

Abstract
Neuroprotection against cerebral ischemia conferred by ischemic preconditioning (IPC) requires translocation of epsilon protein kinase C (εPKC). A major goal in our laboratory is to define the cellular targets by which εPKC confers protection. We tested the hypothesis that εPKC targets the mitochondrial channel ( ) after IPC. Our results demonstrated a rapid translocation of εPKC to rat hippocampal mitochondria after IPC. Because in other tissues εPKC targets channels, but its presence in brain mitochondria is controversial, we determined the presence of the channel-specific subunits (Kir6.1 and Kir6.2) in mitochondria isolated from rat hippocampus. Next, we determined whether channels play a role in the IPC induction. In hippocampal organotypic slice cultures, IPC and lethal ischemia were induced by oxygen-glucose deprivation. Subsequent cell death in the CA1 region was quantified using propidium iodide staining. Treatment with the channel openers diazoxide or pinacidil 48 h prior to lethal ischemia protected hippocampal CA1 neurons, mimicking the induction of neuroprotection conferred by either IPC or εPKC agonist-induced preconditioning. Blockade of channels using 5-hydroxydecanoic acid abolished the neuroprotection due to either IPC or εPKC preconditioning. Both ischemic andεPKC agonist-mediated preconditioning resulted in phosphorylation of the channel subunit Kir6.2. After IPC, selective inhibition of εPKC activation prevented Kir6.2 phosphorylation, consistent with Kir6.2 as a phosphorylation target of εPKC or its downstream effectors. Our results support the hypothesis that the brain channel is an important target of IPC and the signal transduction pathways initiated by εPKC.
Keywords: ischemic tolerance, diazoxide, protein kinase C, organotypic slice culture, cell death, signal transduction
Introduction
ATP-sensitive potassium channels ( channels) are found in many locations within cells, including the plasma membrane and inner mitochondrial membrane (Garlid and Paucek, 2003, Brustovetsky et al., 2005). The mitochondrial ATP-sensitive potassium channel ( channel) performs the unique cellular function of coupling energy metabolism to cellular electrical activity. This coupling maintains the activity of various cell types to meet metabolic demands of the tissue. During global ischemia energy deprivation, massive calcium influx and mitochondrial dysfunction lead to neuronal death in the CA1 region of the hippocampus (Lipton, 1999). Neuronal survival in the hippocampal CA1 region can be improved by ischemic preconditioning (IPC) (Kitagawa et al., 1990, Kirino et al., 1991, Perez-Pinzon et al., 1999). Ischemic preconditioning refers to an endogenous neuroprotective mechanism which is activated by a sublethal ischemic episode, resulting in tissue protection against subsequent lethal ischemic insults (Murry et al., 1986, Kitagawa et al., 1990, Li et al., 1990, Kirino et al., 1991, Perez-Pinzon et al., 1999). Protection of heart against ischemia-reperfusion injury by IPC and channel openers is known to involve the channel (Garlid, 2000, Sato et al., 2006). Brain mitochondria contain seven times more channels than liver or heart mitochondria, which indicates the importance of these channels in neurons (Bajgar et al., 2001). However, the role of the channel in the brain following IPC induction is still not understood clearly.
We recently demonstrated improved mitochondrial function following IPC in an in vivo model of global cerebral ischemia (Dave et al., 2001). The signaling pathway of IPC-induced neuronal survival in the CA1 region of hippocampus requires epsilon protein kinase C (εPKC) activation (Raval et al., 2003, Lange-Asschenfeldt et al., 2004). Interestingly, it has been suggested that PKC translocates to mitochondria and directly modulates the channel leading to cardioprotection (Ohnuma et al., 2002, Jaburek et al., 2006). Because the channel has been implicated to play a role during IPC in brain (Heurteaux et al., 1995, Perez-Pinzon and Born, 1999) and opening of channels prevents cytochrome c release after chemical hypoxia (Liu et al., 2002), we hypothesize that interaction between the channel and εPKC during IPC induction preserves the hippocampal CA1 region after ischemia. Thus, the goal of the present study was to determine whether or not channel activation is required during the triggering phase of IPC. Further, we hypothesize that εPKC mediates neuroprotection via channel activation.
RESULTS
In the context of neuroprotection at 48 hours after ischemic preconditioning, our hypothesis is that ischemic preconditioning rapidly activates a signal transduction pathway requiring the opening of channels. εPKC is a critical mediator of ischemic preconditioning. We first examined the levels of εPKC in mitochondria as a function of time after in vivo ischemic preconditioning. We found a significant increase in the level of εPKC in the mitochondria fraction of hippocampal homogenates at 1 hour after in vivo IPC (Figure 1). This is evidence that εPKC rapidly translocates to mitochondria following IPC. Secondly, we detected the channel-specific subunits Kir6.1 and Kir6.2 in mitochondria isolated from rat pup hippocampus. Figure 2 depicts the presence of Kir6.1 and Kir6.2 in rat hippocampal and liver mitochondria. The presence of Kir6.1 and Kir6.2 in liver mitochondria represents a positive control. The Kir6.1 and Kir6.2 are pore-forming subunits of the channel (Lacza et al., 2003).
Figure 1.
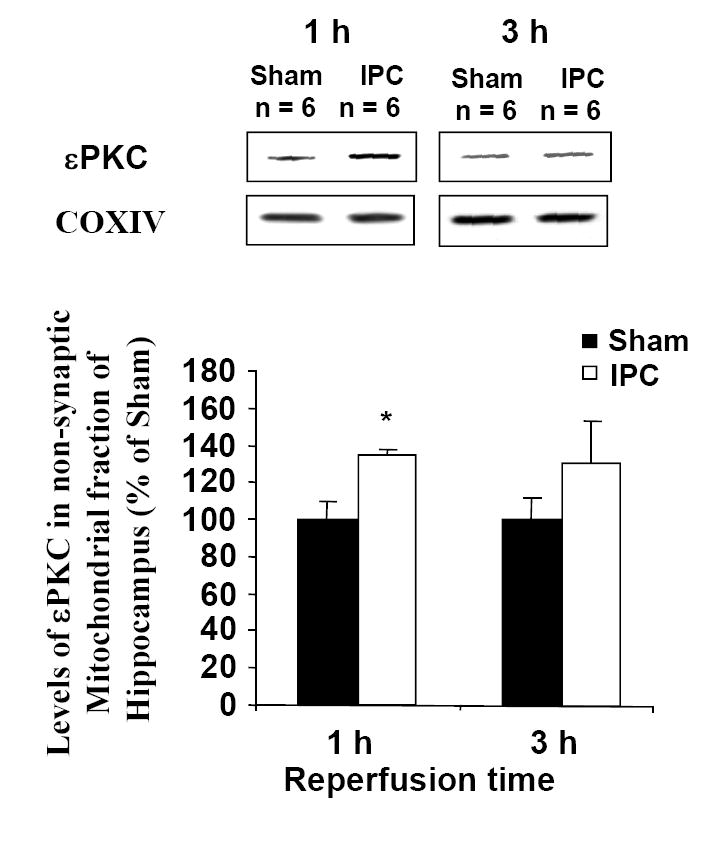
Immunoblot of εPKC isozyme in mitochondrial fraction of hippocampus harvested from rats underwent ischemic preconditioning (bilateral carotid occlusion and systemic hypotension (50 mm Hg) for 2 min) or sham surgery. The hippocampus was harvested at 1 and 3 h following sham/preconditioning. Immunoblots (typical images are shown on top of each bar) were subjected to densitometric analysis, and levels of εPKC are expressed as percentage of control (sham) animals. Results are expressed as mean ± SEM. *, p < 0.05 versus control.
Figure 2.
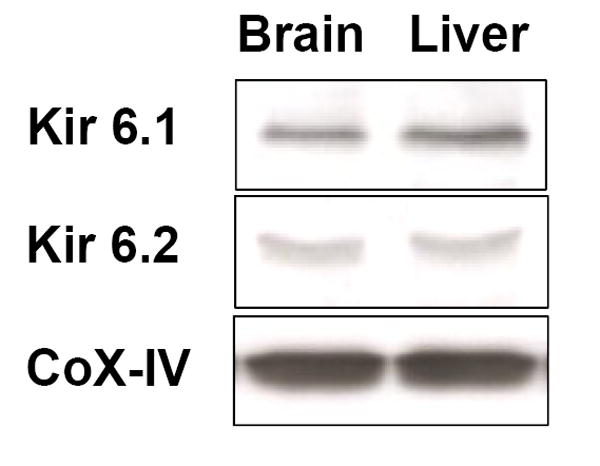
Western blot images showing presence of the pore-forming subunits of the channel, Kir 6.1 and Kir 6.2 in mitochondria isolated from liver and hippocampus (n = 4). The presence of Kir6.1 and Kir6.2 in liver mitochondria represents a positive control.
Next, we asked if ATP-sensitive potassium channels played a role in induction of ischemic preconditioning. Opening of channels with 10 and 50 μM pinacidil, 48 hours prior to lethal ischemia significantly reduced neuronal death in the CA1 region: 35.7 ± 4.7% (n = 8, p<0.001) and 31.9 ± 5.1% (n = 5, p<0.001), respectively; compared to 58.9 ± 7.8% (n = 6) after lethal ischemia without previous exposure to pinacidil. The level of protection conferred by exposure to pinacidil was similar to ischemic preconditioning 31.7 ± 6.0% (n = 6; Figure 3A). In contrast to pinacidil, diazoxide has been demonstrated to specifically open mitochondrial channels (and not plasma membrane channels) when applied at low concentrations (Liu et al., 1998). Our results indicate that diazoxide treatment with a concentration specific for mitochondrial channels 48 h prior to a period of lethal ischemia confers neuroprotection in the CA1 region of hippocampus (Figure 3B). Diazoxide treatment mimicked IPC and there was no difference in degree of neuroprotection between two groups. At higher, non-specific concentrations, diazoxide failed to confer neuroprotection against lethal ischemia.
Figure 3.
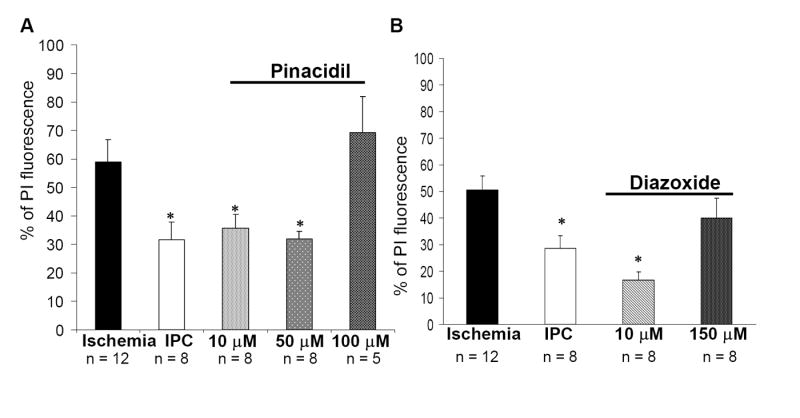
Ischemic preconditioning (IPC) (15 min of OGD) 48 h prior to test ischemia (40 min of OGD) exerts significant neuroprotection. Incubation of slices with the plasma or mitochondrial channel openers pinacidil or diazoxide for 15 min, 48 h prior to ischemia confers neuroprotection in the CA1 region of hippocampus. Note at higher, non-specific concentrations, diazoxide failed to affect cell death after lethal ischemia. PI fluorescence values measured 24 hr after test ischemia in different experimental groups: (A) (1) ischemia, (2) IPC, (3) pinacidil treated; (B) (1) ischemia, (2) IPC, (3) diazoxide treated. Significance compared with ischemia *, p< 0.01.
To examine the role of opening during IPC induction, we used channel blockers. Slices undergoing IPC were exposed to the general channel blocker tolbutamide or to the specific antagonist 5-hydroxydecanoic acid (5-HD) for 1h. Blockade of channels with tolbutamide or 5-hydroxydecanoic acid (5-HD) after IPC induction abolished neuroprotection in a concentration-dependent manner (Figure 4).
Figure 4.
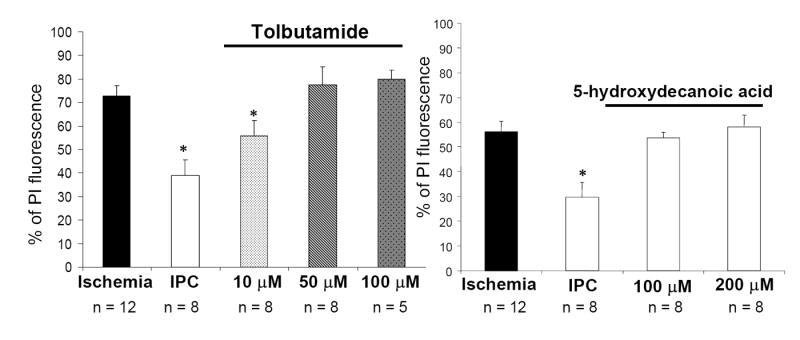
Blockade of channels with tolbutamide or 5-hydroxydecanoic acid (5-HD) after ischemic preconditioning (IPC) (15 min of OGD) induction abolished neuroprotection in a concentration dependent manner PI fluorescence values measured 24 hr after test ischemia in different experimental groups: (A) (1) ischemia, (2) IPC, (3) tolbutamide treated; (B) (1) ischemia, (2) IPC, (3) 5-hydroxydecanoic acid treated. Significance compared with ischemia *, p< 0.01.
We tested the potential role of plasma membrane channel involvement during the induction of ischemic tolerance by pinacidil pretreatment. Hippocampal slices were preconditioned by exposure to the non-specific channel activator pinacidil and channel opening was inhibited by 5-HD during induction of preconditioning. As in our first series of experiments, pharmacological preconditioning with pinacidil induced neuroprotection. However, block of mitochondrial channels with 5-HD during pinacidil treatment abolished this protection, suggesting a requirement for channel opening. These results imply that plasma membrane channels do not play a role in IPC. In a control experiment, 5-HD treatment alone 48 h prior to lethal ischemia did not cause cell death (data not shown).
In addition to the clear role of in the induction of IPC/PPC (Figures 3 and 4), we hypothesized that εPKC-mediated neuroprotection is mediated by channel activation in response to lethal ischemia. To test this hypothesis we preconditioned slices with the εPKC peptide activator. Immediately after εPKC preconditioning, the slices were exposed to the channel-specific antagonist 5-HD (100 μM) for 1h, and 48 h later lethal ischemia was induced. This treatment abolished εPKC-mediated neuroprotection (Figure 5). These results support a direct role for in mediating neuroprotection at the time of lethal ischemia.
Figure 5.
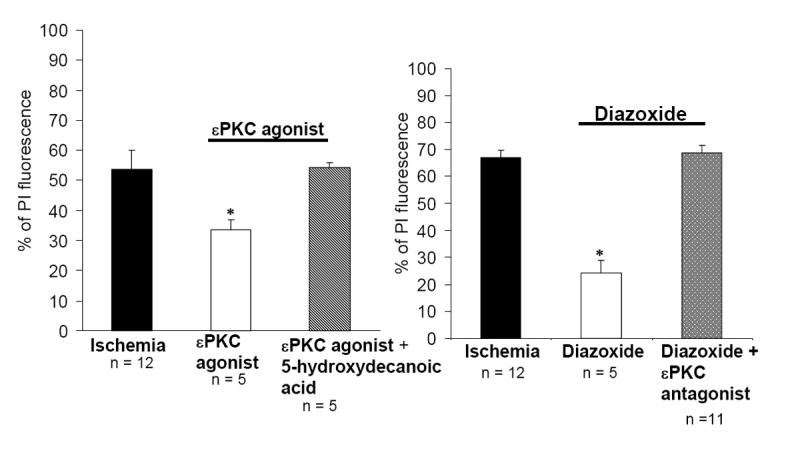
(A) Mitochondrial channel inhibition abolished εPKC agonist-mediated neuroprotection in organotypic hippocampal slices. PI fluorescence values measured 24 hr after test ischemia in different experimental groups: (1) ischemia, (2) pharmacological preconditioning (PPC; εPKC agonist treatment), (3) PPC-εPKC agonist treatment plus 5-hydroxydecanoic acid treated. (B) Inhibition of εPKC activation reduced diazoxide-mediated neuroprotection. PI fluorescence values measured 24 hr after test ischemia in different experimental groups: (1) ischemia (40 min of OGD), (2) pharmacological preconditioning (PPC; diazoxide treatment for 15 min, 48 h prior to ischemia), (3) PPC- diazoxide treatment in presence of plus εPKC antagonist. Significance compared with ischemia *, p< 0.01.
We next asked if preconditioning conferred by activation with diazoxide shared the εPKC signaling pathway with ischemic preconditioning. To precondition slices, we exposed slices to the channel opener diazoxide as above (Figure 3B) and blocked εPKC activation by an isozyme-specific cell-permeable peptide inhibitor (εV1-2). Blockade of εPKC activation during diazoxide preconditioning abolished neuroprotection in CA1 region of hippocampus (Fig 5B). Similar to ischemic preconditioning, these results suggest that pharmacological preconditioning via diazoxide requires εPKC activation.
Because ischemic preconditioning and diazoxide preconditioning share the εPKC signaling pathway, we asked if phosphorylation of Kir6.2 is a common target of ischemic and pharmacological preconditioning. The levels of Kir6.2 phosphorylation were significantly higher following preconditioning: 179% (n = 3, p<0.01) and 204% (n = 3, p<;0.01) in IPC and εPKC agonist-mediated pharmacological preconditioning groups respectively when compared with mitochondria from sham-treated slices (n = 3). Blockade of εPKC activity with the εPKC antagonist εV1-2 (n = 3) prevented Kir6.2 phosphorylation (Figure 6A-B). Thus our results suggest that phosphorylation of Kir6.2 is a target of εPKC and ischemic preconditioning.
Figure 6.
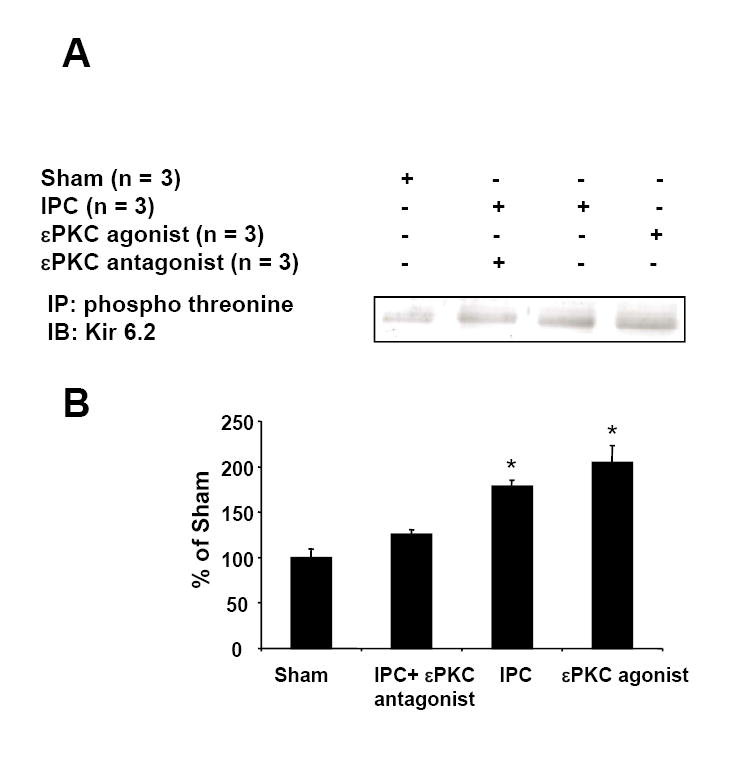
Mitochondria isolated from hippocampal organotypic slices of sham, IPC, εPKC-agonist (ΨεRACK)-mediated preconditioning or IPC plus εPKC antagonist (εV1-2) treated groups were subjected to immunoprecipitation using anti-phospho-threonine antibody. The resulting immunoprecipitates were subjected to Western blot analysis using anti-Kir 6.2 antibody. Typical Western blot images are shown in Figure B. The above Western blots were subjected to densitometric analysis. The extent of Kir 6.2 phosphorylation is shown in Figure B. *, p<0.01 as compared to sham-treated group.
Discussion
The phenomenon of ischemic preconditioning was discovered many years ago and has gained tremendous attention in stroke research (Kitagawa et al., 1990, Kirino et al., 1991, Perez-Pinzon and Born, 1999). Several publications suggested that neuronal preconditioning can also be mimicked pharmacologically; however, the specific mechanisms leading to this state of tolerance remain unknown. Taking into account the results of our previous study where εPKC played a key role in NMDA/adenosine receptor (A1AR)-induced preconditioning (Raval et al., 2003, Lange-Asschenfeldt et al., 2004), we conjectured that signals from different receptors converge at εPKC. We demonstrated increased εPKC content in mitochondrial fraction after IPC (Figure 1), suggesting that higher levels of this anti-apoptotic PKC may protect mitochondrial function after cerebral ischemia. Because not much is known about the downstream signals that ensue following εPKC translocation, we further investigated whether the channel was involved in IPC and εPKC-induced neuroprotection.
Neurons express a variety of plasma-membrane potassium channels that play important roles in regulating neuronal excitability and synaptic transmission (Liu et al., 2002). In contrast, the existence of mitochondrial channels in neurons has been highly controversial. In last few years, many studies have shown that neuronal mitochondria also have functional channels (Bajgar et al., 2001, Lacza et al., 2003). On the other hand, a recent study demonstrates the lack of diazoxide/5HD-sensitive channels in rat brain non-synaptosomal fraction (Brustovetsky et al., 2005). In this context, our results clearly show the presence of mitochondrial channel subunits in the hippocampal synaptosomal fraction (Figure 2). In the context of neuroprotection, many in vivo and in vitro studies have demonstrated beneficial effects of diazoxide against ischemic damage in brain (Liang et al., 2005, Wu et al., 2006). A low concentration of diazoxide is reported to be selective for mitochondrial channels (Liu et al., 1998). In addition to its potential effects on the channel, a major concern is that diazoxide at higher concentration also inhibits succinate dehydrogenase (Schafer et al., 1969). Direct effects of diazoxide on mitochondrial respiration have been observed in isolated heart and liver mitochondria (Drose et al., 2006). Thus, it has been difficult to precisely define the role of channel activation, and to separate these effects from events subsequent to succinate dehydrogenase inhibition or other non-specific effects of diazoxide. Previous studies have found that 3-nitropropionic acid (3-NPA), a specific inhibitor of succinate dehydrogenase, also protected neurons both in vivo and in vitro. It is not known whether effects of succinate dehydrogenase inhibition were additive or counterproductive to channel-related cellular protection, but neuroprotective effects of 3-NPA were substantially less than those reported with diazoxide alone in that study (Riepe et al., 1992, Busija et al., 2005).
Pharmacologically, the channels from myocardium are selectively activated by low concentrations of diazoxide and blocked by 5-hydroxydecanoate (Liu et al., 1998, Yang et al., 2006). The inhibition of pharmacological and ischemic preconditioning by 5HD is well documented. 5-hydroxydecanoate has also been postulated to be a specific inhibitor of mitochondrial ATP-sensitive channels. In contrast, a recent study suggested that a metabolite of 5-HD is 5-hydroxydecanoyl-CoA (5-HD-CoA), which is a substrate for the first step of ß-oxidation (Hanley et al., 2005). Thus, a non-specific effect of 5-HD would be retarded beta-oxidation of fatty acids. Owing to the complex metabolic effects of 5-HD, its use remains a pitfall of our study. However, in combination with our other data the effects of 5-HD are consistent with a role for channels in neuroprotection. For example, we also employed the channel opener diazoxide which has been demonstrated at a low concentration to be selective for (Liu et al., 1998). Our results are consistent with the requirement for channel activation during the induction of ischemic preconditioning. Consistent with a specific role of channels, the higher dosage of diazoxide treatment was not protective. This observation does not support a prominent role for plasma membrane channels. On the other hand, the channel agonist pinacidil (which activates both plasma membrane and mitochondrial channels) significantly reduced neuronal death in the CA1 region. Thus, the role of plasma membrane channels cannot be ruled out in the induction of neuroprotection conferred by IPC. In this context, it is interesting to note that Kir6.2 knockout mice show greater ischemic damage and less membrane hyperpolarization during ischemia (Sun et al., 2006), so the relative neuroprotective contributions of plasma membrane and mitochondrial channels remain to be elucidated. In addition, one of diazoxide’s putative effects mediating neuroprotection involves changes in blood flow. Our use of cultured organotypic hippocampal slices to eliminate the effects of blood flow provides convincing evidence that IPC/PPC-εPKC agonist activates a survival pathway via opening of channels.
It is well documented in heart that the increased channel activity leads to the generation of reactive oxygen species (ROS) (Busija et al., 2005, Roth et al., 2006). ROS are important intracellular signaling molecules and are increased during sublethal oxidative stress (a preconditioning stimulus). Our present findings suggest that the critical time for channels to open is during the trigger phase of preconditioning, i.e. the 48 hours prior to a lethal ischemic insult. Opening of these channels allows potassium to enter the mitochondrial inner matrix, which then causes generation and release of ROS from the respiratory chain. ROS activates phospholipase C and PKC, which, in turn, might amplify the preconditioning stimulus. Murry and colleagues first demonstrated that administration of radical scavengers blocked the beneficial effects of early ischemic preconditioning (Murry et al., 1986). Evidence for an essential role of ROS in the establishment of late preconditioning was reported by Sun and colleagues (Sun et al., 2006). Thus, generation of ROS via channel activation during the initiation of preconditioning represents an essential trigger for early and delayed neuroprotection (Figure 7). Moderate increments in ROS during ischemic preconditioning might lead to mild uncoupling of mitochondrial proteins and prevent consequences of mitochondrial oxidative stress during reperfusion, and thus confer neuroprotection.
Figure 7.
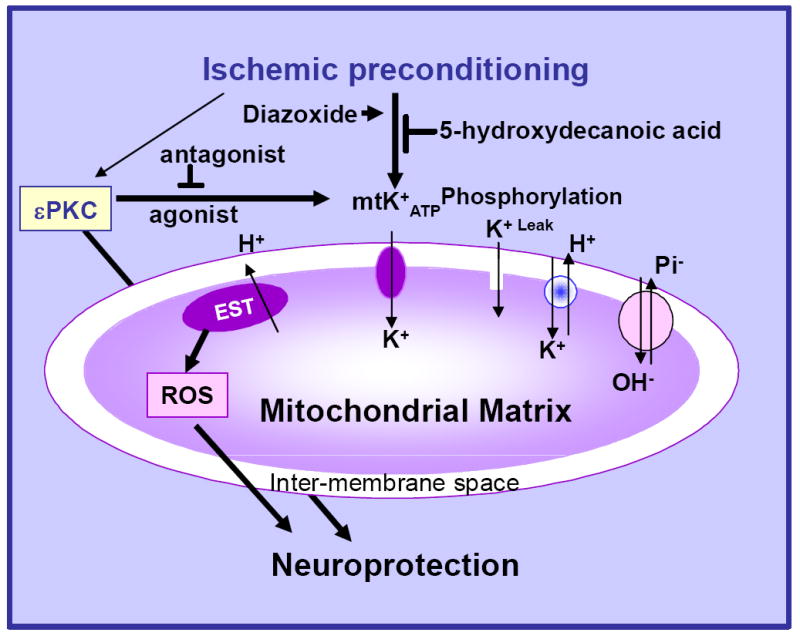
A schematic model describing the sequence of events after induction of ischemic preconditioning in the organotypic hippocampal slices.
In conclusion, the present study clearly demonstrates the presence of ATP-sensitive potassium channel-specific subunits (Kir6.1 and Kir6.2) in hippocampal mitochondria. Most importantly, both ischemic preconditioning and pharmacological preconditioning with the εPKC activating peptide (ΨεRACK) ultimately resulted in phosphorylation of the channel subunit Kir6.2 in mitochondrial fractions. We demonstrated increased εPKC content in the mitochondrial fraction 1h after IPC, suggesting that higher levels of this anti-apoptotic PKC might be responsible for improved mitochondrial function and resistance to cerebral ischemic damage as reported earlier by our group (Dave et al., 2001). We also demonstrated that neuroprotection conferred by ischemic preconditioning was lost in the presence of channel antagonists. Overall, these results suggest that the signal transduction pathway that ensues after IPC/εPKC agonist-mediated preconditioning requires opening of the mitochondrial channel.
Experimental procedure
All animal protocols were approved by the Animal Care and Use Committee of the University of Miami.
Animal model
Male Sprague Dawley rats weighing 250 to 300 g were fasted overnight and then anesthetized with 4% isoflurane and 70% nitrous oxide (in a balance of oxygen) by inhalation. The femoral arteries were cannulated for blood pressure measurements and for arterial sampling of blood gases. Arterial blood gases (178 pH/blood gas analyzer, Ciba-Corning), plasma glucose levels (One Touch glucose monitor, Lifescan), and hematocrit were measured intermittently throughout the experiment. Our goal was to maintain blood gases in the normal range (arterial pCO2 = 35 to 40 mm Hg, pO2 = 120 to 140 mm Hg). If blood gases were not maintained within this range throughout the period of surgery and data collection, the rats were euthanized and not included in the analysis. After endotracheal intubation and artificial ventilation with 1% isoflurane and 70% nitrous oxide (in a balance of oxygen), the rats were immobilized with vecuronium (0.75 mg/kg intravenously). Both common carotid arteries were exposed by a midline ventral incision and gently dissected free of surrounding nerve fibers. Ligatures of polyethylene (PE-10) tubing, contained within a double-lumen Silastic tubing, were passed around each carotid artery. Brain temperature was monitored with a 33-gauge thermocouple implanted in the temporalis muscle (Dietrich et al., 1993). The temperature was maintained at 36° to 37°C throughout the experiment by a small lamp placed above the animal’s head.
Induction of ischemic preconditioning
Prior to preconditioning, blood was gradually withdrawn from the femoral vein into a heparinized syringe to reduce systemic blood pressure to 50 mm Hg. Cerebral ischemia then was produced by tightening the carotid ligatures bilaterally. We have previously shown that this procedure promotes loss of ion homeostasis (anoxic depolarization as measured with potassium-selective microelectrodes), as well as histopathologic changes (Perez-Pinzon et al., 1997). To allow post-ischemic reperfusion, the carotid ligatures were removed, and the shed blood was reinjected into the femoral vein. This infusion usually restored mean arterial blood pressure to 130 to 140 mm Hg. The vessels were inspected to verify that perfusion was re-established. The duration of ischemia for preconditioning was 2 minutes. Two experimental groups were used: Sham: and IPC. Sham surgery was performed on the animals for IPC: two minutes of ischemia (IPC) was induced.
Preparation of organotypic slice cultures
Neonatal (9-11 days old) Sprague-Dawley rats were anesthetized by intraperitoneal injection of ketamine (1.0 mg/pup). Animals were decapitated and the brains quickly removed. Organotypic hippocampal slice cultures were prepared as described previously (Xu et al., 2002). In brief, transverse slices (400 μm) were dissected from the hippocampi and placed in Gey’s Balanced Salt Solution (all chemicals were obtained from Sigma St. Louis, MO, or specified otherwise) supplemented with 6.5 mg/ml glucose at 4°C. After one hour, two slices were placed onto one 30 mm diameter membrane insert (Millicell-CM, Millipore) and inserts were transferred to six-well culture plates with 1 ml of culture medium per well. The culture medium consisted of 50% Minimum Essential Medium, 25% Hank’s Balanced Salt Solution, 25% Heat-Inactivated Horse Serum (all media were purchased from Gibco/Life Technologies) supplemented with 6.5 mg/ml glucose and 1 mM glutamine. The slice cultures were placed in an incubator (equilibrated at 36° C, 95 % O2, 5 % CO+, humidity 100 %) for 14-15 days before experiments were performed.
Experimental Design
The organotypic slices were divided into 5 major groups:
Group-I: Sham. Slices were incubated for 15 min in HBSS solution supplied with an equimolar concentration of glucose instead of sucrose (Sham - IPC) and were subsequently transferred back to regular media. After 48 h the same procedure was performed for the extended duration of 40 min (sham OGD – sham ischemia).
Group-II: ‘Test’ ischemia. Sham IPC was induced as in group-I followed after 48 h by ‘test’ ischemia (40 min of OGD).
Group-III: Ischemic preconditioning (IPC). Slices underwent IPC (15 min of OGD) and 48 h later ‘test’ ischemia (40 min of OGD).
Group-IV: Pharmacological preconditioning (PPC) by selective pharmacological agents. (A) εPKC agonist (tat-ΨεRACK cell-permeable peptide 200 nm, KAI Pharmaceuticals Inc., 270 Littlefield Ave South San Francisco, CA 94080), slices were exposed to εPKC agonist for 15 min as described previously (Raval et al., 2003) (B) general channel opener pinacidil (15 min; 10, 50 and 100 μM) and (C) channel-specific agonist diazoxide ( -specific: 15 min; 50 μM; non-specific: 150 μM (Liu et al., 1998)) hypothesized to emulate IPC. Slices underwent PPC and 48 h later ‘test’ ischemia (40 min of OGD). For control experiments slices were exposed to εPKC agonist or pinacidil or diazoxide for 15 min. Slices underwent sham-OGD (as described for group-I).
Group-V: Pharmacological blockade of IPC. IPC and PPC experiments were carried out followed by general or mitochondria-specific channel inhibitors treatment, (A) tolbutamide (1h of treatment following IPC/PPC; 5, 10 and 15 μM) (B) 5-hydroxydecanoic acid (HD; 1h of treatment following IPC/PPC; 50, 100 and 200 μM) during and after the preconditioning procedure. The slices were subjected to ‘test’ ischemia 48 h later. For control experiments, slices were exposed to tolbutamide or 5-hydroxydecanoic acid for 1h. Slices underwent sham-OGD (as described for group-I).
To provide better controls in these experiments, all six-well plates used contained at least one well for the sham, IPC and ischemia groups. In addition, for statistical purposes, each insert had two slices obtained from two different pups. Thus, every slice represents a different animal.
For the isolation of mitochondria, immunoprecipitation and western blotting experiments, hippocampal organotypic slices were harvested at 1 h of sham, IPC, εPKC agonist-mediated preconditioning or IPC+εPKC antagonist (tat –εV1-2 cell-permeable peptide; described previously (Chen and Mochly-Rosen, 2001)) treatment. For one sample preparation, one hundred slices were pooled together and three such samples per experimental group were used for analysis.
Induction of ischemia (oxygen/glucose deprivation – OGD)
As we described previously, slices were washed three times with aglycemic Hank’s Balanced Salt Solution (AHBSS) (pH 7.4) with the following constituents: CaCl2 1.26 mM, KCl 5.37 mM, KH2PO4 0.44 mM, MgCl2 0.49 mM, MgSO4.7H2O 0.41 mM, NaCl 136.9 mM, NaHCO3 4.17 mM, Na2HPO4 0.34 mM, sucrose 15 mM (Xu et al., 2002). The slices were then transferred into an airtight chamber which was equilibrated with 95 % N2 / 5 % CO2 gas (preheated to 37° C and 100% humidity) blown through the chamber for 5 min (4 L/min) to achieve anoxic conditions. Then, the chamber was sealed and remained incubated for 10 min (for a total of 15 min – preconditioning) or 35 min (for a total of 40 min ischemic insult). Following OGD, slices were placed back in the incubator in plates containing normal culture medium.
Assessment of neuronal cell death by propidium iodide staining technique
To determine the extent of neuronal damage in the organotypic slice culture, we used the propidium iodide (PI) method, described previously (Xu et al., 2002, Raval et al., 2003). The organotypic slices in all the experimental groups were incubated in culture medium supplemented with 2 μg/ml PI (Sigma St. Louis, MO) for 1 h prior to imaging. Images were taken using an inverted fluorescence microscope (Olympus IX 50), equipped with a light-intensifying SPOT CCD camera (Diagnostic Instruments Inc., Sterling Heights, MI), and SPOT Advanced software was used to assess the proportion of cell death. Images of the cultured slices were taken (1) as baseline prior to the preconditioning procedure; (2) 24 h after the ‘test’ ischemic insult to assess ischemic damage; and (3) 24 h after NMDA treatment to assess maximum damage to neuronal cells. The hippocampal CA1 subfield was chosen as the region of interest, and quantification was performed using Scion Image software. The percentage of relative optical intensity (ROI) served as an index of neuronal cell death.
Isolation of mitochondria
Mitochondria from hippocampus/liver of 9-11 day old rat pups and hippocampal organotypic slices were isolated according to the procedure described previously (Sciamanna and Lee, 1993). In brief, rat pups were anesthetized using 5% isoflurane (70 % N2O – 30 % O2). Pups were decapitated and the hippocampus was immediately immersed in cold (4°C) isolation medium, consisting of 250 mM sucrose, 10 mM HEPES (pH 7.4), 1 mg/ml bovine serum albumin (fraction V) (BSA), 0.5 mM ethylenediaminetetraacetic acid (EDTA) and 0.5 mM ethylene glycol bis (-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA) dipotassium salt. The tissue was homogenized in a hand-operated glass Teflon homogenizer by seven up-and-down strokes. For hippocampal organotypic slices, slices were scraped off from the membrane and washed with isolation media twice. Washed slices were homogenized using a pestle (Kimble-Knotes, Vineland, NJ, USA). The homogenates were diluted to yield 10 % (W/V) homogenate, and centrifuged at 2000× g for 3 minutes in a Sorvall RC5 centrifuge. The supernatants were placed inside a nitrogen cell bomb and put under a pressure of 1200 psi for 7.5 min (Brown et al., 2004). The samples obtained following nitrogen compression were centrifuged at 12,000 × g for 8 min. The resulting supernatant was discarded and the pellet was resuspended in the isolation medium. The suspension was centrifuged at 12,000 × g for 10 min. The resulting pellet was resuspended in 0.25 M sucrose and centrifuged at 12,000 × g for 10 min. The pellet was suspended in 0.25 M sucrose and was used as the source of total mitochondria. During standardization procedures we determined the purity of mitochondria by confirming the absence of synaptophysin and calregulin, markers of synaptosomes and endoplasmic reticulum (microsomes), respectively.
Adult rat hippocampal mitochondria were isolated according to previously published procedures (Dunkley et al., 1988) with minor modification. In brief, rats were decapitated under isoflurane anesthesia at various time points after IPC or sham operation. The hippocampus was removed immediately and immersed into cold (4°C) isolation medium. The average weight of hippocampus in all animal groups was approximately 120 mg (pooled from both hemispheres). To improve yield of mitochondria, the pellet was rehomogenized and diluted to 10% (W/V). This step is a modification to the previously published procedures (Dunkley et al., 1988).
Immunoprecipitation
Immunoprecipitation was carried out to study phosphorylation of Kir6.2. The mitochondria were isolated from hippocampal organotypic slices after 1 h of sham, IPC, εPKC agonist-mediated preconditioning or IPC+εPKC antagonist (εV1-2, KAI Pharmaceuticals Inc., 270 Littlefield Ave South San Francisco, CA 94080) treatment. The total mitochondrial fraction (500 μg) was dissolved in 0.3% SDS, sonicated twice times for 5 second each, then immediately adjusted to 0.1% SDS concentration in an immunobuffer consisting of 50 mM Tris-HCl, 1% Triton X-100, 1% CHAPS and 0.5% NP-40 (pH 7.4) and incubated for 20 min at room temperature. Immunoprecipitation was carried out using protein A sepharose beads (Sigma St. Louis, MO) and anti-phospho-threonine mouse monoclonal antibodies (Sigma St. Louis, MO) as per manufacturer’s instructions. The samples were washed twice with immunobuffer following overnight incubation of samples with protein A sepharose beads (Sigma St. Louis, MO) and anti-phospho-threonine. The resulting pellet was used for immunoblotting using anti-Kir6.2 antibody.
Protein analysis and Western blotting
Proteins were measured using the Bio-Rad protein assay kit (Hercules, CA, U.S.A.). Forty micrograms of protein from each sample was separated by 12% SDS-PAGE. Protein was transferred to Immobilon-P (Millipore, MA, U.S.A.) membrane and incubated with the primary antibody anti-Kir 6.1 (1: 1000; Santa Cruz, CA, USA), anti- Kir 6.2 (1:1000; Sigma, MO, USA), anti-COX-IV (cytochrome oxidase subunit four – mitochondrial marker; 1:250, Sigma, MO, USA). Immunoreactivity was detected using enhanced chemiluminescence (ECL Western blotting detection kit, Amershampharmacia biotech, UK). Western blot images were digitized at 8-bit precision by means of a charge-coupled-device-based (CCD) camera (8–12 bit, Xillix Technologies Corp., Vancouver, Canada) equipped with a 55 mm Micro-Nikkor lens (Nikon, Japan). The camera was interfaced to an advanced image-analysis system (MCID Model M2, Imaging Research, Inc., St. Catherines, Ont., Canada). The digitized immunoblots were subjected to densitometric analysis using MCID software.
Statistical analysis
The results are expressed, as mean ± SEM. Statistical significance was determined with an ANOVA test followed by a Bonferroni’s post-hoc test.
Acknowledgments
Supported by PHS grants NS34773, NS05820, NS045676, and NS054147. We thank Dr. Brant Watson for critical reading of this manuscript.
Abbreviations
- BSA
bovine serum albumin
- εPKC
epsilon protein kinase C
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol bis (-aminoethyl ether)-N,N,N’,N’-tetraacetic acid
- HBSS
hank’s balanced salt solution
- HD
5-hydroxydecanoic acid
- IPC
ischemic preconditioning
- OGD
oxygen/glucose deprivation
- channel
mitochondrial ATP-sensitive potassium channel
- MEM
minimum essential medium
- PKC
protein kinase C
- PPC
pharmacological preconditioning
- ROI
relative optical intensity
- Slice cultures
organotypic hippocampal slice cultures
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bajgar R, Seetharaman S, Kowaltowski AJ, Garlid KD, Paucek P. Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J Biol Chem. 2001;276:33369–33374. doi: 10.1074/jbc.M103320200. [DOI] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Dorenbos KA, Modafferi EA, Geddes JW, Steward O. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. J Neurosci Methods. 2004;137:299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- Brustovetsky T, Shalbuyeva N, Brustovetsky N. Lack of manifestations of diazoxide/5-hydroxydecanoate-sensitive KATP channel in rat brain nonsynaptosomal mitochondria. J Physiol. 2005;568:47–59. doi: 10.1113/jphysiol.2005.091199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija DW, Katakam P, Rajapakse NC, Kis B, Grover G, Domoki F, Bari F. Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res Bull. 2005;66:85–90. doi: 10.1016/j.brainresbull.2005.03.022. [DOI] [PubMed] [Google Scholar]
- Chen C, Mochly-Rosen D. Opposing effects of delta and xi PKC in ethanol-induced cardioprotection. J Mol Cell Cardiol. 2001;33:581–585. doi: 10.1006/jmcc.2000.1330. [DOI] [PubMed] [Google Scholar]
- Dave KR, Saul I, Busto R, Ginsberg MD, Sick TJ, Perez-Pinzon MA. Ischemic preconditioning preserves mitochondrial function after global cerebral ischemia in rat hippocampus. J Cereb Blood Flow Metab. 2001;21:1401–1410. doi: 10.1097/00004647-200112000-00004. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Busto R, Alonso O, Globus MY, Ginsberg MD. Intraischemic but not postischemic brain hypothermia protects chronically following global forebrain ischemia in rats. J Cereb Blood Flow Metab. 1993;13:541–549. doi: 10.1038/jcbfm.1993.71. [DOI] [PubMed] [Google Scholar]
- Drose S, Brandt U, Hanley PJ. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J Biol Chem. 2006;281:23733–23739. doi: 10.1074/jbc.M602570200. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Heath JW, Harrison SM, Jarvie PE, Glenfield PJ, Rostas JA. A rapid Percoll gradient procedure for isolation of synaptosomes directly from an S1 fraction: homogeneity and morphology of subcellular fractions. Brain Res. 1988;441:59–71. doi: 10.1016/0006-8993(88)91383-2. [DOI] [PubMed] [Google Scholar]
- Garlid KD. Opening mitochondrial K(ATP) in the heart--what happens, and what does not happen. Basic Res Cardiol. 2000;95:275–279. doi: 10.1007/s003950070046. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P. Mitochondrial potassium transport: the K(+) cycle. Biochim Biophys Acta. 2003;1606:23–41. doi: 10.1016/s0005-2728(03)00108-7. [DOI] [PubMed] [Google Scholar]
- Hanley PJ, Drose S, Brandt U, Lareau RA, Banerjee AL, Srivastava DK, Banaszak LJ, Barycki JJ, Van Veldhoven PP, Daut J. 5-Hydroxydecanoate is metabolised in mitochondria and creates a rate-limiting bottleneck for beta-oxidation of fatty acids. J Physiol. 2005;562:307–318. doi: 10.1113/jphysiol.2004.073932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C, Lauritzen I, Widmann C, Lazdunski M. Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci U S A. 1995;92:4666–4670. doi: 10.1073/pnas.92.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaburek M, Costa AD, Burton JR, Costa CL, Garlid KD. Mitochondrial PKC epsilon and mitochondrial ATP-sensitive K+ channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circ Res. 2006;99:878–883. doi: 10.1161/01.RES.0000245106.80628.d3. [DOI] [PubMed] [Google Scholar]
- Kirino T, Tsujita Y, Tamura A. Induced tolerance to ischemia in gerbil hippocampal neurons. J Cereb Blood Flow Metab. 1991;11:299–307. doi: 10.1038/jcbfm.1991.62. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- Lacza Z, Snipes JA, Kis B, Szabo C, Grover G, Busija DW. Investigation of the subunit composition and the pharmacology of the mitochondrial ATP-dependent K+ channel in the brain. Brain Res. 2003;994:27–36. doi: 10.1016/j.brainres.2003.09.046. [DOI] [PubMed] [Google Scholar]
- Lange-Asschenfeldt C, Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon protein kinase C mediated ischemic tolerance requires activation of the extracellular regulated kinase pathway in the organotypic hippocampal slice. J Cereb Blood Flow Metab. 2004;24:636–645. doi: 10.1097/01.WCB.0000121235.42748.BF. [DOI] [PubMed] [Google Scholar]
- Li GC, Vasquez JA, Gallagher KP, Lucchesi BR. Myocardial protection with preconditioning. Circulation. 1990;82:609–619. doi: 10.1161/01.cir.82.2.609. [DOI] [PubMed] [Google Scholar]
- Liang HW, Xia Q, Bruce IC. Reactive oxygen species mediate the neuroprotection conferred by a mitochondrial ATP-sensitive potassium channel opener during ischemia in the rat hippocampal slice. Brain Res. 2005;1042:169–175. doi: 10.1016/j.brainres.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu D, Lu C, Wan R, Auyeung WW, Mattson MP. Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J Cereb Blood Flow Metab. 2002;22:431–443. doi: 10.1097/00004647-200204000-00007. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Ohnuma Y, Miura T, Miki T, Tanno M, Kuno A, Tsuchida A, Shimamoto K. Opening of mitochondrial K(ATP) channel occurs downstream of PKC-epsilon activation in the mechanism of preconditioning. Am J Physiol Heart Circ Physiol. 2002;283:H440–447. doi: 10.1152/ajpheart.00434.2001. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Born JG. Rapid preconditioning neuroprotection following anoxia in hippocampal slices: role of the K+ ATP channel and protein kinase C. Neuroscience. 1999;89:453–459. doi: 10.1016/s0306-4522(98)00560-0. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Vitro TM, Dietrich WD, Sick TJ. The effect of rapid preconditioning on the microglial, astrocytic and neuronal consequences of global cerebral ischemia. Acta Neuropathol (Berl) 1999;97:495–501. doi: 10.1007/s004010051019. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:175–182. doi: 10.1097/00004647-199702000-00007. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA. Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci. 2003;23:384–391. doi: 10.1523/JNEUROSCI.23-02-00384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riepe M, Hori N, Ludolph AC, Carpenter DO, Spencer PS, Allen CN. Inhibition of energy metabolism by 3-nitropropionic acid activates ATP-sensitive potassium channels. Brain Res. 1992;586:61–66. doi: 10.1016/0006-8993(92)91371-k. [DOI] [PubMed] [Google Scholar]
- Roth S, Dreixler JC, Shaikh AR, Lee KH, Bindokas V. Mitochondrial potassium ATP channels and retinal ischemic preconditioning. Invest Ophthalmol Vis Sci. 2006;47:2114–2124. doi: 10.1167/iovs.05-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Costa AD, Saito T, Ogura T, Ishida H, Garlid KD, Nakaya H. Bepridil, an antiarrhythmic drug, opens mitochondrial KATP channels, blocks sarcolemmal KATP channels, and confers cardioprotection. J Pharmacol Exp Ther. 2006;316:182–188. doi: 10.1124/jpet.105.094029. [DOI] [PubMed] [Google Scholar]
- Schafer G, Wegener C, Portenhauser R, Bojanovski D. Diazoxide, an inhibitor of succinate oxidation. Biochem Pharmacol. 1969;18:2678–2681. [PubMed] [Google Scholar]
- Sciamanna MA, Lee CP. Ischemia/reperfusion-induced injury of forebrain mitochondria and protection by ascorbate. Arch Biochem Biophys. 1993;305:215–224. doi: 10.1006/abbi.1993.1414. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Miki T, Seino S, French RJ. Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol. 2006;95:2590–2601. doi: 10.1152/jn.00970.2005. [DOI] [PubMed] [Google Scholar]
- Wu L, Shen F, Lin L, Zhang X, Bruce IC, Xia Q. The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci Lett. 2006;402:184–189. doi: 10.1016/j.neulet.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Xu GP, Dave KR, Vivero R, Schmidt-Kastner R, Sick TJ, Perez-Pinzon MA. Improvement in neuronal survival after ischemic preconditioning in hippocampal slice cultures. Brain Res. 2002;952:153–158. doi: 10.1016/s0006-8993(02)02988-8. [DOI] [PubMed] [Google Scholar]
- Yang Y, Liu X, Long Y, Wang F, Ding JH, Liu SY, Sun YH, Yao HH, Wang H, Wu J, Hu G. Activation of mitochondrial ATP-sensitive potassium channels improves rotenone-related motor and neurochemical alterations in rats. Int J Neuropsychopharmacol. 2006;9:51–61. doi: 10.1017/S1461145705005547. [DOI] [PubMed] [Google Scholar]