

Abstract
Objective
The function of neutrophils as primary mediators of innate immunity depends on the activity of granule proteins and critical components of the NADPH oxidase complex. Expression of their cognate genes is regulated during neutrophil differentiation by a complex network of intracellular signaling pathways. In this study we have investigated the role of two members of the calcium/calmodulin-dependent protein kinase (CaMK) signaling cascade, CaMKI-like kinase (CKLiK) and CaMKKα, in regulating neutrophil differentiation and functional activation.
Materials and Methods
Mouse myeloid cell lines were used to examine the expression of a CaMK cascade in developing neutrophils and to examine the effects of constitutive activation versus inhibition of CaMKs on neutrophil maturation.
Results
Expression of CaMKKα was shown to increase during neutrophil differentiation in multiple cell lines, whereas expression of CKLiK increased as multipotent progenitors committed to promyelocytes but then decreased as cells differentiated into mature neutrophils. Expression of constitutively active CKLiKs did not affect morphologic maturation, but caused dramatic decreases in both respiratory burst responses and chemotaxis. This loss of neutrophil function was accompanied by reduced secondary granule and gp91phox gene expression. The CaMK inhibitor KN93 attenuated cytokine-stimulated proliferative responses in promyelocytic cell lines, and inhibited the respiratory burst. Similar data were observed with the CaMKKα inhibitor, STO-609.
Conclusions
Overactivation of a cascade of CaMKs inhibits neutrophil maturation, suggesting that these kinases play an antagonistic role during neutrophil differentiation, but at least one CaMK is required for myeloid cell expansion and functional activation.
Keywords: Ca2+/calmodulin-dependent protein kinases, neutrophil, respiratory burst, chemotaxis
Introduction
Neutrophils are an essential component of the innate immune response and mobilize in response to tissue infections or inflammation. However, the destructive nature of proteolytic enzymes and reactive oxygen species (ROS) released by activated neutrophils can have devastating affects on uninfected or inflamed tissues. For example, neutrophils responding to pro-inflammatory cytokines contribute to the pathogenesis of multiple forms of chronic synovitis (e.g. rheumatoid arthritis, reviewed in [1]), and play a role in cardiac injury during reperfusion of ischemic tissue following acute myocardial infarction (reviewed in [2]). Understanding the signaling pathways that control the proper differentiation and functional activation of neutrophils is critical to designing clinically relevant strategies to regulate neutrophil numbers and/or functional responses. One signaling pathway that may play a significant role in neutrophil biology is the Ca2+/calmodulin-dependent protein kinase (CaMK) cascade. CaMKs are multifunctional serine/threonine kinases that contain a carboxyl-terminal autoinhibitory domain located adjacent to a Ca2+/calmodulin (Ca2+/CaM) binding site [3,4]. In the absence of Ca2+/CaM, these kinases are folded such that the autoinhibitory domain inactivates the catalytic domain [5]. Upon binding Ca2+/CaM, the autoinhibitory domain is released and the catalytic domain becomes active. Full activation of several family members, including CaMKI and CaMIV, also requires phosphorylation by CaMKK, thereby establishing a CaMK cascade [6,7].
Multiple studies have demonstrated the importance of Ca2+ signaling in mediating neutrophil functional responses [8–11], but whether the CaMKs activated by Ca2+ play a positive or negative role in the maturation and/or functional activation of neutrophils is currently unclear. Studies by our and Dr. Paul Coffer’s laboratories have shown that i) both CaMKKα and the CaMKI homologue, CaM kinase I-like Kinase (CKLiK), are upregulated during neutrophil development, ii) CKLiK activity is enhanced by CaMKKα in vitro, and iii) CKLiK activity is required for certain neutrophil functional responses [12–14]. However, other studies have demonstrated that CaMK inhibitors KN-93 and KN-62 can induce the differentiation of multiple leukemic myeloid cells (e.g. HL-60 and NB4) towards mature neutrophils [15–17]. Furthermore, recent studies by Dr. Steven Collin’s group demonstrated that CaMKIIγ directly inhibits RARα transcriptional activity [16,17]. Together these studies suggest that at least one CaMK, e.g. CKLiK, is required for functional responses, but that certain CaMKs can play antagonistic roles during neutrophil maturation.
In the present study, we investigated the possible roles for CaMKKα and its putative target CKLiK in murine neutrophil differentiation using the well-characterized myeloid EML/EPRO cell line [18,19]. We first evaluated the expression of mouse CaMKKα and CKLiK in EML cells as they differentiate towards promyelocytic EPRO cells and then into mature neutrophils. We then compared this expression to that seen during the differentiation of a newly generated model of neutrophil differentiation, SCF ER-Hoxb8 cells [20]. Constitutively active forms of CaMKKα or CKLiK were then overexpressed in EML cells, and changes in the differentiation and functional activation of derived cells were examined. Our data demonstrate that constitutive activation of either CaMK does not affect morphologic maturation but inhibits the respiratory burst. We also demonstrate that expression of constitutively active CKLiK inhibits chemotaxis and late neutrophil-specific gene expression. Our observed effects of CaMK inhibitors on the growth and function of mouse myeloid cell lines also demonstrate that, although CaMKs may play an antagonistic role during late neutrophil maturation, they are nonetheless required for cytokine-induced proliferative responses and activation of the respiratory burst. These studies provide novel evidence for a role of a CaMK cascade in regulating neutrophil differentiation, and may provide insight into effective means of modulating neutrophil maturation and their destructive properties at sites of inflammation.
Materials and Methods
Cell lines and bone marrow cells
EML, EPRO and MPRO cells were grown and induced with all-trans retinoic acid (ATRA) as previously described [19,21]. SCF ER-Hoxb8 cells were maintained in OptiMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Gemini Bio-products, Woodland, CA), 1% CHO-conditioned medium as a source of stem cell factor and 1 µM β-estrodiol (Sigma-Aldrich, St Louis, MI); inductions were performed as described elsewhere [20]. Human NB4 and HL-60 cells were maintained and differentiated as previously described [19]. COS-1, HEK293 and Phoenix cells were maintained in DMEM medium supplemented with 10% FBS. Bone marrow cells isolated from femurs of Swiss Webster mice were initially cultured in IMDM (Invitrogen) supplemented with 20% horse serum plus murine SCF and IL-3 (50 ng/mL each, Peprotech, Rocky Hill, NJ), and human G-CSF (10 ng/mL, Amgem, Thousand Oaks, CA) for 5 days, and then cultured in just G-CSF for 2 days. All medium was supplemented with 5 U/mL penicillin, 5 µg/mL streptomycin sulfate and 25 ng/mL Amphotericin B, and cells were maintained at 37°C in a humid atmosphere of air plus 5% CO2.
Northern analyses
Total RNA was isolated from cell lines and bone marrow with TRIzol reagent (Life Technologies, Rockville, MD) according to the manufacturer’s specifications, and RNA was electrophoresed, blotted and probed as described previously [22].
Cloning of mouse CKLiK
An EPRO cell library was screened with an IMAGE Consortium clone that encoded a portion of the mouse CKLiK gene (clone number 513897, Research Genetics, Huntsville, AL) according to [23]. The resulting ~4kb cDNA was sequenced at the Keck DNA Sequencing Facility (Yale University) and internal sequences matched the published murine CKLiK α-isoform and mouse CaMKIδ (Genbank accession no. NM_177343).
Transient transfections and luciferase assays
COS-1 cells were transfected using FuGene reagent (Roche Diagnostics, Indianapolis, IN) with 7 µL of Fugene/35 mm plate (Corning Life Sciences, Acton, MA) together with 0.6 µg of the CaMK expression vector, 0.3 µg of the GAL4-CREB expression vector, 0.6 µg of the 5XGAL4-TATA-luciferase vector and 0.2 µg of pCMVβgal (Clontech, Palo Alto, CA), a plasmid that provides a control for transfection efficiency. After 48 hours, cells were washed with 1X phosphate buffered saline (PBS), lysed and assayed for luciferase activity using the Luciferase Assay System (Promega, Madison, WI) according to the manufacturer’s specifications. Luciferase levels were normalized to β-galactosidase expression levels as previously described [24]. For transfections of HEK293 cells, 2 µg of the expression vectors containing wild-type and mutant CKLiKs together with 6 uL of Fugene were transfected into HEK293 cells on 60 mm plates. Cells were then harvested for Western analyses 48 hours after transfection as described below.
Western analyses
For whole cell lysates, cells were lysed and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis as previously described [19]. For cellular localization of FLAG-tagged CKLiK, nuclear extracts were generated from transfected HEK293 cells essentially as previously described [24], with the only modification being cytoplasmic proteins were obtained after cell lysis in buffer A (10 mmol/L HEPES-KOH (pH 7.9), 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L dithiothreitol (DTT), and 0.5 mmol/L phenylmethylsulfonyl fluoride (PMSF) with 0.1% Nonidet P-40). Antibodies against FLAG and actin were from Stratagene (La Jolla, CA) and Santa Cruz Biotechnologies (Santa Cruz, CA), respectively. Antibodies for CaMKK, gp91phox, p47phox and p67phox were all from BD Transduction Laboratories (San Diego, CA).
Generation of EML cells expressing mutant CKLiK and CaMKKα
The truncated CKLiK (CKLiK296) was generated by PCR amplifying a fragment from EPRO cDNA using forward oligonucleotides with an EcoRI site (underlined) upstream from the start codon (5’-CCGGAATTCCATGGCCCGGGAGAACGGC-3’), and reverse oligonucleotides with a stop codon beyond amino acid 296 plus a XhoI site (5’-CCGCTCGAGTCACTGGGCACTGACAGATTC-3’). The amplified product was then EcoRI/XhoI-digested and ligated into EcoRI-SalI sites of the retroviral vector pBABE-puro [25]. The Lys52 to alanine mutation in the ATP binding site of CKLiK296 was generated using the QuikChange Site Directed Mutagenesis kit (Stratagene) with the following primers: forward 5’-GGAAGCTCTTCGCAGTGGCGTGCATCCCG-3’ and reverse 5’-CGGGATGCACGCCACTGCGAAGAGCTTCC-3’. The FLAG-tagged, full-length cDNA CKLiK385, generously provided by Yamada et al. [26], was mutated using QuikChange with the following primer pairs: for the calmodulin binding domain mutation 289IHES to EDDD, forward 5’-CAGCCCTTAGCAAAAACGAGGACGATGATGTCAGTGCCCAGATCCGG-3’ and reverse 5’- CCGGATCTGGGCACTGACATCATCGTCCTCGTTTTTGCTAAGGGCTG-3’; and for the Phe311 to alanine mutation, forward 5’-TGGAGACAAGCGGCCAACGCCACGGCAG-3’ and reverse 5’-CTGCCGTGGCGTTGGCCGCTTGTCTCCA-3’. The mutant construct was then excised from pCMV-Tag 2C (Stratagene) by digestion with NotI and HindIII, overhangs were filled in with Klenow fragment (New England Biolabs, Beverly, MA), and the product was ligated into pMSCV-puro (Clontech, La Jolla, CA) at HpaI sites. The CaMKK413-KD construct was generated by mutating Lys157 to alanine with the primers 5’-GGGAAGACAGACACTATGCAATGGCAGTTCTTTCC-3’ and 5’-GGAAAGAACTGCCATTGCATAGTGTCTGTCTTCCC-3’. Correct sequences for all mutant constructs were verified. EML cells were transduced with pBABE-puro and pMSCV-puro vectors by first transiently transfecting Pheonix cells with each vector containing mutant CaMKs or vector alone using FuGene reagent (Roche Diagnostics). After 24 hour incubation, the transfected Phoenix cells were overlaid with EML cells in growth medium at 1 × 105 cells/mL. Following 24 hours of co-culture, EML cells were remove, allowed to recover for 24 hours, and then selected in puromycin (1 µg/mL) for a mimimum of 18 days to derive stably transduced lines.
Assays for function of differentiated cell lines
Respiratory burst, chemotaxis and phagocytosis assays were all performed as previously detailed [19]. For analyses of CaMK inhibitors, each inhibitor was added 24 hours prior to the assay at the indicated concentrations. KN-93 and STO-609 were both obtained from Calbiochem (San Diego, CA).
Results
Characterization of a mouse CaMK cascade in myeloid cell lines
A 4kb cDNA with 96% homology to human CKLiK was isolated from an EPRO cell-derived cDNA library [23]. Expression of CaMKKα and CKLiK was then evaluated by northern blot analysis of EML, EPRO and ATRA-induced EPRO cells. Consistent with our previous studies, expression of CaMKKα is nearly absent in uninduced EML and EPRO cells, but is upregulated during ATRA-induced differentiation of EPRO cells (Fig. 1A, left panel) [12]. By comparison, CKLiK expression is detected in EML cells and is substantially upregulated after differentiation into promyelocytic EPRO cells (Fig. 1A, right panel). CKLiK expression is then downregulated after 48 hours of ATRA induction in EPRO cells. To further assess changes in CKLiK expression during early vs. late stages of neutrophil differentiation, transcript levels were then analyzed in EPRO cells induced for 2 and 4 hours and for 24–72 hours (Fig. 1B). Interestingly, CKLiK expression initially increases, but then declines after 24 hours of ATRA induction, coincident with morphologic maturation and secondary granule protein gene expression [21]. Expression levels of CaMKKα and CKLiK were also examined in the myeloid progenitor SCF ER-Hoxb8 cells, which differentiate into mature neutrophils upon estrogen withdrawal [20]. Similar to EML/EPRO cells, expression of CaMKKα increased during the differentiation of Hoxb8-ER-SCF cells, with peak levels coinciding with morphologic maturation (not shown) and activation of lactoferrin expression (Fig. 1C). In contrast, expression of CKLiK appears to be downregulated after 48 hours of estrogen withdrawal. Finally, expression of CaMKKα and CKLiK were assessed in mouse bone marrow cells cultured ex vivo in medium containing G-CSF (Figure 1D). Consistent with our results in cell lines, CKLiK expression declined during G-CSF-induced differentiation, whereas CaMKKα expression initially increased but then also declined.
Figure 1. Expression of CaM kinase transcripts in multiple models of neutrophil differentiation.
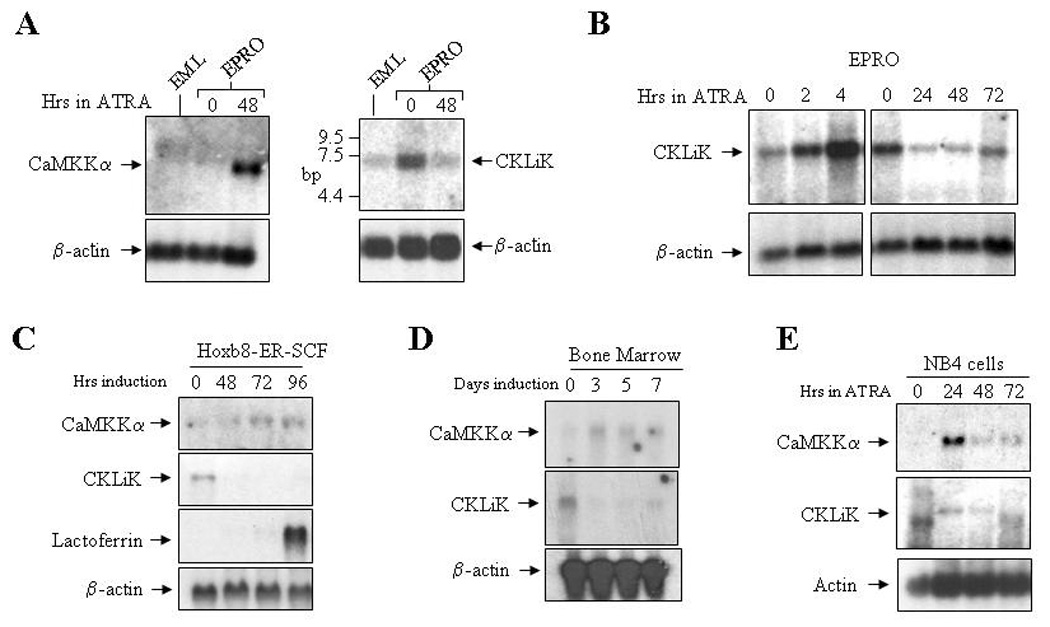
(A) Northern blots were generated using total RNA (5 µg) from uninduced EML and EPRO cells, and EPRO cells induced with 10 µM ATRA for 48 hours. Blots were sequentially hybridized with 32P-labelled cDNAs for the kinase shown and mouse β-actin as a control. (B) Northern analyses were performed using total RNAs from uninduced EPRO cells and cells induced with ATRA for 2 and 4 hours (left panels), or for 24, 48 and 72 hours (right panels). (C) A northern blot was generated using total RNA from SCF ER-Hoxb8 cells grown in the presence of β-estrodiol (1 µM, time 0), and cells grown after β-estrodiol withdrawal to induce neutrophil differentiation (48–96 hours). The blot was sequentially hybridized with 32P-labelled cDNA probes for the two kinases, lactoferrin (to demonstrate neutrophil differentiation) and β-actin. (D) Expression of CaMKKα and CKLiK were assessed in bone marrow-derived stem cells induced with SCF, IL-3 and G-CSF for 3 and 5 days, and then cultured in G-CSF alone. (E) A northern analysis identifies the expression of human CaMKKα and CKLiK in ATRA-induced NB-4 cells.
To compare our observations with the expression of both kinases in human myeloid cells, we also performed northern blot analysis of uninduced versus ATRA-induced human promyelocytic NB4 cells. Our results indicate that both CaMKKα and CKLiK are initially activated by ATRA, but then levels decline after 48 hours of induction (Fig. 1E). We note that a lower band is visible in uninduced NB4 cells and after 72 hours of induction, but expression of the predicted 7 kb fragment (upper band) is only seen in induced cells. We also detected expression of both CaMKKα and CKLiK in leukemic HL-60 cells, but expression did not appear to change during ATRA induction (data not shown).
Constitutively active forms of CKLiK and CaMKKα inhibit specific functional responses in differentiated EPRO cells
To determine whether the observed downregulation of CKLiK is important for neutrophil development, we expressed constitutively active forms of CKLiK using retroviral expression vectors in EML cells and tested for neutrophil differentiation and function activation. We used two different forms of CKLiK, a truncated version similar to the human form previously shown to confer constitutive activity, CKLiK296 [13], and a second FLAG-tagged, full-length version (CKLiK385-CA) with internal mutations that mimic Ca2+/CaM binding. The mutations in CKLiK385-CA do not affect sequences in a putative NES-like domain, previously suggested to regulate cytoplasmic localization of CKLiK and other CaMK family members (see Fig. 2A) [27]. A catalytically inactive, truncated form of CKLiK (CKLiK296-KD, for “kinase dead”) was also generated, which contains an alanine substitution at Lys52, a putative ATP binding site that is essential for catalytic activity in conserved catalytic domains of CaMKI and CaMKIV [28–30]. Cytoplasmic localization of the full-length, constitutively active form of CKLiK was then demonstrated using a Western blot comparing nuclear versus cytoplasmic lysates from HEK293 cells transfected with either the FLAG-tagged, wild-type CKLiK or CKLiK385- CA (Fig. 2B).
Figure 2. Expression and constitutive activity of mutant CKLiK proteins.
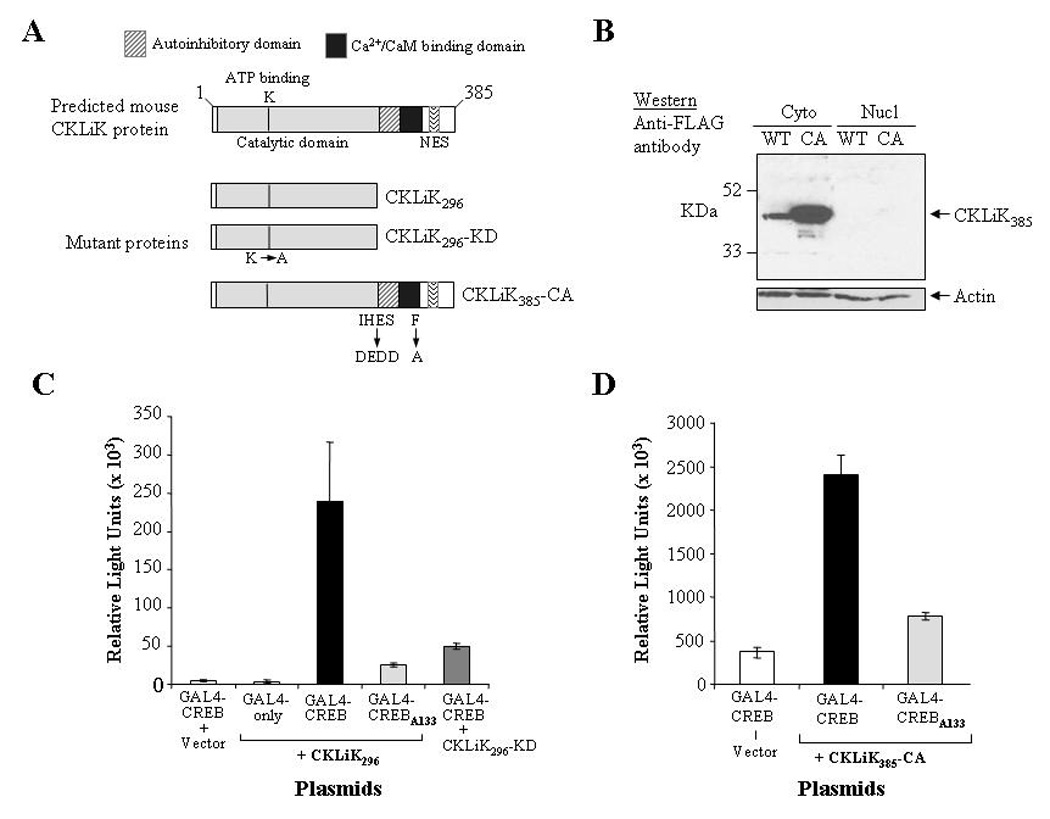
(A) Depicted are the wild-type form of CKLiK and the two constitutively active forms, CKLiK296 (truncated at Gln296) and CKLiK385-CA (internal mutations that mimic Ca2+/CaM binding). Also shown is the “kinase dead” form of the truncated CKLiK (CKLiK296-KD) that contains a mutation in the ATP binding site (Lys52). (B) Results of a Western blot containing cytoplasmic versus nuclear extracts from HEK293 cells transfected with wild-type (WT) or constitutively active (CA) versions of CKLiK demonstrate abundant expression in the cytoplasm, whereas nuclear expression was undetectable. The blot was probed with an anti-FLAG antibody to detect CKLiK expression and with an anti-actin antibody to demonstrate amounts of protein in each lane. (C and D) Activation of CREB by each mutant form of CKLiK was tested by transfecting COS-1 cells with the expression vectors for each mutant kinase together with the two reporter plasmids, pCMV-GAL4-CREBΔb-zip (GAL4-CREB) and p5XGAL4-E1b-luciferase. Control vectors also used were one lacking the CREB activation domain (GAL4-only) and one with a Ser133 to Ala mutation in the CREB motif (GAL4-CREBA133). Levels of luciferase activity were normalized to levels of β-galactosidase expressed from co-transfected plasmids. Values shown are the averages ± SD from triplicate transfections performed in parallel and are representative of at least 3 independent experiments.
To confirm constitutive activity of both the full-length and truncated forms of CKLiK, COS-1 cells were transfected with each mutant construct together with two reporter expression vectors that detect CaMK activity via CREB phosphorylation (pCMV-GAL4-CREBΔb-zip and p5XGAL4-E1b-luciferase, kindly provided by Dr. Richard Maurer, Oregon Health Sciences University) [31]. Figure 2C demonstrates that expression of CKLiK296 activates reporter gene expression, whereas an empty vector or a form of CREB that lacks the GAL4 binding site (GAL4-only) exhibited minimal reporter expression. CKLiK296 appears to primarily activate CREB by phosphorylation of Ser133, since an alanine substitution (GAL4-CREBA133) caused significantly reduced CREB activation. Importantly, the catalytically inactive form of CKLiK296, CKLiK296-KD, also yielded significantly reduced levels of CREB activation as compared to the constitutively active version. CKLiK385-CA also demonstrated constitutive activation of CREB that was dependent on Ser133 (Fig. 2D).
EML cells were transduced with the full-length, constitutively active form of CKLiK (CKLiK385-CA) or either truncated form (CKLiK296 and CKLiK296-KD). Cells transduced with empty vectors were generated for negative controls. Ectopic expression of CKLiK385-CA in lysates from two independently transduced pools of EML cells was confirmed using an anti-FLAG antibody (Fig. 3A). The lower band in the Western blot is a non-specific signal similar to that observed by Yamada et al in MEL cells [26]. Ectopic expression of CKLiK296 and CKLiK296-KD were confirmed in multiple clonal sublines of transduced EML cells by northern blot analysis (Fig. 3B). To analyze possible functions of CaMKKα during neutrophil differentiation, EML cells were transduced with a constitutively active form of rat CaMKKα (CaMKK413, kindly provided by Dr. Tom Soderling, Oregon Health Sciences University) and a kinase dead form with a lysine to alanine substitution at the ATP binding site (CaMKK413-KD). Both transcript and protein expression were confirmed in multiple clonal sublines (Fig. 3C).
Figure 3. Ectopic expression of mutant CKLiK and CaMKKα in EML cell lines.
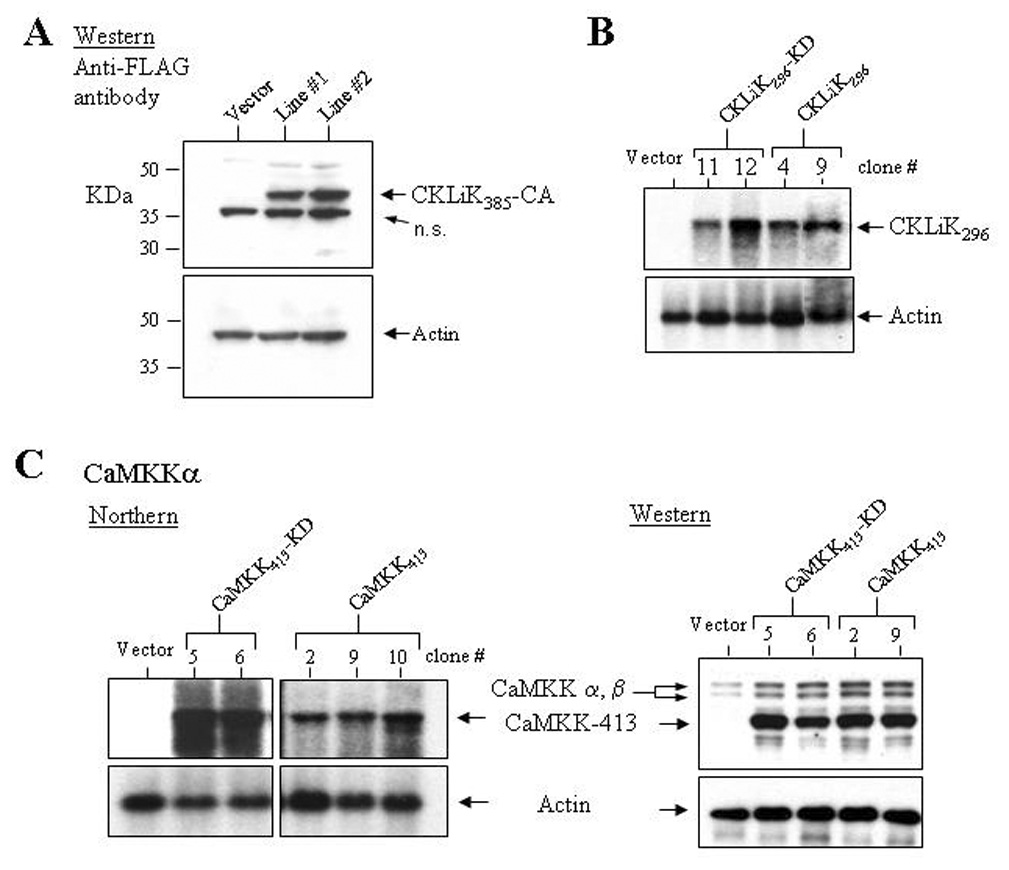
(A) A Western blot containing protein lysates from two independently generated EML cell lines stably transduced with CKLiK385-CA or an empty vector was probed with an anti-FLAG antibody. Below is shown the same blot probed with an anti-actin antibody. (B) Ectopic expression of CKLiK296 and CKLiK296-KD in clonal sublines of transduced EML cells was demonstrated by northern analyses. (C) Ectopic expression of constitutively active and kinase dead forms of CaMKKα (CaMKK413 and CaMKK413-KD, respectively) in EML cells was confirmed by northern assays and a Western blot probed with anti-CaMKK. Shown in the Western blot is expression of the endogenous CaMKKα and β isoforms, and the shorter, truncated forms. Levels of actin expression are shown below each blot.
To test for possible effects of CaMK overactivation on neutrophil maturation or function, EML cells expressing the constitutively active forms of CKLiK or CaMKKα were induced with ATRA to the EPRO stage and then to mature neutrophils. Ectopic expression of either type of constitutively active CKLiK or CaMKKα did not affect morphologic maturation; after 3–5 days of ATRA induction, all transduced cells exhibited multilobulated nuclei characteristic of mature mouse neutrophils (data not shown). The growth profiles of EML or EPRO cells transduced with each mutant kinase were also unaffected (data not shown). However, EPRO cells expressing CKLiK385-CA had severely reduced respiratory burst activity as compared to cells transduced with the empty vector (Fig. 4A). Reduced oxidative burst was also observed in cells overexpressing CKLiK296, but not in cells expressing the catalytically inactive form CKLiK296-KD (Fig. 4B). Interestingly, ectopic expression of CaMKK413 also inhibited the oxidative burst (Fig. 4C), but did not affect morphologic maturation (data not shown).
Figure 4. Expression of constitutively active CaM kinases inhibits the respiratory burst in differentiated EPRO cells.
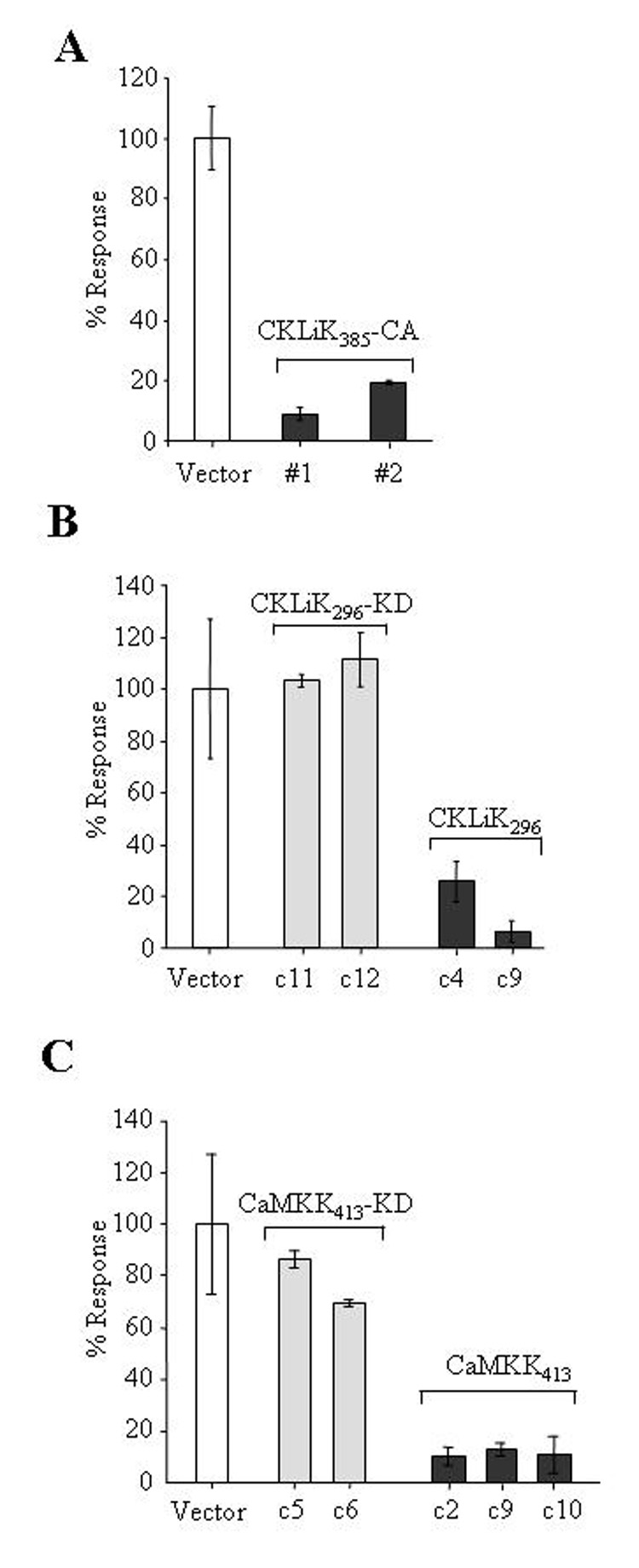
(A) EPRO cells expressing CKLiK385-CA demonstrate reduced levels of the respiratory burst as compared to cells expressing the empty vector. Assays were performed on ATRA-induced cells stimulated with PMA (4 µg/mL) using an enhanced luminol reagent to detect O2− production. Shown are the percentages of peak light units emitted over 10 sec. during a 5 min. period of analysis as compared to control cells expressing the empty vector, set at a 100% response. (B) Expression of the truncated CKLiK (CKLiK296) but not the kinase dead form (CKLiK296-KD) inhibits the respiratory burst. Shown are the responses of two clonal sublines of each type of transduced EPRO cells as compared to cells expressing the empty vector. (C) Expression of the constitutively active CaMKKα (CaMKK413) inhibits the respiratory burst to levels similar to that observed by the constitutively active CKLiKs. Data shown for all assays are given as averages ± SD from triplicate samples analyzed in parallel and are representative of at least three independent experiments.
Chemotaxis was next analyzed using ATRA-induced cells and the murine neutrophil chemoattractant KC. Ectopic expression of either CKLiK385-CA or CKLiK296 significantly inhibited chemotaxis in EPRO cells, whereas cells expressing the kinase dead form of CKLiK296 or the empty vector exhibited responses near that of normal peripheral blood neutrophils (Figs. 5A and B, see figure legends for statistical analyses). In contrast, EPRO cells expressing CaMKK413 exhibited normal chemotaxis (Fig. 5C). Phagocytosis and degranulation were also examined, but no substantial abnormalities were identified (data not shown).
Figure 5. Chemotaxis is inhibited by overexpression of constitutively active CKLiK.
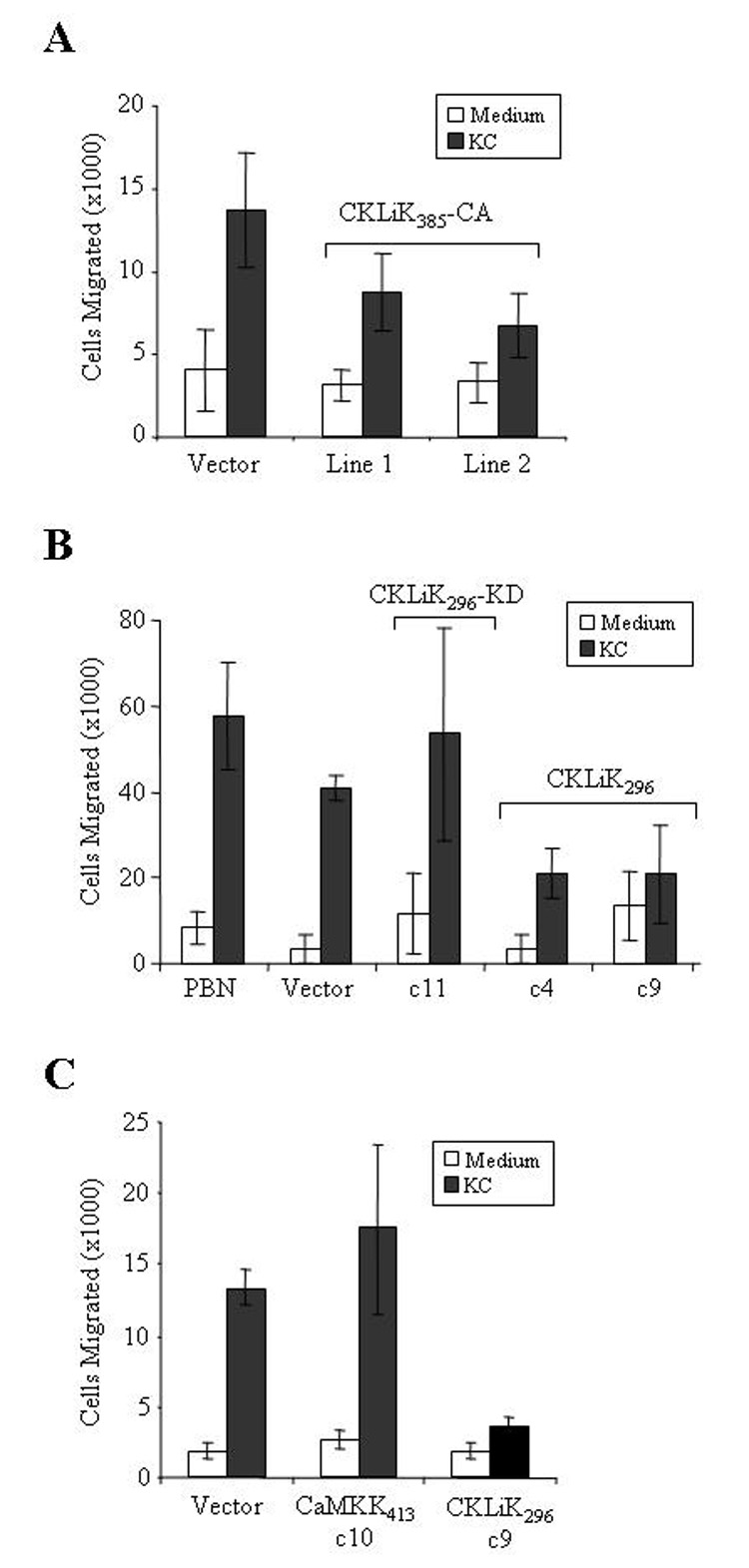
(A) Expression of the full-length, constitutively active CKLiK inhibits chemotaxis. Chemotaxis assays were performed using ATRA induced cells and transwell plates with 3 µm membranes. Graphed are the average total number of cells ± SD that migrated after a 2 hour incubation into the bottom chamber containing medium only or KC. P values generated by the Student’s two sample t-test (Excel, Microsoft Corporation, Redmond, WA) for the differences between chemotaxis by cells expressing the vector versus CKLiK385-CA are as follows: vector vs. line #1, p = 0.004; vector vs. line #2, p = 0.0006. Data shown are from triplicate experiments performed in parallel. (B) EPRO cells that express CKLiK296 exhibit reduced chemotaxis as compared to cells expressing the empty vector or CKLiK296-KD, or to peripheral blood neutrophils (PBN). Statistical analyses yielded the following results: vector vs. CKLiK296-KD c11, p = 0.15 (not statistically different); vector vs. CKLiK296 c4, p = 0.005; vector vs. CKLiK296 c9, p = 0.009. (C) Ectopic expression of CaMKK413 does not disrupt chemotaxis, as demonstrated by levels of cell migration similar to that observed by cell expressing the empty vector. P values: vector vs. CaMKK413 c10, p = 0.23 (not statistically different), vector vs. CKLiK296 c9, p = 0.00008.
Constitutive activation of CKLiK inhibits late neutrophil-specific gene expression in promyelocytes
We next performed northern analysis of secondary granule protein gene expression in ATRA-induced EPRO cells expressing the constitutively active CaMKs. EPRO cells expressing either an empty vector control or the catalytically inactive form of CKLiK exhibited appropriately increased expression of two secondary granule protein genes, lactoferrin and neutrophil gelatinase, and of gp91phox, a critical component of the NADPH oxidase complex (Fig. 6A). Increased expression of these genes was also observed in ATRA-induced EPRO cells expressing CaMKK413 (Fig. 6A, left panel). By comparison, EPRO cells expressing CKLiK296 exhibited severely reduced levels of multiple neutrophil-specific genes (Fig. 6A, left and right panels; not shown are similar results that were observed in clone #4). Interestingly, levels of C/EBPε were also reduced in ATRA-induced EPRO-CKLiK296 cells, but little change was detected in the levels of PU.1 expression. EPRO cells expressing the constitutively active, full-length version of CKLiK yielded similar results (Fig. 6B, left panel). We also examined levels of protein expression of three different NADPH oxidase components upon ATRA-induction of EPRO-CKLiK385-CA cells. Consistent with our observed effects on transcript expression, gp91phox protein expression was undetectable in cells expressing CKLiK385-CA, but ATRA-induced expression of either p47phox or p67phox was unaffected (Fig. 6B, right panel).
Figure 6. Neutrophil-specific gene expression is inhibited by constitutively active CKLiK.
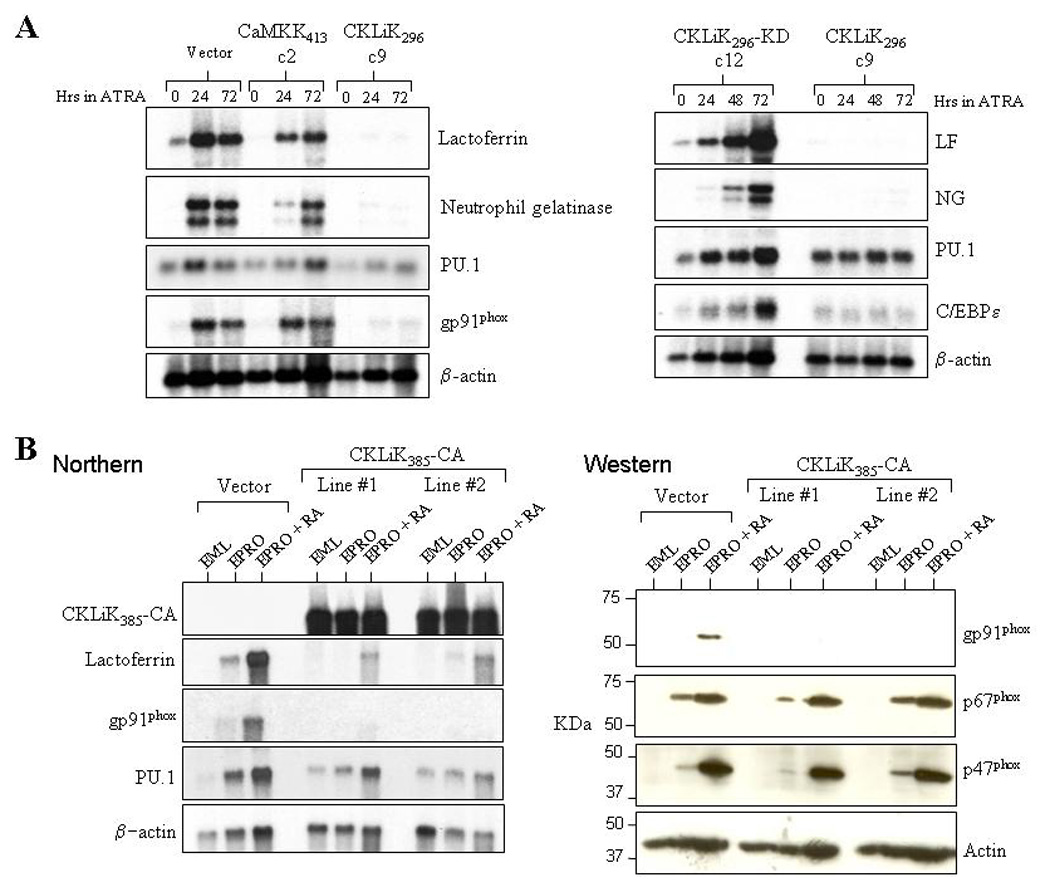
(A) Shown are the results of two northern assays that were performed using total RNAs from uninduced and ATRA-induced EPRO cells expressing the empty vector, constitutively active versions of CaMKKα (CaMKK413, clone 2) or CKLiK (CKLiK296, clone 9), or the kinase dead version of CKLiK296 (CKLiK296-KD, clone 12). The blots were sequentially probed with 32P-labelled cDNAs for the secondary granule genes lactoferrin (LF) and neutrophil gelatinase (NG), the transcriptional regulator PU.1, and either the NADPH oxidase component gp91phox (left panel) or the neutrophil-specific transcriptional regulator C/EBPε (right panel). A β-actin probe was also used to demonstrate amounts of RNA in each lane. (B) A northern assay (left panel) demonstrates that two independently generated lines of EPRO cells that express CKLiK385-CA exhibit reduced lactoferrin and gp91phox transcript expression as compared to cells expressing the empty vector. A Western assay was also performed (right panel), which demonstrates that expression of gp91phox proteins are undetectable in EPRO-CKLiK385-CA cells whereas expression of either p47phox or p67phox is normally upregulated. Levels of actin expression are shown in the bottom panels of each figure.
Effects of CaMK inhibitors on murine neutrophil growth, differentiation and functional activation
The function of neutrophils is dependent on multiple calcium signaling pathways, and recent studies have demonstrated that CaMK inhibitors such as KN93, which interfere with Ca2+/CaM binding, can affect the proliferation and differentiation of human leukemic cells [15–17]. Since these inhibitors disrupt CaMKI and CaMKIV activities [32,33], and may therefore affect CKLiK activities in neutrophils, we tested whether KN-93 and the recently identified CaMKKα inhibitor STO-609 also affect the growth or functional responses of our cell lines. In agreement with previous studies on human leukemic cell lines, KN93 inhibited the growth of both EPRO and SCF ER-Hoxb8 cells in a dose dependent fashion (Fig. 7A). Similar effects were observed in the closely related mouse cell line MPRO cells (data not shown). However, KN-93 did not affect the growth of EML cells (Fig. 7A, right panel). The CaMKK inhibitor STO-609 also significantly inhibited EPRO and Hoxb8ER-SCF cell growth, but in contrast to KN-93, it inhibited EML cell growth (Fig. 7B). EPRO cells exposed to KN-93 were also assessed for morphologic maturation similar to that found in MPRO cells exposed to KN-62 [16,17]. As shown in Figure 7C, a small but significant number of EPRO cells exposed to either KN-93 or KN-62 showed maturation based on changes in nuclear morphology. However, in contrast to studies on MPRO cells, changes in CD11b expression were not detected (data not shown). None of the cell lines showed maturation in the presence of STO-609. To test whether functional responses of mature neutrophils require CaMK activity, ATRA-induced, morphologically mature EPRO and MPRO cells were exposed to either KN-93 or STO-609 for 2 hours prior to performing PMA-stimulated respiratory burst assays (Fig. 7D). KN-93 significantly inhibited ROS production by both EPRO and MPRO cells, and STO-609 reduced the oxidative burst produced by EPRO cells in a dose-dependent fashion.
Figure 7. Effects of CaM kinase inhibitors on neutrophil growth and functional activation.
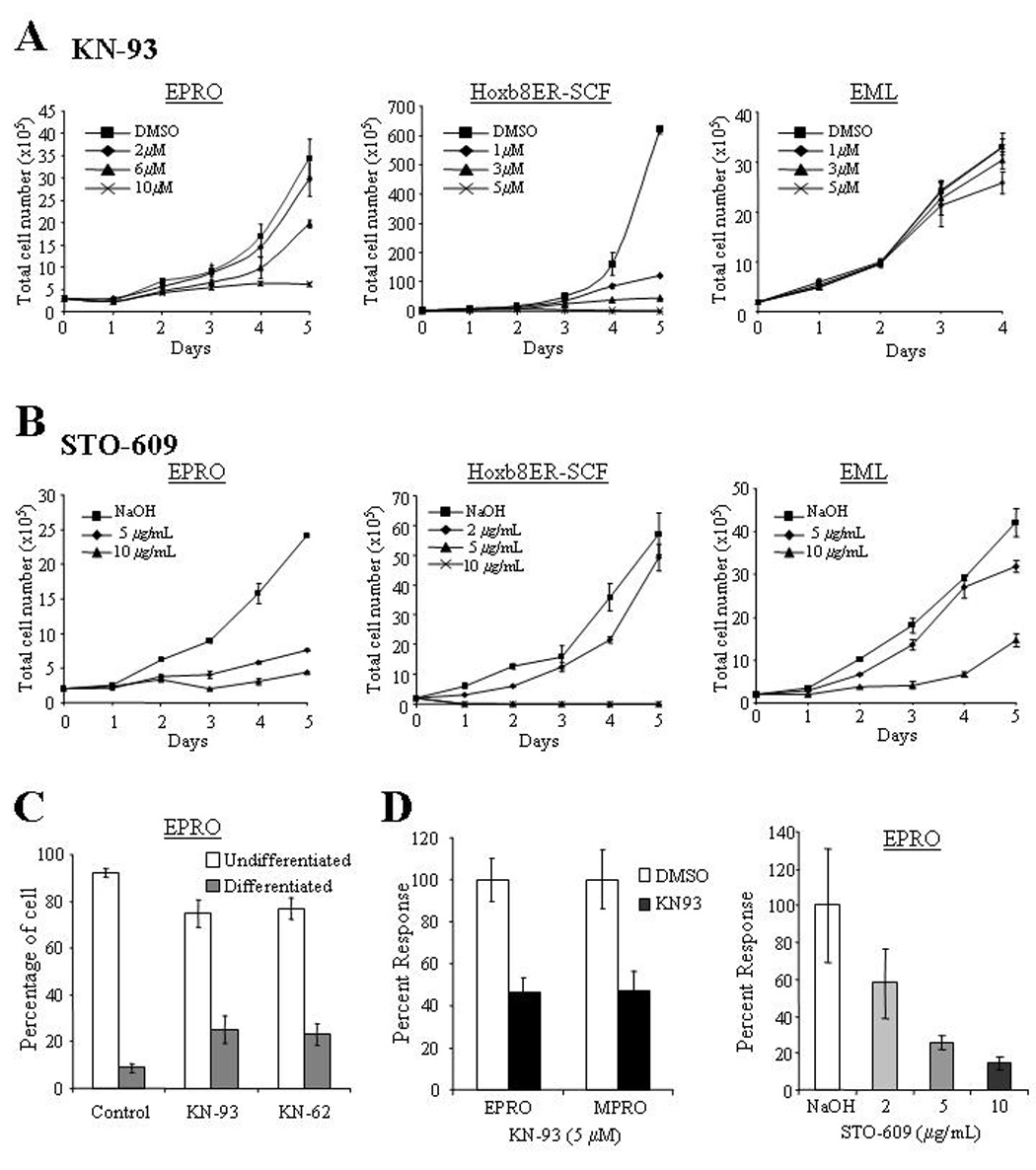
(A) KN93 inhibited the proliferation of both EPRO and SCF ER-Hoxb8 cell lines, whereas the inhibitor did not affect the growth of EML cells. For each assay, cells were diluted to 2 × 105 cells/mL and total numbers of cells that excluded trypan blue after the indicated times were counted using a hemacytometer. Note levels of KN93 used with EPRO cells were two-fold higher than those used in SCF ER-Hoxb8 and EML cells. (B) The CaMKK inhibitor STO-609 significantly reduced the growth profiles of both EPRO and SCF ER-Hoxb8 cells, and inhibited the growth of EML cells. (C) Morphologic maturation of EPRO cells exposed to the CaMK inhibitors KN-62 or KN-93 was assessed by examining cells after 5 days of treatment and counting the number undifferentiated cells with promyelocyte characteristics (e.g. high nuclear to cytoplasmic ratios) versus differentiated cells with kidney shaped or lobulated nuclei. Shown are the results of three independent experiments. P values: KN-62 vs. control, p = 0.001; KN-93 vs. control, p = 0.001. (D) The respiratory burst exhibited by ATRA-induced EPRO or MPRO cells upon PMA stimulation was inhibited by the CaM kinase inhibitor KN-93 (left panel); STO-609 also inhibited the oxidative burst produced by EPRO cells in a dose-dependent fashion. Shown are data from differentiated cells that were incubated with the inhibitors for 24 hours prior to each assay, with data sets given as percentages of peak light units emitted as compared to cells incubated with the diluting reagent. Data shown are the average responses ± SD from three independent experiments.
Discussion
Our expression analyses demonstrate that both CaMKKα and CKLiK are initially upregulated during early promyelocytic differentiation, but that CKLiK is downregulated during intermediate and late stages when secondary granule protein gene expression and neutrophil functional activity peak (Fig. 1). Both the EML/EPRO and SCF ER-Hoxb8 cell lines, as well as the G-CSF-induced bone marrow-derived stem cells, showed CKLiK downregulation, suggesting that loss of CKLiK expression is important to late-stage neutrophil differentiation. These results contrast with those initially reported by Verploegen et al, where continuous CKLiK expression was identified in G-CSF-induced umbilical cord blood-derived CD34+ cells [13]. However, later studies identified low CKLiK protein expression in human peripheral blood despite high level CKLiK activity in Ca2+/calmodulin-stimulated cells [14]. Furthermore, both CKLiK and CaMKKα gene expression levels initially increase but then diminish during ATRA-induced differentiation of human promyelocytic NB4 cells (see Fig. 1D). Since hematopoietic and mesenchymal stem cells derived from umbilical cord blood vs. bone marrow exhibit different gene expression profiles [34–36], these conflicting results may simply be due to the different model systems utilized. Despite these inconsistencies, the previous and current results suggest that expression of both CKLiK and CaMKKα are required throughout normal neutrophil development, but that attenuated expression may be important during late stages of neutrophil maturation.
The expression of constitutively active forms of CKLiK in EML/EPRO cells inhibited the expression of maturation–specific genes, including gp91phox (Fig. 6), which is most likely the underlying cause of the deficient respiratory burst. How this effect also caused a loss of chemotaxis is unclear, but these results support the notion that attenuated expression of CKLiK is important to normal neutrophil development, and suggest that CKLiK may antagonize the activation of neutrophil-specific transcription programs. Importantly, these results are consistent with the recently identified capacity of CaMK inhibitors to induce neutrophil differentiation and of CaMKIIγ to inhibit RAR-regulated differentiation of myeloid leukemic cell lines [15–17]. Identifying the precise mechanism by which CKLiK interferes with neutrophil gene expression will require more detailed analyses, but CKLiK may target a repressor of neutrophil differentiation, such as the CCAAT displacement protein (CDP/cut) or the transcription factor PU.1. Both factors can repress neutrophil gene expression, and their activities have been suggested to be modulated by serine phosphorylation [24,37–41]. Interestingly, CDP represses the expression of C/EBPε, lactoferrin and gp91phox, all three of which were inhibited in cells expressing the constitutively active CKLiK. Alternatively, CKLiK may inhibit the actions of a positive regulator of neutrophil gene expression similar to proposed functions of CaMKIIγ, such as RARα. A caveat to these mechanisms is that CKLiK protein expression is undetectable in the nucleus (Fig. 2 and [13]), thus the effects on transcription may be indirect.
The proposed antagonistic role of CKLiK during neutrophil differentiation seems to conflict with recently identified positive roles for CKLiK in neutrophil functions, including chemotaxis and the respiratory burst [14]. Furthermore, the calmodulin inhibitor W7 disrupts these same neutrophil functions, and our data demonstrate that the respiratory burst is inhibited by KN-93 (Fig. 7 and [11]). A possible model to explain this paradox is that CKLiK performs distinct functions during different stages of neutrophil development, and that protein concentrations determine whether CKLiK acts to support or antagonize neutrophil development and functional responses. In this model, increased CKLiK activity during early myeloid development suppresses transcriptional activators of maturation-specific gene expression, which helps to maintain the progenitors in a proliferative state. This would explain why CKLiK expression levels initially increase in promyelocytes, and why CaMK inhibitors suppress the proliferation of neutrophil progenitors (Fig. 1, Fig. 7) CKLiK may also help to delay the expression of certain genes that encode proteins targeted to stage-specific granules, including the secondary granule proteins lactoferrin and neutrophil gelatinase. As the progenitors commit to terminal differentiation, CKLiK expression is downregulated, which facilitates the transcriptional activation of genes critical to mature neutrophil function. Despite this decrease, sufficient levels are then maintained in mature cells to support key Ca2+/calmodulin-dependent functional responses. A similar model could be envisioned for CaMKIIγ, and CKLiK plus CaMKIIγ may be part of a complex signaling network that regulates both neutrophil differentiation and function. Precedence for this model is provided by two other regulators of hematopoiesis: the Src family kinase Lyn can positively and negatively regulate cytokine signaling in B-cells and in myeloid lineages, and concentrations of PU.1 determine whether it cooperates with or antagonizes the function of C/EBPα to regulate neutrophil maturation [42–44].
The capacity of CaMKK413 to inhibit the respiratory burst but not other functional responses or differentiation may seem surprising, given the more extensive inhibitory effects of constitutively active CKLiK. However, our model of how CKLiK functions in maturing neutrophils may explain these observations, and why STO609 inhibited the respiratory burst. Previous data indicate that CKLiK is a target of CaMKKα [13], therefore constitutive CaMKKα activities in EPRO-CaMKK413 cells may push endogenous CKLiK activities to the point that neutrophil function is disrupted. Since only the respiratory burst was affected by CaMKK413, this suggests that the respiratory burst is more sensitive to regulation by the CaMKKα-CKLiK cascade than other neutrophil functions. The lack of effects on gp91phox gene expression suggests that cascade overactivation can antagonize functions of NADPH oxidase components via post-translational modifications. Indeed, serine phosphorylation regulates the activities of several components, including p47phox, p67phox and Rap1, and threonine phosphorylation of p40phox inhibits NADPH oxidase function [45–48]. Despite this antagonistic role, the cascade is required in mature neutrophils but CKLiK expression levels may be insufficient to positively regulate NADPH oxidase components in the absence of CaMKKα activities. This would explain why STO-609 abrogates the respiratory burst. Alternatively, CaMKKα may directly target another factor that in turn regulates neutrophil function, such as protein kinase B (Akt), which plays positive roles in respiratory burst activation and is directly regulated by CaMKKα [11,49,50].
In addition to potential roles in neutrophil development and function, our analyses of CaMK inhibitors indicate that both CaMKKα and at least one CaMK are essential to cytokine-induced proliferation of myeloid progenitors at multiple stages of differentiation. How CaMKs might regulate myeloid cell growth is unknown, but previous studies demonstrate that CaMKII controls cell cycle progression of several cell line models [33,51–53]. Further investigations are required to determine whether the effects of KN-93 on cell growth are due to specific affects on CKLiK activities or to effects on other CaMKs, and whether STO-609 affects CaMK-dependent or -independent functions of CaMKKα. We note that EPRO cells showed a much less dramatic maturation response to KN-62 as compared to previous studies on MPRO cells, but we found that EPRO cells were less sensitive to the growth inhibition caused by KN-93 as compared to MPRO cells (data not shown); therefore differences between these two cell lines may account for the lack of CD11b upregulation. Nonetheless, our results suggest that use of pharmacological inhibitors of CaMKs in the therapy of myelogenous leukemias must proceed with caution, since such inhibitors may have undesirable effects on the expansion and functions of normal neutrophils in treated patients. On the other hand, these inhibitors may be useful in controlling the production of oxygen radicals by neutrophils at sites of inflammation that are not accompanied by pathogen infection, e.g. in the cartilage of joints afflicted by rheumatoid arthritis.
Acknowledgments
This work was supported by National Institutes of Health Awards RO1-KD53471 and P01-HL63357 (N.B.) and KO1-DK60565 (P.G.). We thank Dr. Toshiyuki Yamada for the FLAG-tagged, full length CKLiK plasmid, Dr. Richard Maurer for CREB-GAL4 expression vectors, and Dr. Thomas Soderling for the rat CaMKK413 plasmid.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu H, Pope RM. Phagocytes: Mechanisms of inflammation and tissue destruction. Rheum. Dis. Clin. North Am. 2004;30:19–39. doi: 10.1016/S0889-857X(03)00107-8. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 3.Soderling TR. The ca-calmodulin-dependent protein kinase cascade. Trends Biochem. Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- 4.Corcoran EE, Means AR. Defining Ca2+/calmodulin-dependent protein kinase cascades in transcriptional regulation. J. Biol. Chem. 2001;276:2975–2978. doi: 10.1074/jbc.R000027200. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg J, Nairn AC, Kuriyan J. Structural basis for the autoinhibition of calcium/calmodulin-dependent protein kinase I. Cell. 1996;84:875–887. doi: 10.1016/s0092-8674(00)81066-1. [DOI] [PubMed] [Google Scholar]
- 6.Tokumitsu H, Enslen H, Soderling TR. Characterization of a Ca2+/calmodulin-dependent protein kinase cascade. Molecular cloning and expression of calcium/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 1995;270:19320–19324. doi: 10.1074/jbc.270.33.19320. [DOI] [PubMed] [Google Scholar]
- 7.Anderson K, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, Means AR. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase beta. J. Biol. Chem. 1998;273:31880–31889. doi: 10.1074/jbc.273.48.31880. [DOI] [PubMed] [Google Scholar]
- 8.Korchak HM, Vosshall LB, Zagon G, Ljubich P, Rich AM, Weissmann G. Activation of the neutrophil by calcium-mobilizing ligands. I. A chemotactic peptide and the lectin concanavalin A stimulate superoxide anion generation but elicit different calcium movements and phosphoinositide remodeling. J. Biol. Chem. 1988;263:11090–11097. [PubMed] [Google Scholar]
- 9.Korchak HM, Vosshall L, Haines KA, Wilkenfeld C, Lundquist KF, Weissmann G. Activation of the human neutrophil by calcium-mobilizing ligands. II. correlation of calcium, diacyl glycerol, and phosphatidic acid generation with superoxide anion generation. J. Biol. Chem. 1988;263:11098–11105. [PubMed] [Google Scholar]
- 10.Pittet D, Lew DP, Mayr GW, Monod A, Schlegel W. Chemoattractant receptor promotion of Ca2+ influx across the plasma membrane of HL-60 cells. A role for cytosolic free calcium elevations and inositol 1,3,4,5-tetrakisphosphate production. J. Biol. Chem. 1989;264:7251–7261. [PubMed] [Google Scholar]
- 11.Verploegen S, van Leeuwen CM, van Deutekom HW, Lammers JW, Koenderman L. Coffer PJ. Role of Ca2+/calmodulin regulated signaling pathways in chemoattractant induced neutrophil effector functions. comparison with the role of phosphotidylinositol-3 kinase. Eur. J. Biochem. 2002;269:4625–4634. doi: 10.1046/j.1432-1033.2002.03162.x. [DOI] [PubMed] [Google Scholar]
- 12.Lawson N, Zain M, Zibello T, Picciotto MR, Nairn AC, Berliner N. Modulation of a calcium/calmodulin-dependent protein kinase cascade by retinoic acid during neutrophil maturation. Exp. Hematol. 1999;27:1682–1690. doi: 10.1016/s0301-472x(99)00108-3. [DOI] [PubMed] [Google Scholar]
- 13.Verploegen S, Lammers JW, Koenderman L, Coffer PJ. Identification and characterization of CKLiK, a novel granulocyte ca(++)/calmodulin-dependent kinase. Blood. 2000;96:3215–3223. [PubMed] [Google Scholar]
- 14.Verploegen S, Ulfman L, van Deutekom HW, van Aalst C, Honing H, Lammers JW, Koenderman L, Coffer PJ. Characterization of the role of CaMKI-like kinase (CKLiK) in human granulocyte function. Blood. 2005;106:1076–1083. doi: 10.1182/blood-2004-09-3755. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto-Yamaguchi Y, Okabe-Kado J, Kasukabe T, Honma Y. Induction of differentiation of human myeloid leukemia cells by immunosuppressant macrolides (rapamycin and FK506) and calcium/calmodulin-dependent kinase inhibitors. Exp. Hematol. 2001;29:582–588. doi: 10.1016/s0301-472x(01)00626-9. [DOI] [PubMed] [Google Scholar]
- 16.Si J, Mueller L, Collins SJ. CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J. Clin. Invest. 2007;117:1412–1421. doi: 10.1172/JCI30779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Si J, Mueller L, Schuler A, Simon J, Collins SJ. The retinoic acid receptor/CaMKII interaction: Pharmacologic inhibition of CaMKII enhances the differentiation of myeloid leukemia cells. Blood Cells Mol. Dis. doi: 10.1016/j.bcmd.2007.05.009. In press. [DOI] [PubMed] [Google Scholar]
- 18.Tsai S, Bartelmez S, Sitnicka E, Collins S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 1994;8:2831–2841. doi: 10.1101/gad.8.23.2831. [DOI] [PubMed] [Google Scholar]
- 19.Gaines P, Chi J, Berliner N. Heterogeneity of functional responses in differentiated myeloid cell lines reveals EPRO cells as a valid model of murine neutrophil functional activation. J. Leukoc. Biol. 2005;77:669–679. doi: 10.1189/jlb.1004567. [DOI] [PubMed] [Google Scholar]
- 20.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods. 2006;3:287–293. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 21.Lawson ND, Krause DS, Berliner N. Normal neutrophil differentiation and secondary granule gene expression in the EML and MPRO cell lines. Exp. Hematol. 1998;26:1178–1185. [PubMed] [Google Scholar]
- 22.Maun NA, Gaines P, Khanna-Gupta A, Zibello T, Enriquez L, Goldberg L, Berliner N. G-CSF signaling can differentiate promyelocytes expressing a defective retinoic acid receptor: Evidence for divergent pathways regulating neutrophil differentiation. Blood. 2004;103:1693–1701. doi: 10.1182/blood-2002-10-3247. [DOI] [PubMed] [Google Scholar]
- 23.Lawson ND, Khanna-Gupta A, Berliner N. Isolation and characterization of the cDNA for mouse neutrophil collagenase: Demonstration of shared negative regulatory pathways for neutrophil secondary granule protein gene expression. Blood. 1998;91:2517–2524. [PubMed] [Google Scholar]
- 24.Khanna-Gupta A, Zibello T, Kolla S, Neufeld EJ, Berliner N. CCAAT displacement protein (CDP/cut) recognizes a silencer element within the lactoferrin gene promoter. Blood. 1997;90:2784–2795. [PubMed] [Google Scholar]
- 25.Morgenstern JP, Land H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamada T, Suzuki M, Satoh H, Kihara-Negishi F, Nakano H, Oikawa T. Effects of PU.1-induced mouse calcium-calmodulin-dependent kinase I-like kinase (CKLiK) on apoptosis of murine erythroleukemia cells. Exp. Cell Res. 2004;294:39–50. doi: 10.1016/j.yexcr.2003.10.023. [DOI] [PubMed] [Google Scholar]
- 27.Stedman DR, Uboha NV, Stedman TT, Nairn AC, Picciotto MR. Cytoplasmic localization of calcium/calmodulin-dependent protein kinase I-alpha depends on a nuclear export signal in its regulatory domain. FEBS Lett. 2004;566:275–280. doi: 10.1016/j.febslet.2004.04.042. [DOI] [PubMed] [Google Scholar]
- 28.Hanks SK, Quinn AM, Hunter T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 29.Chatila T, Anderson KA, Ho N, Means AR. A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J. Biol. Chem. 1996;271:21542–21548. doi: 10.1074/jbc.271.35.21542. [DOI] [PubMed] [Google Scholar]
- 30.Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J. Neurosci. 2004;24:3786–3794. doi: 10.1523/JNEUROSCI.3294-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8:2527–2539. doi: 10.1101/gad.8.21.2527. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Mora OG, LaHair MM, McCubrey JA, Franklin RA. Calcium/calmodulin-dependent kinase I and calcium/calmodulin-dependent kinase kinase participate in the control of cell cycle progression in MCF-7 human breast cancer cells. Cancer Res. 2005;65:5408–5416. doi: 10.1158/0008-5472.CAN-05-0271. [DOI] [PubMed] [Google Scholar]
- 33.Kahl CR, Means AR. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003;24:719–736. doi: 10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- 34.Ng YY, van Kessel B, Lokhorst HM, Baert MR, van den Burg CM, Bloem AC, et al. Gene-expression profiling of CD34+ cells from various hematopoietic stem-cell sources reveals functional differences in stem-cell activity. J.Leukoc.Biol. 2004;75:314–323. doi: 10.1189/jlb.0603287. [DOI] [PubMed] [Google Scholar]
- 35.Panepucci RA, Siufi JL, Silva WA, Jr, Proto-Siquiera R, Neder L, Orellana M, et al. Comparison of gene expression of umbilical cord vein and bone marrow-derived mesenchymal stem cells. Stem Cells. 2004;22:1263–1278. doi: 10.1634/stemcells.2004-0024. [DOI] [PubMed] [Google Scholar]
- 36.Wagner W, Wein F, Seckinger A, Frankhauser M, Wirkner U, Krause U, et al. Comparative characteristics of mesenchymal stem cells from human bone marrow, adipose tissue, and umbilical cord blood. Exp.Hematol. 2005;33:1402–1416. doi: 10.1016/j.exphem.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 37.Skalnik DG, Strauss EC, Orkin SH. CCAAT displacement protein as a repressor of the myelomonocytic-specific gp91-phox gene promoter. J. Biol. Chem. 1991;266:16736–16744. [PubMed] [Google Scholar]
- 38.Lievens PM, Donady JJ, Tufarelli C, Neufeld EJ. Repressor activity of CCAAT displacement protein in HL-60 myeloid leukemia cells. J. Biol. Chem. 1995;270:12745–12750. doi: 10.1074/jbc.270.21.12745. [DOI] [PubMed] [Google Scholar]
- 39.Catt D, Hawkins S, Roman A, Luo W, Skalnik DG. Overexpression of CCAAT displacement protein represses the promiscuously active proximal gp91(phox) promoter. Blood. 1999;94:3151–3160. [PubMed] [Google Scholar]
- 40.Rieske P, Pongubala JM. AKT induces transcriptional activity of PU.1 through phosphorylation-mediated modifications within its transactivation domain. J.Biol.Chem. 2001 Mar 16;276:8460–8468. doi: 10.1074/jbc.M007482200. [DOI] [PubMed] [Google Scholar]
- 41.Dahl R, Simon MC. The importance of PU.1 concentration in hematopoietic lineage commitment and maturation. Blood Cells Mol.Dis. 2003;31:229–233. doi: 10.1016/s1079-9796(03)00152-9. [DOI] [PubMed] [Google Scholar]
- 42.Harder KW, Parsons LM, Armes J, Evans N, Kountouri N, Clark R, et al. Gain- and loss-of-function Lyn mutant mice define a critical inhibitory role for Lyn in the myeloid lineage. Immunity. 2001;15:603–615. doi: 10.1016/s1074-7613(01)00208-4. [DOI] [PubMed] [Google Scholar]
- 43.Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity. 2005;22:9–18. doi: 10.1016/j.immuni.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 44.Dahl R, Walsh JC, Lancki D, Laslo P, Iyer SR, Singh H, et al. Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat.Immunol. 2003;4:1029–1036. doi: 10.1038/ni973. [DOI] [PubMed] [Google Scholar]
- 45.Sahyoun N, McDonald OB, Farrell F, Lapetina EG. Phosphorylation of a Ras-related GTP-binding protein, Rap-1b, by a neuronal Ca2+/calmodulin-dependent protein kinase, CaM kinase Gr. Proc.Natl.Acad.Sci.U.S.A. 1991;88:2643–2647. doi: 10.1073/pnas.88.7.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benna JE, Dang PM, Gaudry M, Fay M, Morel F, Hakim J, et al. Phosphorylation of the respiratory burst oxidase subunit p67(phox) during human neutrophil activation. Regulation by protein kinase C-dependent and independent pathways. J.Biol.Chem. 1997;272:17204–17208. doi: 10.1074/jbc.272.27.17204. [DOI] [PubMed] [Google Scholar]
- 47.Dang PM, Elbim C, Marie JC, Chiandotto M, Gougerot-Pocidalo MA, El-Benna J. Anti-inflammatory effect of interleukin-10 on human neutrophil respiratory burst involves inhibition of GM-CSF-induced p47PHOX phosphorylation through a decrease in ERK1/2 activity. FASEB J. 2006;20:1504–1506. doi: 10.1096/fj.05-5395fje. [DOI] [PubMed] [Google Scholar]
- 48.Lopes LR, Dagher MC, Gutierrez A, Young B, Bouin AP, Fuchs A, et al. Phosphorylated p40PHOX as a negative regulator of NADPH oxidase. Biochemistry. 2004;43:3723–3730. doi: 10.1021/bi035636s. [DOI] [PubMed] [Google Scholar]
- 49.Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- 50.Chen Q, Powell DW, Rane MJ, Singh S, Butt W, Klein JB, McLeish KR. Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J. Immunol. 2003;170:5302–5308. doi: 10.4049/jimmunol.170.10.5302. [DOI] [PubMed] [Google Scholar]
- 51.Rasmussen G, Rasmussen C. Calmodulin-dependent protein kinase II is required for G1/S progression in HeLa cells. Biochem Cell Biol. 1995;73:201–207. doi: 10.1139/o95-024. [DOI] [PubMed] [Google Scholar]
- 52.Tombes RM, Grant S, Westin EH, Krystal G. G1 cell cycle arrest and apoptosis are induced in NIH 3T3 cells by KN-93, an inhibitor of CaMK-II (the multifunctional Ca2+/CaM kinase) Cell Growth Differ. 1995;6:1063–1070. [PubMed] [Google Scholar]
- 53.Morris TA, DeLorenzo RJ, Tombes RM. CaMK-II inhibition reduces cyclin D1 levels and enhances the association of p27kip1 with Cdk2 to cause G1 arrest in NIH 3T3 cells. Exp. Cell Res. 1998;240:218–227. doi: 10.1006/excr.1997.3925. [DOI] [PubMed] [Google Scholar]