

Abstract
Activation of nuclear factor-κB (NF-κB), a key feature of the neurotrophin signaling, has been shown to be critical for neuronal survival under pathologic settings. However, the precise mechanism by which neurotrophins activate NF-κB is not well understood. Here we report that the Ankyrin-rich Membrane Spanning (ARMS/Kidins220) protein, a novel transmembrane substrate of tropomyosin receptor kinase B (TrkB), plays an important role in NF-κB signaling elicited by brain-derived neurotrophic factor (BDNF). Accordingly, depletion of ARMS by specific RNA interference, or disruption of ARMS-TrkB interaction with expression of dominant-negative ARMS mutant, abolished BDNF-induced signaling to NF-κB. Our data further suggests that ARMS may promote NF-κB signaling via activation of mitogen-activated kinase (MAPK) and IκB kinase (IKK), thereby facilitating phosphorylation of RelA (major NF-κB subunit) at an IKK-sensitive site. The results shown here identify ARMS as a major factor that links neurotrophin signaling to NF-κB.
INTRODUCTION
The neurotrophin family of proteins plays an important role in determining neuronal survival, differentiation and synaptic plasticity in the nervous system via binding to two discrete receptor subtypes: the Trk (tropomyosin receptor kinase) family of receptor tyrosine kinases and the p75NTR (p75 neurotrophin receptor) (reviewed in (Chao et al., 2006)). The latter receptor subtype binds to all neurotrophins with similar affinity, whereas the Trk receptors (TrkA, TrkB and TrkC) are engaged preferentially by nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3), respectively. Upon ligand binding, Trk receptors dimerize and become catalytically active by autophosphorylation at specific tyrosine residues, which serve as interaction sites for adaptor molecules that contain PTB/SH2 domains (Hennigan et al., 2007). The consequences of Trk receptor activation include a number of signaling cascades, such as Ras/Raf/MAPK (mitogen-activated protein kinase) (Thomas et al., 1992), PI3K (phosphoinositide 3-kinase) (Atwal et al., 2000), and phospholipase C-γ pathways (Vetter et al., 1991), leading to the activation of a number of transcription factors, including nuclear factor κB (NF-κB) (Maggirwar et al., 1998; Ramirez et al., 2001).
NF-κB represents a family of transcription factors that regulate diverse genes involved in the activation and survival of multiple cell types, including neurons (Nagy et al., 2007; Ramirez et al., 2001). In mammals, the NF-κB family includes RelA, RelB, c-Rel, NF-κB1 (or p50), and NF-κB2 (or p52), which form different homo- and hetero-dimers. The NF-κB members are normally sequestered in the cytoplasm as inactive complexes by physical interaction with specific inhibitors, including IκBα and related proteins (Bonizzi and Karin, 2004). Activation of NF-κB involves phosphorylation-triggered degradation of IκBα molecules and nuclear translocation of NF-κB complexes, particularly the p50/RelA and RelA/RelA dimers. A multi-subunit IκB kinase (IKK) complex responds to diverse cellular stimuli, such as neurotrophic factors, neurotransmitters, cytokines and oxidative stress, and mediates the phosphorylation of various components of NF-κB signaling, including IκBα as well as NF-κB family members such as RelA (Ghosh and Karin, 2002; Malek et al., 2007). Following activation, NF-κB performs a key role as a mediator of transcription-dependent changes in the neuronal structure and function, not only during development, but also in the pathological setting (Mattson and Meffert, 2006).
Following neurotrophin binding, a set of adaptor molecules, such as Shc, Grb2, Gab1 and FRS2, dock on the Trk receptors at two phosphorylated tyrosine residues, Y490 and Y785, and promote activation of several intracellular signaling pathways (Chao, 2003). Among these pathways, sustained activation of MAPK is known to be a unique feature of neurotrophin signaling (Marshall, 1995). Recent studies implicated a novel tetraspanning membrane protein substrate of Trk receptors, Ankyrin-Rich Membrane Spanning (ARMS) protein (Kong et al., 2001) (also known as a substrate for protein kinase D, Kidins220 (Iglesias et al., 2000)) (referred hereafter as ARMS), in mediating neurotrophin signals in a manner that selectively triggers a sustained activation of MAPK pathway in neurons (Arevalo et al., 2006; Arevalo et al., 2004). ARMS is a large protein (220 kilodalton) that interacts with all three Trk receptors and p75NTR (but not with EGF receptor (Kong et al., 2001)) through its transmembrane domains. Its structural features include multiple conserved phosphorylation sites, ankyrin repeats, a SAM domain and a C-terminal PDZ domain. However, it does not contain distinguishing domains of adaptor molecules, like SH2, SH3 or PH domains. Further investigations elucidated that ARMS also plays an important role in promoting neuronal differentiation via direct interaction with the kinesin-1 motor complex in the cells (Bracale et al., 2007).
In the present study we show that ARMS plays a critical role in the BDNF-mediated NF-κB signaling pathway in a manner that promotes survival of neurons. Moreover, we show that the sequential events, such as transmembrane interaction of ARMS with TrkB, followed by ARMS-dependent activation of MAPK and IKK, are crucial for BDNF to trigger NF-κB activation. Consistently, an intact expression of ARMS appears to be necessary for BDNF-induced phosphorylation of the NF-κB subunit RelA at IKK-sensitive site (Serine 536). Together, these findings establish ARMS as a key component of Trk signaling that regulates NF-κB activation in neurons, a pathway which controls neuronal survival and function.
RESULTS
ARMS is required for neuroprotective activity of BDNF
BDNF prevents neuronal apoptosis induced by a wide range of toxins, including molecules encoded by HIV-1 (Bachis and Mocchetti, 2005; Ramirez et al., 2001; Sui et al., 2006b). Since a previous report suggests that an unusual ankyrin-rich membrane spanning protein (ARMS) acts as a major platform for prolonged MAP kinase signaling by neurotrophins (Arevalo et al., 2004), we sought to determine if ARMS plays a role in mediating the neuroprotective effects of BDNF. To do this, we first constructed a clone of lentivirus vector, in which ARMS-specific shRNA was placed directly under the control of the U6 promoter derived from human RNA polymerase III (Qin et al., 2003)(Fig. 1A). This vector also encodes for enhanced green fluorescence protein (GFP) regulated separately by an ubiquitin C promoter. The virus was then packaged, purified and used to infect primary cultures of rat cortical neurons. As determined by fluorescence microscopy, when used at multiplicity of infection (MOI) of 10, nearly 80% of neurons were infected by this virus (Fig. 1B). Importantly, under these conditions, expression of targeted shRNA in cortical neurons resulted in a marked reduction in endogenous ARMS, but not α-tubulin levels (lanes marked as ‘ARMS shRNA-GFP’, Fig. 1C). Whereas infection with empty vector (this contains only GFP; Fig. 1C) failed to alter ARMS levels in neurons.
Figure 1.
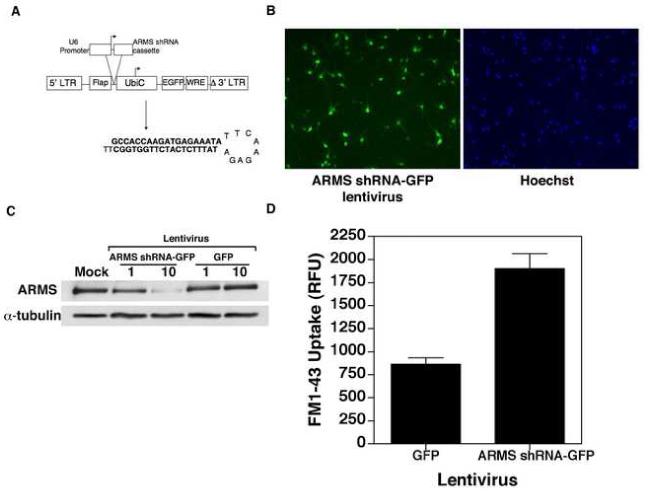
(A) Generation of ARMS shRNA-GFP expressing lentivirus. The ARMS shRNA cassette was subcloned into a plasmid (pBluescript) downstream of the human U6 RNA polymerase III promoter. The plasmid region containing the cassette and the promoter was then subcloned into the FG12 lentiviral vector, which also encodes for GFP under the control of Ubiquitin C (UbiC) promoter. (B) Analysis of the efficiency of lentiviral transduction of primary cortical neurons. Primary cortical neurons were transduced with ARMS shRNA-GFP lentivirus (MOI 10) as outlined in Materials and Methods. Panel on the left-hand indicates GFP-positive cells, whereas the right-hand panel shows Hoechst 33342 staining of the same microscopic field (C) Analysis of ARMS expression following lentivirus infection. Cell lysates were prepared from infected cortical neurons and immunoblot analyses were performed using antibodies specific to ARMS and α-tubulin. ARMS expression was reduced by approximately 80%. (D) Functional validation of ARMS shRNA-GFP expressing lentivirus. Primary cortical neurons were infected with indicated lentiviruses for 7 days. On DIV9, 10μM of FM1-43 was added to the medium for 15 minutes. After washing, the cells were resuspended in HBSS plus 100mM KCl for depolarization. The release of FM1-43 was monitored and recorded over 60 minutes at 1 minute intervals. As previously reported (Cortes et al., 2007), synaptic activity was increased in the absence of ARMS.
Recently, it was proposed that ARMS might function as a homeostatic regulator of overall synaptic strength in neurons (Cortes et al., 2007). In particular, these studies have used ARMS-specific shRNA to show an inverse relationship between synaptic activity and ARMS levels in cultured neurons. Therefore, to functionally validate our ARMS shRNA lentivirus vector, we tested its effect on activity-dependent synaptic vesicle uptake in neurons. To do this, cortical neurons were infected at DIV 2 (day in vitro) with lentivirus vectors expressing ARMS-shRNA or GFP alone, and vesicular activity was analyzed at DIV 9 (at which time the expression of postsynaptic density protein 95 and ARMS in uninfected neurons reached peak levels, see Fig. 5A) using the styryl dye, FM1-43, as previously described (Bradley and Sporns, 1999). Briefly, 10 μM FM1-43 was added directly to the culture medium of vector-transduced neurons for 15 minutes at 37°C. Following loading, the cells were suspended in a depolarizing solution of HBSS containing 100 mM KCl, and release of FM1-43 was monitored at one minute intervals for a total of one hour (to allow time for complete recycling of all FM1-43 labeled vesicles), using 438 excitation and 605 emission filters. Here, the total loss of FM1-43 signal during the release period equals the total spontaneous activity-dependent vesicular uptake of FM1-43 in each well over the loading period. This absolute FM1-43 uptake value reflects the total activity of the culture and is equivalent to the synaptic or vesicular release probability of the culture. Thus, in this assay, we found that the vesicular activity (as reflected by FM1-43 uptake) was elevated in those cells that had reduced levels of ARMS (Fig. 1D), thereby implicating this protein in the regulation of activity-dependent vesicular uptake.
Figure 5.
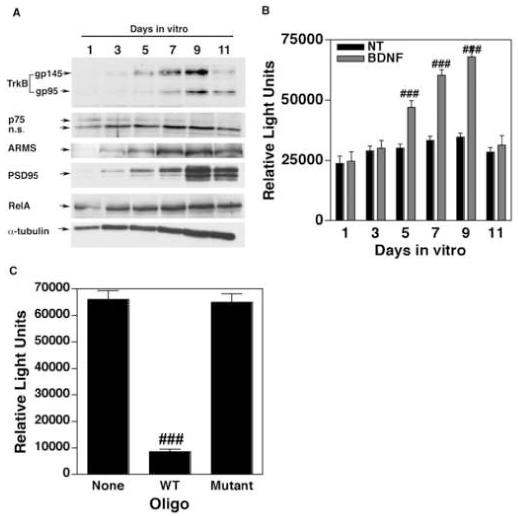
(A) Expression of TrkB and ARMS expression is developmentally regulated. Whole cell lysates of primary cortical neurons were prepared from cultures incubated for various days in vitro (DIV) and immunoblot analyses were performed as indicated. (B) The extent of NF-κB activation induced by BDNF correlates with the expression of TrkB and ARMS. TransAM NF-κB DNA binding ELISAs were performed using nuclear extracts of primary cortical neurons treated with BDNF on different DIV. The data (expressed as relative light units) is the mean of three independent observations (bars indicate SEM). ### equals p<0.001. (C) Specificity of NF-κB ELISA. Nuclear extracts derived from neuronal cultures (DIV 9) treated with BDNF (100 ng/ml) were used for TransAM ELISA in the presence of 20× excess of oligonucleotides containing either WT or Mutant NF-κB consensus sequences. These assays demonstrate that the measured activity is indeed specific to NF-κB. ### equals p<0.001 as compared to ‘None’.
To investigate whether ARMS is required for neuroprotection conferred by BDNF, we incubated cortical neurons for 7 days with lentivirus vectors encoding either GFP alone (control) or GFP together with ARMS shRNA (ARMS shRNA-GFP). The cells were then treated with gp120 (5nM) in the absence or presence of BDNF (100ng/ml) for 72h, and neuronal survival was measured by staining the cells with Hoechst 33342 dye. Here, the neurons that had a diffuse nuclear Hoechst staining and a round, intact nuclei were scored healthy, whereas neurons demonstrating bright nuclear staining and shrunken, distorted nuclei were considered apoptotic. As expected (Sui et al., 2006a), gp120 treatment led to a high level of apoptosis in neurons that are expressing either GFP or ARMS shRNA-GFP (Fig. 2A and B). Interestingly, this effect of gp120 was profoundly inhibited by pre-incubation with BDNF in control neurons (expressing GFP alone), but not in neurons expressing ARMS shRNA-GFP (Fig. 2A and B), suggesting that the intact expression of ARMS is necessary for BDNF-induced neuroprotection.
Figure 2.
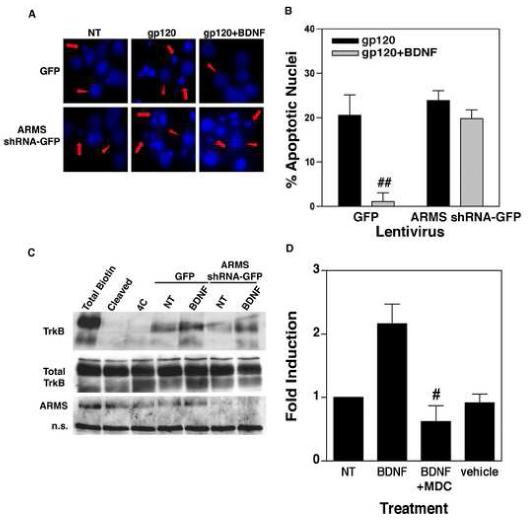
(A) Neuroprotective effect of BDNF is mediated by ARMS. Primary cortical neurons infected with ARMS shRNA-GFP or GFP lentivirus were treated with BDNF (100 ng/ml) for 18 hours, followed by incubation of the cells with gp120 (5nM) for an additional 72h. Staining of the cultures with Hoechst 33342 dye revealed a higher percentage of apoptotic nuclei in gp120-exposed cells (middle panels). Whereas, co-administration of BDNF inhibited gp120-induced apoptosis in control cells (cells expressing GFP alone, upper right panel) but not in ARMS shRNA-GFP expressing cells (lower right panel). Apoptotic cells are marked with arrows, whereas arrow-heads show healthy cells. (B) Quantitative analysis of data from three independent experiments that examined survival of neurons as outlined above. The data are presented as Mean ± SEM of percent apoptotic neurons. ## indicates P<0.01 as compared to gp120-treated control cells (GFP, filled bar). (C) Loss of ARMS does not affect BDNF-induced internalization of TrkB. Surface proteins of cells infected with ARMS shRNA-GFP or GFP expressing lentivirus were labeled by EZ Link Sulfo NHS-SS-biotin. TrkB internalization was then initiated by incubation with BDNF (100 ng/ml) for 30 minutes at 37°C. Glutathione cleavage buffer removed the remaining biotin on the cell surface. The biotinylated, internalized TrkB was precipitated by streptavidin followed by immunoblot using a TrkB antibody. Both full-length (145 kD) and truncated (95 kD) TrkB receptors are shown. Total surface biotinylated TrkB (Total biotin) was measured in cells held on ice; the efficiency of biotin cleavage is demonstrated in (Cleaved); and spontaneous TrkB internalization induced by BDNF is shown (4C). Cells expressing either GFP lentivirus or ARMS shRNA-GFP expressing lentivirus exhibit similar levels of basal and BDNF-induced TrkB internalization. (D) Receptor internalization is required for BDNF-induced NF-κB transcriptional activity. DIV7 primary cortical neurons were transfected with NF-κB luciferase plasmid for 40 hours. Cells were then stimulated with BDNF (100ng/mL) alone, BDNF plus the internalization inhibitor, MDC (25μM), or vehicle alone for 8 hours. ‘NT’ represents non-treated cells. Analysis of luciferase gene expression (presented as a fold induction) shows that inhibition of receptor internalization blocks NF-κB activity induced by BDNF. The bars represents SEMs and the data is representative of three separate experiments. # indicates statistical significance p<0.05 as compared to BDNF-treated cells.
Neurotrophins are best known for their ability to initiate trophic effects via clathrin-mediated internalization of the neurotrophin-Trk receptor complex. However, the mechanisms that govern the internalization of Trk receptors remain largely unclear. Recent observations that ARMS localizes with Trk molecules in the endosomal compartment led to the suggestion that ARMS may take part in regulation of Trk receptor internalization (Arevalo et al., 2006). Therefore, to understand how ARMS promotes BDNF-induced neuronal survival, we examined the role of ARMS in regulation of BDNF receptor (TrkB) internalization. Neurons were infected with lentivirus vectors (ARMS shRNA-GFP, and control GFP) as outlined above and then incubated with BDNF (100 ng/ml) on ice for 30 min. The cell surface proteins were labeled with EZ-Link Sulfo-NHS-SS-Biotin (0.5mg/ml) for 30 min. Following this, receptor internalization was initiated by placing the cultures in a 37°C incubator. After 60 min. placing the cultures on ice terminated the internalization, and the remaining biotinylated surface proteins were de-biotinylated by cleavage of the NHS-SS-biotin disulfide bond with glutathione on ice for 15 min. Whole cell lysates were then subjected to precipitation of internalized biotinylated proteins by streptavidin and fractionated by gel electrophoresis (Fig. 2C). Immunoblotting was then performed using TrkB-specific antibodies. The total surface biotinylated TrkB was determined in cells held on ice without glutathione cleavage, and background internalization was analyzed in cells that were de-biotinylated without initiation of internalization (see lanes ‘total biotin’ and ‘4C’ in Fig. 2C; lane ‘cleaved’ shows an additional control in which de-biotinylation followed by internalization of TrkB was performed). Spontaneous internalization of TrkB was detected in cells not treated with BDNF (lanes marked as ‘NT’), while incubation with BDNF led to an increase in TrkB internalization, regardless of ARMS expression. These results indicate that ARMS might not be involved in BDNF-induced internalization of TrkB. Our data also suggests that the ability of ARMS to promote BDNF-mediated neuronal survival may involve signaling events downstream to TrkB internalization.
ARMS is involved in activation of NF-κB by BDNF
Since previous studies have implicated BDNF-induced activation of NF-κB as being critical for BDNF-mediated neuronal protection against HIV-1 neurotoxins (Ramirez et al., 2001; Saha and Pahan, 2007), first, we tested whether the internalization of TrkB is necessary for BDNF to activate NF-κB in neurons. Cortical neurons (DIV 7) were transiently transfected with a luciferase reporter plasmid driven by the NF-κB enhancer. After 40 hours, the cells were treated with BDNF (100 ng/ml) alone or in the presence of monodansylcadaverine (MDC, 25 μM), a widely used drug known to inhibit clathrin-mediated receptor internalization in a variety of cell types (Du et al., 2003; Schutze et al., 1999). The cells were then harvested after 8h for luciferase assays. As shown in Figure 2D, BDNF treatment resulted in high luciferase gene expression, indicating activation of NF-κB in these cells. This effect was completely inhibited by administration of MDC, suggesting that the Trk receptor internalization is an essential event in BDNF-induced NF-κB signaling mechanisms.
Next, we performed an analogous experiment in which the cortical neurons were co-transfected with a plasmid vector encoding nothing (empty vector) or vector expressing ARMS shRNA (controlled by U6 promoter), along with an NF-κB luciferase reporter plasmid. After 40 hours, the cells were either left untreated or treated with BDNF (100 ng/ml) for 8h and harvested for luciferase assays. As shown in Figure 3A, the expression of ARMS shRNA in neurons resulted in reduction of both basal as well as BDNF-induced NF-κB activity, thereby implicating ARMS in this process.
Figure 3.
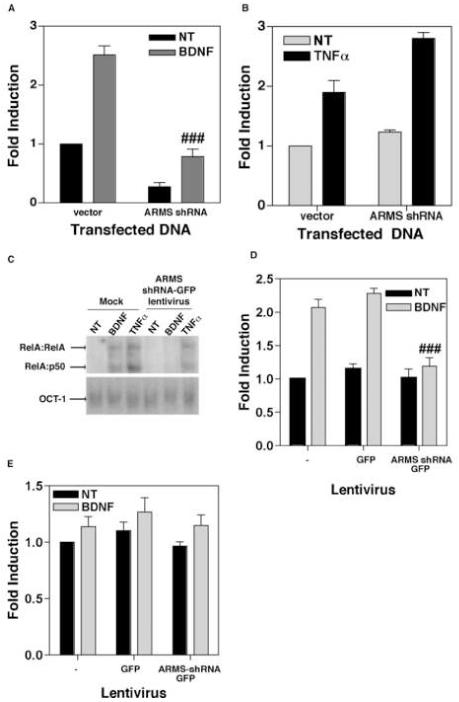
(A) Depletion of ARMS reduces basal as well as BDNF-stimulated transcriptional activity of NF-κB. Primary cortical neurons were transfected with an NF-κB luciferase reporter plasmid plus either empty vector (vector) or plasmid expressing ARMS shRNA (ARMS shRNA). 40 hours post-transfection, the cells were treated with BDNF (100ng/mL) for 8 hours. Data shows that the loss of ARMS results in a reduction of NF-κB driven gene expression. (B) Analogous experiments were performed in HEK293 cells that were treated with TNFα (20ng/mL). Expression of ARMS shRNA had no effect on NF-κB elicited by TNFα. (C) Primary cortical neurons were either mock infected of infected with ARMS shRNA-GFP lentivirus. After 7 days, cells were exposed to either nothing, BDNF (100ng/mL), or TNFα (20ng/mL) for 2 hours. EMSA was then performed to analyze DNA binding activity of NF-κB and Oct-1(control transcription factor). Both BDNF-, and TNFα-treatment led to increased binding of NF-κB complexes (RelA:RelA and RelA:p50 are indicated) in mock infected cells. Cells expressing ARMS shRNA-GFP exhibited increased activity of NF-κB only in response to TNFα, but not BDNF. In all the cases, activity of Oct-1 remained unaltered. (D) Quantitative analysis of the DNA binding activity of NF-κB after expression of ARMS shRNA. A TransAM NF-κB DNA binding ELISA was performed using nuclear extracts of GFP and ARMS shRNA-GFP infected primary cortical neurons. BDNF was unable to stimulate DNA binding activity in cells expressing the ARMS shRNA. (E) The loss of ARMS does not effect DNA binding activity of NF-YA in a TransAM NF-YA DNA binding ELISA. The results presented here are derived from two (C), three (B and D), and four (A) sets of experiments. Data represents Mean ± SEM of fold induction as a function of the luciferase activity in untreated (and vector-transfected) cells; ### shows P<0.01 (A) and P<0.05 (D), as compared to BDNF-treated control groups. Vehicle-treated cells are shown as ‘NT’.
To test whether ARMS also participates in NF-κB signaling triggered by other stimuli, we performed similar luciferase assays in which HEK293 cells were first transfected with ARMS shRNA together with an NF-κB luciferase reporter and then exposed to TNFα (20 ng/ml). Here, the HEK293 cells were chosen mainly for three reasons: 1) these cells harbor a low level of endogenous ARMS protein that can facilitate TrkA signaling (Arevalo et al., 2004), 2) TNFα is able to activate NF-κB in these cells (Kouba et al., 2001), and 3) long-term exposure to TNFα is toxic to the neurons, but not to HEK293 cells (Rickle et al., 2006). As shown in Figure 3B, an expression of ARMS shRNA in HEK293 cells failed to inhibit basal, as well as TNFα-induced NF-κB activity.
To validate these findings in primary cortical neurons, we resorted to measuring early events of NF-κB activation (such as DNA binding activity of nuclear NF-κB) following short-term exposure to TNFα. In these assays, cortical neurons (DIV 2) were either not infected (Fig. 3C; lanes marked as ‘Mock’) or infected with lentiviral vectors encoding ARMS shRNA-GFP. After 7 days, the neurons were exposed to vehicle (denoted as ‘NT’), BDNF (100 ng/ml) or TNFα (20 ng/ml) for 2h and nuclear extracts were prepared. NF-κB DNA-binding activity was then analyzed by performing electrophoresis mobility shift assays (EMSA). Here, we used a radio-labeled double-stranded oligonucleotide probe containing a consensus NF-κB-binding site (or a control probe containing an Oct-1-binding site) in combination with an excess of double-stranded poly(dI-dC), to eliminate non-specific binding activity (Ramirez et al., 2001). EMSA revealed a low level of DNA-binding activity of NF-κB in untreated cells (Fig. 3C, ‘NT’). Incubation of mock-infected cells (no virus added) with BDNF and TNFα led to an enhanced binding of two complexes containing predominantly RelA subunit of NF-κB (Fig. 3C, antibody-based supershift analyses were performed as previously described (Ramirez et al., 2001), data is not shown). Whereas, the treatment of ARMS shRNA expressing neurons with BDNF, but not with TNFα, failed to activate RelA-containing complexes of NF-κB. In contrast, in all these experimental conditions, the DNA-binding activity of the control transcription factor Oct-1 was not altered (Fig. 3C, lower panel).
To quantitate RelA DNA-binding activity, and to confirm whether BDNF indeed fails to activate RelA-containing complexes in neurons that are expressing ARMS shRNA, we subjected the nuclear extracts of vector-transduced neurons to ELISA analyses as previously described (Renard et al., 2001; Sui et al., 2006b). Here, the binding of the specific transcription factor to the target oligonucleotide was detected by incubation with antibodies specific to RelA or to NF-YA (ubiquitously expressed control transcription factor). These antibodies recognize an epitope that is accessible only when these transcription factors are bound to their target DNA. As shown in Figure 3D, a nearly 2 fold increase in DNA-binding activity of RelA complexes was stimulated by BDNF (100 ng/ml, 2h) in uninfected cells. This activity was also seen in control cells that were infected with empty lentivirus vector encoding only GFP. However, no induction of RelA activity was observed in BDNF-treated ARMS shRNA-GFP vector-infected cells. These assays also revealed a generally modest increase in NF-YA DNA-binding activity, irrespective of ARMS expression in these cells (Fig. 3E). Collectively, these results indicate that the effect of ARMS on NF-κB activation is specific and also limited to signaling pathways induced by certain stimuli (i.e. neurotrophins and not TNFα).
Over-expression of ARMS enhances NF-κB activity
To confirm whether ARMS plays a role in BDNF-induced NF-κB signaling, reciprocal experiments were performed. In this case, full-length ARMS protein was over-expressed in cortical neurons; these cells also received NF-κB luciferase reporter plasmid (control cells received Oct-1 luciferase reporter). BDNF (100 ng/ml) was added to the cultures for 8h and luciferase activity was measured. As shown in Figure 4A, over-expression of ARMS resulted in a dose-dependent increase in basal as well as inducible activity of NF-κB. In contrast, Oct-1 activity was not changed following ARMS over-expression (Fig. 4B).
Figure 4.
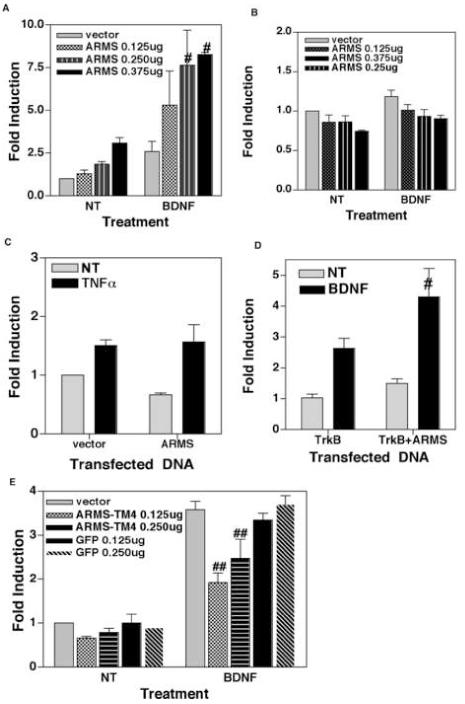
(A) Overexpression of ARMS augments BDNF-induced activation of NF-κB. DIV 7 cortical neurons were co-transfected with NF-κB luciferase reporter plasmid and either empty vector or increasing amounts of full-length ARMS plasmid. The recipient cells were then treated with BDNF (100ng/mL) for 8 hours and luciferase activity was measured. Data is presented as the mean (fold induction) of 3 independent observations, the bars represents SEM. # denotes p<0.05 as compared to untreated cells (NT). (B) Similar luciferase assays were performed to measure Oct-1 activity in cortical neurons. The results (Mean of fold induction ± SEM of 3 experiments) show that the over-expression of ARMS has no effect on Oct-1 activity. (C) Overexpression of ARMS does not influence TNFα mediated activation of NF-κB. NF-κB-dependent luciferase assays were performed in which HEK293 cells were treated with TNFα (20ng/mL) for 8 hours. Results revealed similar levels of NF-κB activity in untreated as well as TNFα-treated cells. (D) Expression of TrkB and ARMS is sufficient for BDNF to induce NF-κB activity. HEK293 cells were co-transfected with NF-κB luciferase reporter plasmid and TrkB alone, or TrkB plus full-length ARMS. After this, the recipient cells were treated as indicated. # denotes p<0.05 derived from three sets of experiments, shown as Mean of fold induction ± SEM. (E) Interaction between TrkB and ARMS is required for BDNF to induce NF-κB. Neuronal cells were co-transfected with an NF-κB luciferase reporter and indicated plasmids. ARMS-TM4 encodes transmembrane region (amino acids 2041 to 2673) of ARMS. Luciferase activity is presented as a mean of fold induction ± SEM. ## equals p<0.01.
We also tested whether the effects of ARMS on NF-κB activation are limited to BDNF-exposed cells. Analogous experiments were performed in which HEK293 cells were treated with TNFα for 8h. These assays revealed that the over-expression of ARMS has no effect on TNF-α-stimulated activation of NF-κB (Fig. 4C), confirming our earlier findings that ARMS promotes BDNF-mediated activation of NF-κB in neuronal cells.
BDNF activates NF-κB via transmembrane interaction of ARMS with TrkB receptor
ARMS is known to interact directly with Trk receptors (Arevalo et al., 2004). Therefore, to test whether an ARMS:TrkB interaction promotes BDNF-mediated activation of NF-κB, we performed additional luciferase reporter assays. In these experiments, the neurotrophin receptor (TrkB) signaling mechanism was reconstituted in HEK293 cells by transfecting these with TrkB alone or together with ARMS (note that HEK293 does not express TrkB (Daniel et al., 2007)). The cells were also co-transfected with NF-κB reporter plasmid. Following this, the cells were either left untreated or treated with BDNF (100 ng/ml) for 8h and luciferase activity was measured. As shown in Figure 4D, BDNF-treatment led to an increase in NF-κB activity in TrkB-expressing cells; this effect of BDNF was significantly enhanced in cells expressing both TrkB and ARMS, suggesting that the ARMS:TrkB interaction might be important for BDNF-signaling.
ARMS and TrkB receptors interact with each other via engaging transmembrane domains of each protein. This interaction can be disrupted by expressing the fourth membrane-spanning segment of ARMS resulting in a concomitant loss in the sustained activation of MAP kinase mediated by TrkB receptors upon BDNF treatment (Arevalo et al., 2004). In light of these findings, we sought to determine whether the transmembrane interaction between ARMS and TrkB is necessary for BDNF to activate the NF-κB signaling pathway. To do this, NF-κB-, or Oct-1-, dependent luciferase assays were performed in which cortical neurons were transfected with NF-κB (or Oct-1) reporter plasmids together with a plasmid encoding the fourth transmembrane segment of ARMS (amino acid residues from 2041 to 2673; hereafter referred to as ARMS-TM4) and treated with BDNF (100 ng/ml) for 8h. Control cells received reporter plasmids and a vector encoding an irrelevant protein, GFP. In these assays, the basal level of NF-κB activity remained unaltered in cortical neurons transfected with ARMS-TM4 (or GFP) expression plasmid (Fig. 4E). Whereas, transfection with ARMS-TM4, but not GFP, showed a significant reduction in BDNF-stimulated NF-κB activity. In contrast, Oct-1 activity was not changed following ARMS-TM4 expression (data not shown). These results suggest that ARMS functionally interacts with TrkB in cortical neurons and that the disruption of this interaction results in aborted BDNF-dependent signaling events critical for NF-κB activation.
Ability of BDNF to activate NF-κB correlates with the endogenous levels of ARMS and TrkB in developing neurons
To investigate the role of endogenous ARMS and TrkB in mediating BDNF-induced activation of NF-κB, we analyzed the levels of ARMS, TrkB and NF-κB (RelA) in developing neurons. At the same time, we also examined whether the relative levels of TrkB and ARMS during development governs the responsiveness of neurons to BDNF in terms of activating NF-κB. Briefly, whole-cell lysates were prepared from dissociated cortical neurons cultured for various days in vitro (ranging from DIV 1 to 11) and immunoblot assays were performed. In parallel, neurons (at DIV 1 to 11) were also exposed to BDNF (100ng/ml) for 2h and nuclear extracts were prepared, which were then subjected to ELISA for measuring the DNA-binding activity of RelA (NF-κB subunit). High levels of TrkB and ARMS were found in neurons at DIV 9, at which time expression of postsynaptic density protein 95 (PSD-95) and α-tubulin (markers of neuronal development) were also elevated (Fig. 5A). Consistent with Kim et al (Kim et al., 2004), we found that the other neurotrophin receptors, such as p75 and TrkA, were either absent (TrkA, data not shown) or reduced (p75; this faint band was found co-migrating with p75NTR protein that was derived from transfected COS7 cells, data not shown) in developing neurons. In contrast, uniform expression of RelA was detected in neurons (especially from DIV 3 to 11). Interestingly, the DNA-binding activity of RelA was higher in cortical neurons that were treated with BDNF at DIV 9 (Fig. 5B). In this assay, the DNA-binding activity appears to be specific to RelA, because, (1) no change in the activity of NF-YA (control transcription factor (Dugast and Weber, 2001)) was observed in neurons (DIV 1 to 11) despite BDNF treatment (data not shown), and (2) pre-incubation of nuclear extract (derived from BDNF-treated neurons at DIV 9) with excess amount of oligonucleotide that contains an intact NF-κB responsive element (WT, Fig. 5C) significantly decreased the binding of RelA to the target probe. Taken together, these results suggest that the ability of BDNF to activate NF-κB is associated with the increased expression of TrkB and ARMS during development.
BDNF-mediated activation of NF-κB is blocked by K252a
We performed additional experiments to determine the role of ARMS and TrkB in mediating NF-κB activation. Since the alkaloid-like compound K252a blocks BDNF-induced phosphorylation of ARMS in TrkB-expressing cells (Kong et al., 2001), we tested whether this drug also inhibits activation of NF-κB following exposure to BDNF. In these experiments, cortical neurons (DIV 9) were transiently transfected with a plasmid vector encoding a RelA-GFP fusion protein and treated with either BDNF (100ng/ml) alone or together with K252a (50 nM) for 1h. After this, the nuclear translocation of RelA-GFP was analyzed using fluorescent microscopy. As expected, BDNF treatment led to the translocation of RelA-GFP to the nucleus, which was completely blocked by co-administration of K252a (Fig. 6A).
Figure 6.
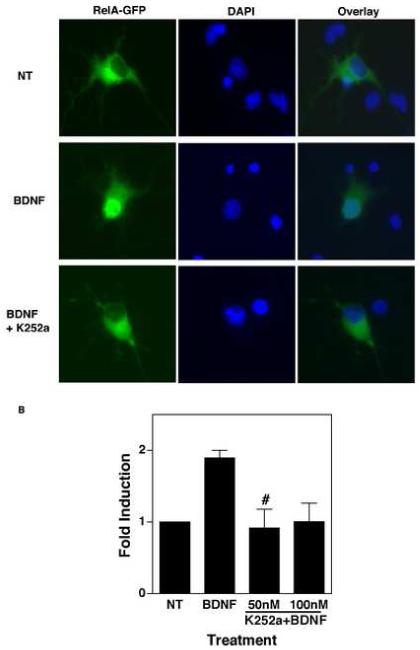
(A) BDNF induces the nuclear translocation of RelA. DIV 7 cortical neurons were transiently transfected with a plasmid encoding RelA-GFP. Fourty hours later, neurons were treated with BDNF (100 ng/ml) alone, or together with K252a (50 nM) for 1 hour, and examined under a fluorescent microscope for mobilization of RelA-GFP from the cytoplasm to the nucleus. (B) BDNF induces NF-κB activity in a TrkB-dependent manner. DIV 7 cortical neurons were transfected with an NF-κB luciferase reporter plasmid and were treated with BDNF (100ng/ml), or BDNF plus K252a (50 nM), for 8 hours. The results shown are mean (of fold induction ± SEM) of three independent observations. # equals p<0.05 as compared to BDNF-treated cells.
Next, we quantitatively measured the effect of K252a on activation of endogenous NF-κB elicited by stimulation of TrkB and ARMS. This was carried out by analyzing transcriptional activation of NF-κB-dependent luciferase reporter in BDNF-treated neurons using methods analogous to earlier experiments. As shown in Figure 6B, we found that the K252a treatment caused significant reduction in BDNF-induced NF-κB activity (Fig. 6B), but not in Oct-1 activity (data not shown).
NF-κB activation by BDNF involves ARMS-dependent activation of MAPK and IKK
To understand better how ARMS plays a role in BDNF-mediated activation of NF-κB, we examined the activity of various kinases in ARMS-depleted neurons. To do this, cortical neurons (DIV 2) were infected with lentiviral vectors encoding either ARMS shRNA-GFP or GFP alone. Seven days later, the cells were exposed to BDNF (100ng/ml) to stimulate NF-κB signaling, and cell lysates were prepared to examine the phosphorylation of cellular ERK1 and 2 (at activation loop residues Thr202/Tyr204 as a measure of MEK1/2 activation) and AKT (at Thr308 as an indicator of PDK1 activation) by using phosphospecific antibodies and immunoblot assays. The results of this analysis (presented in Figure 7A) revealed that BDNF treatment resulted in rapid phosphorylation of ERK1/2 and AKT in control neurons (GFP lentivirus-infected). Whereas, in ARMS shRNA-GFP-infected neurons, BDNF failed to induce phosphorylation of ERK1/2, but not of AKT, indicating that ARMS is essential for BDNF to activate MAPK pathway in neurons.
Figure 7.
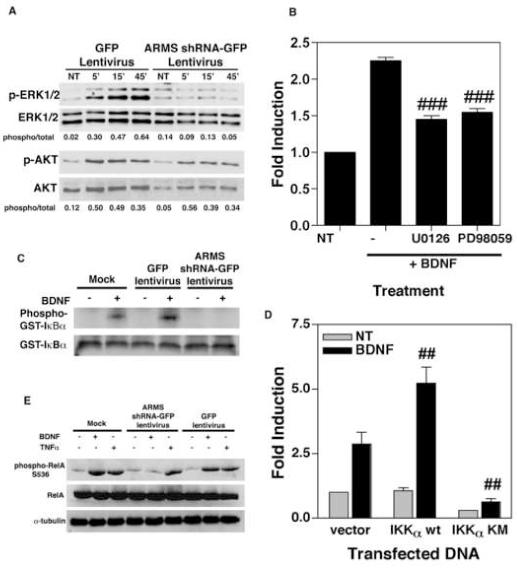
(A) ARMS regulates MAPK activation. Following lentiviral infection, neurons were treated with BDNF (100 ng/ml) and whole cell lysates were prepared at indicated times (NT denotes non-treated cells). These lysates were then analyzed for the phosphorylation status of ERK1/2 and AKT by using specific antibodies. Increased immunoreactivity to phospho ERK1/2-specific antibodies, but not to that of AKT, was observed in control cells (GFP-expressing cells) that were treated with BDNF. BDNF failed to elicit such an effect in ARMS-depleted cells (ARMS shRNA-GFP expressing cells). (B) MAPK is involved in the activation of NF-κB by BDNF. NF-κB luciferase assays were performed in which neurons were treated with BDNF (100ng/mL), and MAPK blockers, U0126 (10μM) or PD98059 (25μM), for 8 hours. Results (Mean ± SEM) are representative of three independent observations. ### equals p<0.001 as compared to BDNF-treated groups. (C) ARMS interferes with IKK activation. ARMS-depleted, and control neurons were treated with BDNF (100ng/mL) for 30 minutes and IKK complexes were purified using IKKα-specific antibodies. The immune-complexes were then incubated with GST-IκBα and γ [32P] ATP for 30 min. at room temperature. The incorporation of 32P in GST-IκBα was then measured by SDS-PAGE and autoradiography. Strong phosphorylation of GST-IκBα in response to BDNF was observed in mock-, and GFP-infected samples, but not in those infected with ARMS shRNA-GFP lentivirus. (D) Role of IKK in BDNF-mediated activation of NF-κB. Luciferase assays were performed in DIV 7 neurons following transfection with NF-κB luciferase reporter and IKKα expressing plasmid (WT, wild type; KM, dominant negative mutant). BDNF-induced luciferase transcription was completely blocked in IKKα KM-expressing cells, indicating a key role for IKK in BDNF-elicited NF-κB signaling. The data (Mean ± SEM) is derived from three parallel experiments. ## denotes (p<0.01) statistically significant inhibition of NF-κB activity by IKKα KM. (E) ARMS mediates phosphorylation of RelA at IKK-sensitive site. Immunoblot analysis was carried out by using lysates of infected neurons and phospho (Ser536)-specific antibodies to RelA. As shown, both BDNF (100ng/mL) and TNFα (20ng/mL) exerted enhanced phosphorylation of RelA at serine 536 in mock, as well as GFP infected cells. This effect was observed only in TNFα-exposed ARMS-depleted cells.
Next, we tested the role of MEK1/2 in BDNF-stimulated induction of NF-κB by performing luciferase reporter assays in which cortical neurons were also treated with MEK1/2 inhibitors. Figure 7B shows that the stimulation of NF-κB activity by BDNF was reduced upon treatment with U0126 (10μM) and PD98059 (25μM), suggesting that the MAPK pathway plays an important role in BDNF signaling towards NF-κB activation. The effect of U0126 and PD98059 appears to be limited to BDNF-stimulated NF-κB, because these drugs had no effect on Oct-1 activity (data not shown).
Since the MAPK pathway is known to stimulate NF-κB activity via IKK activation in other models (Kupfer et al., 2007; Lee et al., 1998; Tuosto et al., 2000), we measured IKK activity in BDNF-treated ARMS-depleted neurons. In these assays, cortical neurons were infected as above with lentiviruses encoding ARMS shRNA-GFP, GFP alone or mock infected (no virus added). At DIV 9, the neurons were exposed to BDNF for 30 min, and whole cell lysates were prepared. IKK complexes were then immunoprecipitated from cell lysates by addition of a specific monoclonal antibody recognizing the IKKα isoform, followed by incubation with protein A-agarose beads. These IKKα-bound beads were then incubated with substrate (GST-IκBα) and [γ-32P]ATP for 30 min. at 30°C, and the radiolabeled products were separated by SDS-PAGE, transferred to a membrane filter, and visualized by autoradiography. Figure 7C shows that IKK activity, as measured by using GST-IκBα substrate, was strongly induced upon BDNF-treatment of mock-infected neurons or neurons infected with control GFP lentivirus (lanes ‘Phospho-GST-IκBα’; and compare signal intensity in the paired ‘-’ and ‘+’ samples, which correspond to BDNF-treated or untreated cells, respectively). In contrast, BDNF-treatment did not induce an up-regulation of IKK activity in neurons that were infected with the lentiviral vector encoding ARMS shRNA-GFP. This was not the result of a loss of the substrate protein, since this was equivalent in all the reactions (lanes ‘GST-IκBα’).
To explore further the role of IKK in NF-κB signaling triggered by BDNF in neurons, we transfected cortical neurons with vectors encoding either nothing (empty vector), wild-type or dominant negative forms of IKKα (IKKα wt, IKK KM) together with luciferase reporter plasmid driven by NF-κB enhancer and performed luciferase assays. The results, as depicted in Figure 7D, revealed a much higher level of NF-κB activity in IKKα wt-expressing cells stimulated by BDNF. In contrast, BDNF failed to activate NF-κB in neurons expressing the dominant negative form of IKKα (IKKα KM), suggesting that the activation of IKK is essential for BDNF to activate NF-κB.
During NF-κB signaling, multiple mediators like RelA, are phosphorylated by IKK (Hacker and Karin, 2006; Nicholas et al., 2007). Therefore, we investigated whether an intact expression of ARMS render cells responsive to BDNF in terms of phosphorylation of RelA at the IKK-sensitive site (Serine 536). To do this, the same experimental design was used to infect cortical neurons, and immunoblot analyses were conducted. As shown in Figure 7E, BDNF-treatment in mock-infected neurons, or in neurons infected with GFP lentivirus, led to a massive increase in phosphorylation of RelA at Ser 536. This event was not observed in those neurons that had reduced levels of ARMS (ARMS shRNA-GFP-infected cells). In contrast, TNF-α-treatment led to an increase in phosphorylation of RelA, in spite of ARMS expression, presumably due to involvement of other kinases (Sakurai et al., 2003). Thus, BDNF may involve ARMS in NF-κB signaling that proceeds through activation of MAPK cascade and IKK.
DISCUSSION
Despite significant advances in our understanding of neurotrophin signaling, little is known about how neurotrophins regulate the activity of NF-κB. This is in part due to the fact that a majority of neuronal cells express more than one neurotrophin receptor subtype, while each receptor subtype possesses an ability to stimulate NF-κB activity in response to neurotrophins. For example, dissociated cultures of rodent sympathetic neurons contain TrkA and p75NTR on their surface (Wyatt and Davies, 1995), where p75NTR is believed to interact with TrkA to form a high affinity binding site (Hempstead et al., 1991) and to regulate TrkA signaling (Verdi et al., 1994). Both the receptor subtypes are known to independently mediate NGF-elicited activation of NF-κB (Carter et al., 1996; Maggirwar et al., 1998). Similarly, cerebellar granule neurons have been reported to express TrkB and p75NTR, but not TrkA (Courtney et al., 1997), and mediate BDNF-induced NF-κB activation (Ramirez et al., 2001). Whereas, four receptor subtypes (TrkA, TrkB, TrkC and p75NTR) were found in neurons derived from rodent hippocampus (Brann et al., 1999; Cellerino, 1996; Ernfors et al., 1992; Kadari et al., 1997; Ringstedt et al., 1993). Moreover, with a noted exception of NT4-5 and TrkC deficient mice, all neurotrophin and Trk knockout mice exhibit poor viability (Conover and Yancopoulos, 1997). Thus, the mechanism of neurotrophin-mediated activation of NF-κB in cells that express multiple neurotrophin receptors remains difficult to ascertain.
One way of avoiding these limitations is to employ neuronal cells that have relative abundance of a certain neurotrophin receptor subtype, as previously described (Carter et al., 1996). In these studies by Carter and coworkers, p75NTR was implicated in NGF-mediated activation of NF-κB by using rat Schwann cells that expresses p75NTR, but not TrkA. Analogous to this approach, here we have used primary cultures of rodent cortical neurons that predominantly contain TrkB. Under these conditions (DIV 9), we found that the intact expression of ARMS protein is necessary for BDNF to activate NF-κB in neurons. This notion is supported by results that were derived from complementary approaches, such as depletion of ARMS via shRNA expression, over-expression of exogenous ARMS, and by assessment of BDNF-induced NF-κB, as a function of relative levels of endogenous ARMS.
As opposed to TrkB, decreasing amounts of p75NTR protein were detected in developing neurons, which was not correlative to the extent of NF-κB activation. Because ARMS also interacts with p75NTR protein to form a ternary complex with TrkB (Arevalo et al., 2004; Chang et al., 2004), it remained possible that the ability of BDNF to activate NF-κB in neurons might be secondary to the interaction of ARMS with low levels of p75NTR. However, at least two pieces of evidence suggest that this may not be the case. First, over-expression of the fourth transmembrane domain of ARMS (ARMS-TM4), which disrupts ARMS:TrkB complexes, but not ARMS:p75NTR complexes (formation of ARMS:p75NTR complexes require cytoplasmic sequences within ARMS molecules (Chang et al., 2004)) reduced BDNF-mediated activation of NF-κB. Second, an exogenous expression of TrkB and ARMS rendered HEK293 cells responsive to BDNF (note that the endogenous expression of p75NTR protein in these cells was undetectable (Kong et al., 1999)). Thus, we conclude that ARMS might interact with TrkB to initiate a signaling cascade that culminates in NF-κB activation.
ARMS facilitates interaction of Trk receptors with CrkL and Rap-1 proteins thereby promoting neurotrophin-dependent activation of MAPK (Arevalo et al., 2006). Our data corroborate this view and suggest that ARMS might promote NF-κB, in part, through a MAPK mechanism. We also found that the activation of IKK, following BDNF addition, does not occur in ARMS-depleted neurons and that the IKK activity is critical for BDNF to stimulate NF-κB. Although, the precise mechanism by which BDNF regulates IKK activity remains unclear, it is highly likely that the two signaling events (activation of MAPK and IKK) might be linked. It is worth noting that the role of MAPK signaling, especially MEKK (see below), in activation of IKK is widely accepted (Kupfer et al., 2007; Lee et al., 1998; Tuosto et al., 2000). Accordingly, several factors (including, TNFα, phorbol esters and lipopolysaccharides) that stimulate MAPK pathway, also possess the ability to activate NF-κB with similar kinetics (Chang et al., 2005; Huh et al., 2007; Sanchez et al., 2003). The activation of NF-κB is rapid and occurs in the absence of protein synthesis (reviewed in (Ghosh and Karin, 2002)). As such, NF-κB must be tightly regulated at multiple levels within the cell, including its release from the inhibitory proteins (these includes mainly IκBα and IκBβ), its mobilization from the cytoplasm into the nucleus, and its engagement of cognate κB enhancers, leading to changes in target gene expression. Thus the activation of NF-κB can be viewed as occurring in two distinct phases. The first phase comprises the proximal events that lead to the degradation of IκBα and the translocation of NF-κB complexes (mainly RelA/p50) into the nuclear compartment. This initial phase of NF-κB activation proceeds through a kinase cascade culminating in the site-specific phosphorylation, ubiquitination, and subsequent degradation of the IκBα inhibitor by the 26 S proteasome. Although the panel of kinases that are recruited following exposure to a particular stimulus includes NIK (NF-κB-inducing kinase), TAK-1 (Transforming growth factor--activated kinase), and MEKK1 (mitogen-activated protein kinase/extracellular signal-regulated kinase kinase kinase-1), they all appear to converge at the activation of IKK complex, which in turn catalyzes the phosphorylation of the inhibitory proteins at two specific N-terminal serines (Ser32 and Ser36 in IκBα and Ser19 and Ser23 in IκBβ) (Hacker and Karin, 2006). Indeed, neurotrophins are known to stimulate NF-κB in a manner that involves phosphorylation and proteasome-dependent degradation of IκBα (Maggirwar et al., 1998; Ramirez et al., 2001). In keeping with these reports, our results reveal that RelA translocates to the nucleus in BDNF-treated cells, presumably via activation of TrkB (since it was efficiently blocked by addition of K252a). We further show that BDNF stimulates DNA-binding as well as transcriptional activity of RelA, which is largely dependent upon intact expression of ARMS.
The second phase in the activation of NF-κB involves the post-translational modification of the NF-κB subunits themselves by both phosphorylation and acetylation (Hacker and Karin, 2006). Although the former phosphorylation events on IκBα inhibitor are required for release of NF-κB, the latter modifications on RelA appear to enhance its transcriptional activity and the duration of the response. Likewise, our data suggests that the exposure of cells to BDNF elicits a positive effect on the transcriptional activity of NF-κB, which may proceed via ARMS-dependent activation of IKK and phosphorylation of RelA at IKK sensitive site.
In summary, the results shown here identify ARMS as a major factor that links neurotrophin signaling to NF-κB. Given the critical role played by NF-κB in neuronal survival and function, these findings may have therapeutic implications in the context of neurodegenerative disorders.
EXPERIMENTAL METHODS
Primary cortical neuron cultures
Primary neuronal cultures were prepared from embryonic day 18 rats, as previously described (Sui et al., 2006a). Briefly, cortices were dissected, cleared from meninges and other remaining tissues, and then incubated in 2.0 ml Ca++/Mg++-free HBSS (with 10mM HEPES, pH 7.3) with antibiotics (50mg/L penicillin, 50mg/L streptomycin, 100mg/L neomycin) and 0.5ml of 0.25% trypsin for 15 minutes at 37°C. After a washing with HBSS and trituration, dissociated cells were plated in poly-D-lysine coated 24-well tissue culture plates at a density of 1×105 cells per well. The cells were suspended in Neurobasal medium with B27 supplement (Life Technologies, Gaithersburg, MD) until the 4th day in vitro (DIV) and later in the same medium that was devoid of antioxidants, as described (Sui et al., 2006a). The media formulation does not support the growth of glia, therefore resulting in a culture that is 98% pure. Cells were cultured for 9-14 days at 37°C and 5% CO2 and used for experiments at DIV 9, unless otherwise indicated.
Lentivirus production and neuronal infection
HIV-1-based pseudotyped lentivirus vectors encoding short hairpin RNA (shRNA) targeted to rat ARMS transcripts (nucleotides 1027-1046) were generated by placing an oligonucleotide sequence 5′GCCACCAAGATGAGAAATA3′ under the control of the human U6 RNA polymerase III promoter (Figure 1A), using FG12 lentiviral vector (gift of Dr. David Baltimore), as described (Qin et al., 2003). In brief, HEK293T cells were transiently transfected using calcium phosphate and a mixture of 12.5 μg of transfer plasmid containing ARMS shRNA, 12.5 μg packaging construct pCMV4-Δ89.2, and 12.5 μg of VSV-G envelope construct. Vector produced in the trasfected cultures was harvested at 48h post-transfection and filtered through a 0.45μM filter. To obtain highly concentrated virus stock, the supernatant was centrifuged at low speed (1800 g) for 30 min. at 4°C, placed on a 20% sucrose cushion, and then subjected to ultracentrifugation at 113,000 g for 2h at 4°C using a Beckman SW28 rotor. Vector pellet was resuspended in a small amount (1% of original volume) of serum-free DMEM. Titration of vector was performed using HeLa cells infected with 10-fold serial dilutions and virus titer was determined at day 3 post-inoculation by counting the number of GFP-positive cells using flow cytometry. Infection of neurons was initiated by adding virus, at multiplicity of infection (MOI) 10, to the neuronal cultures at DIV2, followed by a 3h adsorption, resuspention in Neurobasal medium and incubation at 37°C under 5% CO2 for the next seven days.
Immunoblot analysis
These assays were performed as previously described (Sui et al., 2007). Whole cell lysates were first prepared using ELB buffer (50mM HEPES pH 7, 250mM NaCl, 0.1% NP-40, 5mM EDTA, 10mM NaF, 0.1mM Na3VO4, 50μM ZnCl2), supplemented with 0.1mM PMSF, 1mM DTT and a mixture of protease and phosphatase inhibitors. Cell debris was removed by centrifugation at 13000rpm. Proteins were then resolved by SDS-PAGE and electrophoretically transferred to Hybond ECL nitrocellulose membrane (Amersham, Arlington Heights, IL). Primary antibodies consisted of either anti-TrkB, anti α-tubulin, anti-p75NTR, anti-PSD95, anti-RelA, anti-ERK1/2 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-GST (Pharmacia); anti-AKT, anti-phospho-AKT, anti-phospho-ERK1/2, anti-phospho RelA (Cell Signaling Technology, Beverly, MA). Polyclonal antibodies to ARMS were raised against the C-terminus region (amino acids 1535-1715) (Cocalico Biologicals, Inc. Reamstown, PA). Immunoreactivity of these antibodies was then verified by comparing with anti-ARMS (clone 892, (Kong et al., 2001)). Horseradish peroxidase-linked secondary antibodies (Amersham, Arlington Heights, IL) were used to detect bound primary antibodies. Immunblots were developed using ECL (Amersham, Arlington Heights, IL) and exposure to X-ray film (Kodak).
FM1-43 uptake assay
Total spontaneous activity-dependent vesicular uptake was assessed using the lipophilic styryl dye N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide (FM1-43; Invitrogen, Carlsbad, CA), as previously described (Perry et al., 2005). Briefly, 10 μM of FM1-43 was directly added into medium of infected cortical neurons (DIV 9) in the absence of any additional depolarizing stimulus for 15 min at 37°C, a length of time that has been determined to label spontaneous activity without saturating the vesicle pool (Perry et al., 2005). Following loading, excess FM1-43 was washed out in HBSS+ for 15 min at room temperature. Wash solution was prechilled to 4°C to limit exocytosis of dye from spontaneous activity. Following washout, cells were incubated at 37°C in a depolarizing solution of HBSS+ with 100 mM KCl added. Immediately after addition of the depolarizing solution, cells were transferred to a Bio-Rad fluoromark plate reader, and release of FM1-43 was monitored over 1h (at 1 min intervals) using 438 excitation and 605 emission filters and Bio-Rad microplate reader III software. However, the majority of activity-dependent FM1-43 signal was lost well before 15 min, as would be expected for exocytotic processes occurring at the synapse. Because background nonvesicular staining by FM1-43 is not released by KCl (Perry et al., 2005), then for each condition, the total loss of FM1-43 signal during the release period equals the total spontaneous activity-dependent vesicular uptake of FM1-43 in each well over the loading period. This absolute FM1-43 uptake value reflects the total activity of the culture, and is equivalent to the synaptic or vesicular release probability of the culture.
Toxicity assay
Primary cortical neurons were infected with lentivirus expressing ARMS shRNA or GFP alone as described above. Cells were then either pre-treated with 100ng/mL recombinant human BDNF (100 ng/ml; Chemicon, Temecula, CA), or left untreated. 18 hours later, the neurons were exposed to gp120 (5nM, AIDS Research and Reference Reagent Program) for an additional 72 hours. Neuronal survival was measured by staining the nuclei with Hoechst 33342 dye (Molecular Probes/ Invitrogen Carlsbad, CA). Neurons exhibiting diffuse Hoechst staining along with a round nucleus were determined to be healthy. In contrast, neurons possessing a shrunken or blebbed nucleus with bright staining were scored as apoptotic.
TrkB receptor internalization assay
Primary cortical neurons were infected with either ARMS shRNA-GFP or GFP expressing lentivirus. On DIV9, cells were either left untreated or were treated with 100ng/mL BDNF for 30 minutes on ice. The unbound BDNF was then washed off using ice-cold PBS containing calcium and magnesium. The surface of the cells was next labeled with 0.5mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Promega, Madison, WI) in PBS for 30 minutes on ice. The cells were then transferred to warm media (37°C) for 60 minutes, which initiates the internalization of surface proteins. Any biotinylated surface proteins remaining on the cell surface were debiotinylated using a glutathione cleavage buffer and subsequent washing. Whole cell lysates were then prepared and internalized biotinylated proteins were precipitated using Immunopure Immobilized Streptavidin beads (Promega, Madison, WI). Proteins were resolved on a 7.5% SDS-PAGE gel and immunoblots were performed by using TrkB-specific antibodies.
Luciferase assay
Cells were transiently transfected with 1.5μg of either NF-κB-luciferase plasmid or Oct-1 luciferase plasmid and various amounts of ARMS (full-length and ARMS TM4), as well as TrkB-expression vector (these are previously described (Arevalo et al., 2004; Kong et al., 2001; Sui et al., 2007)) using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA). Additional plasmids, expressing IKK (WT and KM), were kindly provided by Dr. S.-C. Sun. Forty hours post-transfection, cells were treated with BDNF (100ng/ml), or as indicated for 8 hours. Some experiments also involve incubation of the cells with recombinant human tumor necrosis factor alpha (TNFα; 20ng/ml; R&D Systems, Minneapolis, MN), PD98059 (25 μM,), U0126 (10 μM, both the reagents were from Calbiochem, Gibbstown, NJ), or Monodansylcadaverine (25 μM, Sigma-Aldrich, St. Louis, MO). Cell extracts were then prepared using reporter lysis buffer (Promega, Madison, WI), and luciferase activity was measured using the Promega Luciferase Assay System and a Lumicount Microplate Luminometer (Packard Instrument Co.).
DNA Binding Assay
The DNA-binding of NF-κB was analyzed using specific TransAM ELISA-based kits from Active Motif (Carlsbad, CA) per the manufacturer’s instructions. Briefly, nuclear extracts from DIV9 cortical neurons were incubated in a 96-well plate containing immobilized oligonucleotide with an NF-κB consensus binding site (5′-GGGACTTTCC-3′) for 1 hour, in the presence of excess amounts of herring sperm DNA. A primary antibody to RelA was then added for an additional 1 hour to bind to the activated NF-κB that was bound to the oligonucleotide sequence. After washing, a horseradish peroxidase-conjugated secondary antibody was added for 1 hour. Following 3 washes, the assay was developed using a Lumicount Microplate Luminometer (Packard Instrument Co.) at 450nm absorbance. This assay provides a quantitative measure of NF-κB DNA binding activity.
The specificity of DNA-binding activity was verified by performing competition assays, in which a 20× excess amount of soluble oligonucleotides that contained either intact or mutant consensus binding sequences (also supplied by Active Motif) was coincubated in the above-described assays.
The ELISA results were confirmed by performing an additional independent technique, electrophoresis mobility shift assays (EMSA), as previously described (Ramirez et al., 2001), in which poly(dI-dC) was used to ensure the specificity of binding.
Nuclear translocation of RelA
Primary cortical neurons (DIV 7) were transiently transfected with a plasmid that encodes for a RelA-GFP fusion protein (kindly provided by Dr. Schmid J.A.), using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). 48 hours post-transfection, cells were either left untreated or treated with BDNF (100 ng/ml) in the absence and presence of K252a (50 nM; EMD Biosciences, Gibbstown, NJ) for 1 hour. Cells were then fixed in 4% paraformaldehyde, and the subcellular localization of RelA-GFP was visualized by fluorescence microscopy.
In vitro kinase assays
Whole cell lysates were prepared from mock infected, or infected cortical neurons as indicated. Cell debris was removed by high-speed centrifugation. Lysates were then pre-cleared by incubation with protein A-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) and centrifugation. Cell lysates were next incubated with 1mg of anti-IKKa (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. IKKa was then precipitated using protein G-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). After washing, the beads were incubated with GST-IkBa substrate (100 ng, Santa Cruz Biotechnology, Santa Cruz, CA) and 10mCi of [g-32P]ATP in kinase buffer (50mM Tris-Cl [pH 8], 10mM MgCl2, 5mM dithiothreitol) at 30°C for 30 minutes. Reactions were stopped by the addition of SDS-PAGE loading dye. Phosphorylation levels of IkBa were analyzed by SDS-PAGE and autoradiography.
Statistical analysis
Mean data values, and the standard error of the mean (SEM) were computed for each variable. The data were analyzed by one-way analysis of variance (ANOVA) followed by Bonferroni’s test for multiple comparisons. P<0.05 was designated as being statistically significant.
ACKNOWLEDGEMENTS
We thank Drs. Shao.-Cong Sun., David Baltimore, and Johannes A. Schmid for generously providing us with reagents. We also thank Dr. Seth Perry for valuable help with the vesicular uptake assays. This work was supported by the following NIH grants: RO1 NS054578, P01 MH64570 (SBM); NS21072, HD23315 (MVC); S11NS043499 (YL); T32 AI49105 (LS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Arevalo JC, Pereira DB, Yano H, Teng KK, Chao MV. Identification of a switch in neurotrophin signaling by selective tyrosine phosphorylation. J Biol Chem. 2006;281:1001–1007. doi: 10.1074/jbc.M504163200. [DOI] [PubMed] [Google Scholar]
- Arevalo JC, Yano H, Teng KK, Chao MV. A unique pathway for sustained neurotrophin signaling through an ankyrin-rich membrane-spanning protein. Embo J. 2004;23:2358–2368. doi: 10.1038/sj.emboj.7600253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal JK, Massie B, Miller FD, Kaplan DR. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron. 2000;27:265–277. doi: 10.1016/s0896-6273(00)00035-0. [DOI] [PubMed] [Google Scholar]
- Bachis A, Mocchetti I. Brain-derived neurotrophic factor is neuroprotective against human immunodeficiency virus-1 envelope proteins. Ann N Y Acad Sci. 2005;1053:247–257. doi: 10.1196/annals.1344.022. [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Bracale A, Cesca F, Neubrand VE, Newsome TP, Way M, Schiavo G. Kidins220/ARMS is transported by a kinesin-1-based mechanism likely to be involved in neuronal differentiation. Mol Biol Cell. 2007;18:142–152. doi: 10.1091/mbc.E06-05-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J, Sporns O. BDNF-dependent enhancement of exocytosis in cultured cortical neurons requires translation but not transcription. Brain Res. 1999;815:140–149. doi: 10.1016/s0006-8993(98)01112-3. [DOI] [PubMed] [Google Scholar]
- Brann AB, Scott R, Neuberger Y, Abulafia D, Boldin S, Fainzilber M, Futerman AH. Ceramide signaling downstream of the p75 neurotrophin receptor mediates the effects of nerve growth factor on outgrowth of cultured hippocampal neurons. J Neurosci. 1999;19:8199–8206. doi: 10.1523/JNEUROSCI.19-19-08199.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde YA. Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- Cellerino A. Expression of messenger RNA coding for the nerve growth factor receptor trkA in the hippocampus of the adult rat. Neuroscience. 1996;70:613–616. doi: 10.1016/s0306-4522(96)83001-6. [DOI] [PubMed] [Google Scholar]
- Chang MS, Arevalo JC, Chao MV. Ternary complex with Trk, p75, and an ankyrin-rich membrane spanning protein. J Neurosci Res. 2004;78:186–192. doi: 10.1002/jnr.20262. [DOI] [PubMed] [Google Scholar]
- Chang MS, Chen BC, Yu MT, Sheu JR, Chen TF, Lin CH. Phorbol 12-myristate 13-acetate upregulates cyclooxygenase-2 expression in human pulmonary epithelial cells via Ras, Raf-1, ERK, and NF-kappaB, but not p38 MAPK, pathways. Cell Signal. 2005;17:299–310. doi: 10.1016/j.cellsig.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Chao MV, Rajagopal R, Lee FS. Neurotrophin signalling in health and disease. Clin Sci (Lond) 2006;110:167–173. doi: 10.1042/CS20050163. [DOI] [PubMed] [Google Scholar]
- Conover JC, Yancopoulos GD. Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Rev Neurosci. 1997;8:13–27. doi: 10.1515/revneuro.1997.8.1.13. [DOI] [PubMed] [Google Scholar]
- Cortes RY, Arevalo JC, Magby JP, Chao MV, Plummer MR. Developmental and activity-dependent regulation of ARMS/Kidins220 in cultured rat hippocampal neurons. Dev Neurobiol. 2007 doi: 10.1002/dneu.20542. [DOI] [PubMed] [Google Scholar]
- Courtney MJ, Akerman KE, Coffey ET. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci. 1997;17:4201–4211. doi: 10.1523/JNEUROSCI.17-11-04201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel PB, Lux W, Samson AL, Schleuning WD, Niego B, Weiss TW, Tjarnlund-Wolf A, Medcalf RL. Two conserved regions within the tissue-type plasminogen activator gene promoter mediate regulation by brain-derived neurotrophic factor. Febs J. 2007;274:2411–2423. doi: 10.1111/j.1742-4658.2007.05777.x. [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Zaitsev E, Je HS, Liu XW, Lu B. Regulation of TrkB receptor tyrosine kinase and its internalization by neuronal activity and Ca2+ influx. J Cell Biol. 2003;163:385–395. doi: 10.1083/jcb.200305134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugast C, Weber MJ. NF-Y binding is required for transactivation of neuronal aromatic Lamino acid decarboxylase gene promoter by the POU-domain protein Brn-2. Brain Res Mol Brain Res. 2001;89:58–70. doi: 10.1016/s0169-328x(01)00063-8. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Merlio JP, Persson H. Cells Expressing mRNA for Neurotrophins and their Receptors During Embryonic Rat Development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109(Suppl):S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Hennigan A, O’Callaghan RM, Kelly AM. Neurotrophins and their receptors: roles in plasticity, neurodegeneration and neuroprotection. Biochem Soc Trans. 2007;35:424–427. doi: 10.1042/BST0350424. [DOI] [PubMed] [Google Scholar]
- Huh JE, Yim JH, Lee HK, Moon EY, Rhee DK, Pyo S. Prodigiosin isolated from Hahella chejuensis suppresses lipopolysaccharide-induced NO production by inhibiting p38 MAPK, JNK and NF-kappaB activation in murine peritoneal macrophages. Int Immunopharmacol. 2007;7:1825–1833. doi: 10.1016/j.intimp.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Iglesias T, Cabrera-Poch N, Mitchell MP, Naven TJ, Rozengurt E, Schiavo G. Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J Biol Chem. 2000;275:40048–40056. doi: 10.1074/jbc.M005261200. [DOI] [PubMed] [Google Scholar]
- Kadari A, Windisch JM, Ebendal T, Schneider R, Humpel C. Cell death of adult pyramidal CA1 neurons after intraventricular injection of a novel peptide derived from trkA. J Neurosci Res. 1997;50:402–412. doi: 10.1002/(SICI)1097-4547(19971101)50:3<402::AID-JNR6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Kim DH, Zhao X, Tu CH, Casaccia-Bonnefil P, Chao MV. Prevention of apoptotic but not necrotic cell death following neuronal injury by neurotrophins signaling through the tyrosine kinase receptor. J Neurosurg. 2004;100:79–87. doi: 10.3171/jns.2004.100.1.0079. [DOI] [PubMed] [Google Scholar]
- Kong H, Boulter J, Weber JL, Lai C, Chao MV. An evolutionarily conserved transmembrane protein that is a novel downstream target of neurotrophin and ephrin receptors. J Neurosci. 2001;21:176–185. doi: 10.1523/JNEUROSCI.21-01-00176.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong H, Kim AH, Orlinick JR, Chao MV. A comparison of the cytoplasmic domains of the Fas receptor and the p75 neurotrophin receptor. Cell Death Differ. 1999;6:1133–1142. doi: 10.1038/sj.cdd.4400587. [DOI] [PubMed] [Google Scholar]
- Kouba DJ, Nakano H, Nishiyama T, Kang J, Uitto J, Mauviel A. Tumor necrosis factor-alpha induces distinctive NF-kappa B signaling within human dermal fibroblasts. J Biol Chem. 2001;276:6214–6224. doi: 10.1074/jbc.M004511200. [DOI] [PubMed] [Google Scholar]
- Kupfer R, Lang J, Williams-Skipp C, Nelson M, Bellgrau D, Scheinman RI. Loss of a gimap/ian gene leads to activation of NF-kappaB through a MAPK-dependent pathway. Mol Immunol. 2007;44:479–487. doi: 10.1016/j.molimm.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Lee FS, Peters RT, Dang LC, Maniatis T. MEKK1 activates both IkappaB kinase alpha and IkappaB kinase beta. Proc Natl Acad Sci U S A. 1998;95:9319–9324. doi: 10.1073/pnas.95.16.9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-kappaB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek R, Borowicz KK, Jargiello M, Czuczwar SJ. Role of nuclear factor kappaB in the central nervous system. Pharmacol Rep. 2007;59:25–33. [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- Nagy I, Caelers A, Monge A, Bonabi S, Huber AM, Bodmer D. NF-kappaB-dependent apoptotic hair cell death in the auditory system. Audiol Neurootol. 2007;12:209–220. doi: 10.1159/000101328. [DOI] [PubMed] [Google Scholar]
- Nicholas C, Batra S, Vargo MA, Voss OH, Gavrilin MA, Wewers MD, Guttridge DC, Grotewold E, Doseff AI. Apigenin blocks lipopolysaccharide-induced lethality in vivo and proinflammatory cytokines expression by inactivating NF-kappaB through the suppression of p65 phosphorylation. J Immunol. 2007;179:7121–7127. doi: 10.4049/jimmunol.179.10.7121. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Litzburg A, Zhang D, Dewhurst S, Gelbard HA. HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol. 2005;174:4333–4344. doi: 10.4049/jimmunol.174.7.4333. [DOI] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci U S A. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S, Maggirwar SB. Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem. 2001;78:874–889. doi: 10.1046/j.1471-4159.2001.00467.x. [DOI] [PubMed] [Google Scholar]
- Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res. 2001;29:E21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickle A, Behbahani H, Ankarcrona M, Winblad B, Cowburn RF. PTEN, Akt, and GSK3beta signalling in rat primary cortical neuronal cultures following tumor necrosis factor-alpha and trans-4-hydroxy-2-nonenal treatments. J Neurosci Res. 2006;84:596–605. doi: 10.1002/jnr.20970. [DOI] [PubMed] [Google Scholar]
- Ringstedt T, Lagercrantz H, Persson H. Expression of members of the trk family in the developing postnatal rat brain. Brain Res Dev Brain Res. 1993;72:119–131. doi: 10.1016/0165-3806(93)90165-7. [DOI] [PubMed] [Google Scholar]
- Saha RN, Pahan K. Differential regulation of Mn-superoxide dismutase in neurons and astroglia by HIV-1 gp120: Implications for HIV-associated dementia. Free Radic Biol Med. 2007;42:1866–1878. doi: 10.1016/j.freeradbiomed.2007.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, Saiki I. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–36923. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- Sanchez JF, Sniderhan LF, Williamson AL, Fan S, Chakraborty-Sett S, Maggirwar SB. Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol Cell Biol. 2003;23:4649–4662. doi: 10.1128/MCB.23.13.4649-4662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutze S, Machleidt T, Adam D, Schwandner R, Wiegmann K, Kruse ML, Heinrich M, Wickel M, Kronke M. Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J Biol Chem. 1999;274:10203–10212. doi: 10.1074/jbc.274.15.10203. [DOI] [PubMed] [Google Scholar]
- Sui Z, Fan S, Sniderhan L, Reisinger E, Litzburg A, Schifitto G, Gelbard HA, Dewhurst S, Maggirwar SB. Inhibition of mixed lineage kinase 3 prevents HIV-1 Tat-mediated neurotoxicity and monocyte activation. J Immunol. 2006a;177:702–711. doi: 10.4049/jimmunol.177.1.702. [DOI] [PubMed] [Google Scholar]
- Sui Z, Sniderhan LF, Fan S, Kazmierczak K, Reisinger E, Kovacs AD, Potash MJ, Dewhurst S, Gelbard HA, Maggirwar SB. Human immunodeficiency virus-encoded Tat activates glycogen synthase kinase-3beta to antagonize nuclear factor-kappaB survival pathway in neurons. Eur J Neurosci. 2006b;23:2623–2634. doi: 10.1111/j.1460-9568.2006.04813.x. [DOI] [PubMed] [Google Scholar]
- Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, Maggirwar SB. Functional synergy between CD40 ligand and HIV-1 Tat contributes to inflammation: implications in HIV type 1 dementia. J Immunol. 2007;178:3226–3236. doi: 10.4049/jimmunol.178.5.3226. [DOI] [PubMed] [Google Scholar]
- Thomas SM, DeMarco M, D’Arcangelo G, Halegoua S, Brugge JS. Ras is essential for nerve growth factor- and phorbol ester-induced tyrosine phosphorylation of MAP kinases. Cell. 1992;68:1031–1040. doi: 10.1016/0092-8674(92)90075-n. [DOI] [PubMed] [Google Scholar]
- Tuosto L, Costanzo A, Guido F, Marinari B, Vossio S, Moretti F, Levrero M, Piccolella E. Mitogen-activated kinase kinase kinase 1 regulates T cell receptor- and CD28-mediated signaling events which lead to NF-kappaB activation. Eur J Immunol. 2000;30:2445–2454. doi: 10.1002/1521-4141(200009)30:9<2445::AID-IMMU2445>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. p75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- Vetter ML, Martin-Zanca D, Parada LF, Bishop JM, Kaplan DR. Nerve growth factor rapidly stimulates tyrosine phosphorylation of phospholipase C-gamma 1 by a kinase activity associated with the product of the trk protooncogene. Proc Natl Acad Sci U S A. 1991;88:5650–5654. doi: 10.1073/pnas.88.13.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt S, Davies AM. Regulation of nerve growth factor receptor gene expression in sympathetic neurons during development. J Cell Biol. 1995;130:1435–1446. doi: 10.1083/jcb.130.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]