-3476(03)00302-0. [DOI] [PubMed] [Google Scholar]
- 5.Frei KP, Schiffmann R. Myoclonus in Gaucher disease. Adv Neurol. 2002;89:41–48. [PubMed] [Google Scholar]
- 6.Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, Morrison A, Lwin A, Colegial C, Allman JM, Schiffmann R. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Zupanc ML, Legros B. Progressive myoclonic epilepsy. Cerebellum. 2004;3:156–171. doi: 10.1080/14734220410035356. [DOI] [PubMed] [Google Scholar]
- 8.Wenger DA, Roth S, Kudoh T, Grover WD, Tucker SH, Kaye EM, Ullman MD. Biochemical studies in a patient with subacute neuropathic Gaucher disease without visceral glucosylceramide storage. Pediatr Res. 1983;17:344–348. doi: 10.1203/00006450-198305000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Beutler E, Grabowski GA. Gaucher Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill; 2001. pp. 3635–3668. [Google Scholar]
- 10.Grabowski G, Kolodny E, Weinreb N, Rosenbloom B, Prakash-Cheng A, Kaplan P, Charrow J, Pastores G, Mistry P. Gaucher disease: Phenotypic and Genetic Variation. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill Companies; 2005. [Google Scholar]
- 11.Svennerholm L, Hakansson G, Mansson JE, Nilsson O. Chemical differentiation of the Gaucher subtypes. Prog Clin Biol Res. 1982;95:231–252. [PubMed] [Google Scholar]
- 12.Conradi NG, Sourander P, Nilsson O, Svennerholm L, Erikson A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol (Berl) 1984;65:99–109. doi: 10.1007/BF00690463. [DOI] [PubMed] [Google Scholar]
- 13.Tybulewicz VLJ, Tremblay ML, LaMarca ME, Willemsen R, Stubblefield BK, Winfield S, Zablocka B, Sidransky E, Martin BM, Huang SP, et al. Animal model of Gaucher's disease from targeted disruption of the mouse glucocerebrosidase gene. Nature. 1992;357:407–410. doi: 10.1038/357407a0. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Suzuki K, Reed JD, Grinberg A, Westphal H, Hoffmann A, Doring T, Sandhoff K, Proia RL. Mice with type 2 and 3 Gaucher disease point mutations generated by a single insertion mutagenesis procedure. Proc Natl Acad Sci U S A. 1998;95:2503–2508. doi: 10.1073/pnas.95.5.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanfer JN, Legler G, Sullivan J, Raghavan SS, Mumford RA. The Gaucher mouse. Biochem Biophys Res Commun. 1975;67:85–90. doi: 10.1016/0006-291x(75)90286-7. [DOI] [PubMed] [Google Scholar]
- 16.Xu YH, Quinn B, Witte D, Grabowski GA. Viable mouse models of acid β-glucosidase deficiency: the defect in Gaucher disease. Am J Pathol. 2003;163:2093–2101. doi: 10.1016/s0002-9440(10)63566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finn LS, Zhang M, Chen SH, Scott CR. Severe type II Gaucher disease with ichthyosis, arthrogryposis and neuronal apoptosis: molecular and pathological analyses. Am J Med Genet. 2000;91:222–226. [PubMed] [Google Scholar]
- 18.Sidransky E, Tayebi N, Stubblefield BK, Eliason W, Klineburgess A, Pizzolato GP, Cox JN, Porta J, Bottani A, DeLozier-Blanchet CD. The clinical, molecular, and pathological characterisation of a family with two cases of lethal perinatal type 2 Gaucher disease. J Med Genet. 1996;33:132–136. doi: 10.1136/jmg.33.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tayebi N, Cushner SR, Kleijer W, Lau EK, Damschroder-Williams PJ, Stubblefield BK, Den Hollander J, Sidransky E. Prenatal lethality of a homozygous null mutation in the human glucocerebrosidase gene. Am J Med Genet. 1997;73:41–47. doi: 10.1002/(sici)1096-8628(19971128)73:1<41::aid-ajmg9>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 20.Mumford RA, Raghavan SS, Kanfer JN. Hydrolytic and transglucolytic activities of a partially purified calf brain beta-glucosidase. J Neurochem. 1976;27:943–948. doi: 10.1111/j.1471-4159.1976.tb05159.x. [DOI] [PubMed] [Google Scholar]
- 21.Premkumar L, Sawkar AR, Boldin-Adamsky S, Toker L, Silman I, Kelly JW, Futerman AH, Sussman JL. X-ray structure of human acid-beta-glucosidase covalently bound to conduritol-B-epoxide. Implications for Gaucher disease. J Biol Chem. 2005;280:23815–23819. doi: 10.1074/jbc.M502799200. [DOI] [PubMed] [Google Scholar]
- 22.Grace ME, Newman KM, Scheinker V, Berg-Fussman A, Grabowski GA. Analysis of human acid beta-glucosidase by site-directed mutagenesis and heterologous expression. J Biol Chem. 1994;269:2283–2291. [PubMed] [Google Scholar]
- 23.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 24.Grabowski GA, Osiecki-Newman K, Dinur T, Fabbro D, Legler G, Gatt S, Desnick RJ. Human acid beta-glucosidase. Use of conduritol B epoxide derivatives to investigate the catalytically active normal and Gaucher disease enzymes. J Biol Chem. 1986;261:8263–8269. [PubMed] [Google Scholar]
- 25.Igisu H, Takahashi H, Suzuki K, Suzuki K. Abnormal accumulation of galactosylceramide in the kidney of twitcher mouse. Biochem Biophys Res Commun. 1983;110:940–944. doi: 10.1016/0006-291x(83)91053-7. [DOI] [PubMed] [Google Scholar]
- 26.Liou B, Kazimierczuk A, Zhang M, Scott CR, Hegde RS, Grabowski GA. Analyses of variant acid beta-glucosidases: effects of Gaucher disease mutations. J Biol Chem. 2006;281:4242–4253. doi: 10.1074/jbc.M511110200. [DOI] [PubMed] [Google Scholar]
- 27.Daniels LB, Coyle PJ, Glew RH, Radin NS, Labow RS. Brain glucocerebrosidase in Gaucher's disease. Arch Neurol. 1982;39:550–556. doi: 10.1001/archneur.1982.00510210020005. [DOI] [PubMed] [Google Scholar]
- 28.Hara A, Radin NS. Enzymic effects of beta-glucosidase destruction in mice. Changes in glucuronidase levels. Biochim Biophys Acta. 1979;582:423–433. doi: 10.1016/0304-4165(79)90134-x. [DOI] [PubMed] [Google Scholar]
- 29.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 30.Wolf HK, Buslei R, Schmidt-Kastner R, Schmidt-Kastner PK, Pietsch T, Wiestler OD, Blumcke I. NeuN: a useful neuronal marker for diagnostic histopathology. J Histochem Cytochem. 1996;44:1167–1171. doi: 10.1177/44.10.8813082. [DOI] [PubMed] [Google Scholar]
- 31.Brady RO, Pentchev PG, Gal AE, Hibbert SR, Dekaban AS. Replacement therapy for inherited enzyme deficiency. Use of purified glucocerebrosidase in Gaucher's disease. N Engl J Med. 1974;291:989–993. doi: 10.1056/NEJM197411072911901. [DOI] [PubMed] [Google Scholar]
- 32.Butters TD, Mellor HR, Narita K, Dwek RA, Platt FM. Small-molecule therapeutics for the treatment of glycolipid lysosomal storage disorders. Philos Trans R Soc Lond B Biol Sci. 2003;358:927–945. doi: 10.1098/rstb.2003.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng SH, Smith AE. Gene therapy progress and prospects: gene therapy of lysosomal storage disorders. Gene Ther. 2003;10:1275–1281. doi: 10.1038/sj.gt.3302092. [DOI] [PubMed] [Google Scholar]
- 34.Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, Platt F, Butters T, Dwek R, Moyses C, Gow I, Elstein D, Zimran A. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–1485. doi: 10.1016/S0140-6736(00)02161-9. [DOI] [PubMed] [Google Scholar]
- 35.Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, Parker C, Schiffmann R, Hill SC, Brady RO. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122:33–39. doi: 10.7326/0003-4819-122-1-199501010-00005. [DOI] [PubMed] [Google Scholar]
- 36.Matsuda J, Suzuki O, Oshima A, Yamamoto Y, Noguchi A, Takimoto K, Itoh M, Matsuzaki Y, Yasuda Y, Ogawa S, Sakata Y, Nanba E, Higaki K, Ogawa Y, Tominaga L, Ohno K, Iwasaki H, Watanabe H, Brady RO, Suzuki Y. Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proc Natl Acad Sci U S A. 2003;100:15912–15917. doi: 10.1073/pnas.2536657100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Migita M, Medin JA, Pawliuk R, Jacobson S, Nagle JW, Anderson S, Amiri M, Humphries RK, Karlsson S. Selection of transduced CD34+ progenitors and enzymatic correction of cells from Gaucher patients, with bicistronic vectors. Proc Natl Acad Sci U S A. 1995;92:12075–12079. doi: 10.1073/pnas.92.26.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease. Proc Natl Acad Sci U S A. 2002;99:15428–15433. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meikle PJ, Fuller M, Hopwood JJ. Epidemiology and Screening Policy. In: Futerman AH, Zimran A, editors. Gaucher Disease. Boca Raton: CRC Press, Taylor & Francis Group; 2007. p. 321. [Google Scholar]
- 40.Beck M. Agalsidase alfa--a preparation for enzyme replacement therapy in Anderson-Fabry disease. Expert Opin Investig Drugs. 2002;11:851–858. doi: 10.1517/13543784.11.6.851. [DOI] [PubMed] [Google Scholar]
- 41.Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, Prakash-Cheng A, Rosenbloom BE, Scott CR, Wappner RS, Weinreb NJ. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr. 2004;144:112–120. doi: 10.1016/j.jpeds.2003.10.067. [DOI] [PubMed] [Google Scholar]
- 42.Harmatz P, Whitley CB, Waber L, Pais R, Steiner R, Plecko B, Kaplan P, Simon J, Butensky E, Hopwood JJ. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome) J Pediatr. 2004;144:574–580. doi: 10.1016/j.jpeds.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 43.Mehta A. Agalsidase alfa: specific treatment for Fabry disease. Hosp Med. 2002;63:347–350. doi: 10.12968/hosp.2002.63.6.2005. [DOI] [PubMed] [Google Scholar]
- 44.Miranda SR, He X, Simonaro CM, Gatt S, Dagan A, Desnick RJ, Schuchman EH. Infusion of recombinant human acid sphingomyelinase into niemann-pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. Faseb J. 2000;14:1988–1995. doi: 10.1096/fj.00-0014com. [DOI] [PubMed] [Google Scholar]
- 45.Muenzer J, Lamsa JC, Garcia A, Dacosta J, Garcia J, Treco DA. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): a preliminary report. Acta Paediatr Suppl. 2002;91:98–99. doi: 10.1111/j.1651-2227.2002.tb03115.x. [DOI] [PubMed] [Google Scholar]
- 46.Van den Hout JM, Kamphoven JH, Winkel LP, Arts WF, De Klerk JB, Loonen MC, Vulto AG, Cromme-Dijkhuis A, Weisglas-Kuperus N, Hop W, Van Hirtum H, Van Diggelen OP, Boer M, Kroos MA, Van Doorn PA, Van der Voort E, Sibbles B, Van Corven EJ, Brakenhoff JP, Van Hove J, Smeitink JA, de Jong G, Reuser AJ, Van der Ploeg AT. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics. 2004;113:e448–e457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 47.Altarescu G, Hill S, Wiggs E, Jeffries N, Kreps C, Parker CC, Brady RO, Barton NW, Schiffmann R. The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher's disease. J Pediatr. 2001;138:539–547. doi: 10.1067/mpd.2001.112171. [DOI] [PubMed] [Google Scholar]
- 48.Michelakakis H, Skardoutsou A, Mathioudakis J, Moraitou M, Dimitriou E, Voudris C, Karpathios T. Early-onset severe neurological involvement and D409H homozygosity in Gaucher disease: outcome of enzyme replacement therapy. Blood Cells Mol Dis. 2002;28:1–4. doi: 10.1006/bcmd.2001.0477. [DOI] [PubMed] [Google Scholar]
- 49.Prows CA, Sanchez N, Daugherty C, Grabowski GA. Gaucher disease: enzyme therapy in the acute neuronopathic variant. Am J Med Genet. 1997;71:16–21. [PubMed] [Google Scholar]
- 50.Zimran A, Hadas-Halpern I, Zevin S, Levy-Lahad E, Abrahamov A. Low-dose high-frequency enzyme replacement therapy for very young children with severe Gaucher disease. Br J Haematol. 1993;85:783–786. doi: 10.1111/j.1365-2141.1993.tb03224.x. [DOI] [PubMed] [Google Scholar]
- 51.Zirzow GC, Sanchez OA, Murray GJ, Brady RO, Oldfield EH. Delivery, distribution, and neuronal uptake of exogenous mannose-terminal glucocerebrosidase in the intact rat brain. Neurochem Res. 1999;24:301–305. doi: 10.1023/a:1022578424693. [DOI] [PubMed] [Google Scholar]
- 52.Sinclair GB, Jevon G, Colobong KE, Randall DR, Choy FY, Clarke LA. Generation of a conditional knockout of murine glucocerebrosidase: utility for the study of Gaucher disease. Mol Genet Metab. 2007;90:148–156. doi: 10.1016/j.ymgme.2006.09.008. [DOI] [PubMed] [Google Scholar]