

Abstract
The availability of cysteine is thought to be the rate limiting factor for synthesis of the tripeptide glutathione (GSH), based on studies in rodents. GSH status is compromised in various disease states and by certain medications leading to increased morbidity and poor survival. To determine the possible importance of dietary cyst(e)ine availability for whole blood glutathione synthesis in humans, we developed a convenient mass spectrometric method for measurement of the isotopic enrichment of intact GSH and then applied it in a controlled metabolic study. Seven healthy male subjects received during two separate 10-day periods an l-amino acid based diet supplying an adequate amino acid intake or a sulfur amino acid (SAA) (methionine and cysteine) free mixture (SAA-free). On day 10, l-[1-13C]cysteine was given as a primed, constant i.v. infusion (3μmol⋅kg−1⋅h−1) for 6 h, and incorporation of label into whole blood GSH determined by GC/MS selected ion monitoring. The fractional synthesis rate (mean ± SD; day-1) of whole blood GSH was 0.65 ± 0.13 for the adequate diet and 0.49 ± 0.13 for the SAA-free diet (P < 0.01). Whole blood GSH was 1,142 ± 243 and 1,216 ± 162 μM for the adequate and SAA-free periods (P > 0.05), and the absolute rate of GSH synthesis was 747 ± 216 and 579 ± 135 μmol⋅liter−1⋅day−1, respectively (P < 0.05). Thus, a restricted dietary supply of SAA slows the rate of whole blood GSH synthesis and diminishes turnover, with maintenance of the GSH concentration in healthy subjects.
The tripeptide glutathione [γ-glutamyl-cysteinyl-glycine (GSH)] is present in relatively high concentrations in mammalian cells. It has evolved to serve diverse functions, including detoxification of xenobiotics, protection of cells from oxidative stress, acting as a storage and a transport form of cysteine and affecting cellular thiol redox status, with consequences for the regulation of signal transduction pathways and gene transcription (1, 2). Reduced tissue levels of GSH are thought to compromise cell function, to promote tissue damage, and increase morbidity (3) under various disease conditions, including inflammatory bowel disease (4), critical illness (5), HIV infection (6, 7), and acetaminophen-induced GSH depletion (8), which may result in hepatic and renal failure and ultimately in death. Thus, various therapeutic approaches are being explored as a basis for improving GSH status (9, 10).
The mechanisms responsible for altered GSH homeostasis in human subjects under various conditions have not been fully determined, but this knowledge is important for the design of safe and effective clinical strategies aimed at maintaining or enhancing GSH status in defined physiological conditions. Jahoor et al. (11) have shown, using a tracer approach and noncompartmental modeling, that the absolute synthesis rate and concentration of erythrocyte GSH are reduced in HIV-infected subjects and that this appears to be caused, in part, by a shortage in cysteine availability. Extensive studies in rodents have led to the conclusion that the supply of cysteine rate-limits the synthesis of glutathione (12), although the supply of glutamate/glutamine (13, 14) might also play a role in modulating GSH synthesis in human subjects.
To better characterize in vivo aspects of GSH metabolism and homeostasis in humans, we considered it important to use a tracer paradigm, requiring a convenient procedure for precise measurement of the stable isotope enrichment of the intact tripeptide. With this, it would then be possible to determine for the first time whether relatively short-term changes in the dietary supply of the sulfur amino acids would alter the rate of GSH synthesis in healthy volunteers. The studies presented here expand our understanding of the quantitative dietary and metabolic relationships among the sulfur amino acids and glutathione in normal healthy adults. Further, they provide a better basis for the design and conduct of studies and interpretation of results from investigations in hospitalized and sick patients. These might include those suffering from severe burn injury, in which we (15) have observed that the metabolism of methionine is altered, and cirrhotic patients for which others (16) have reported changes in methionine metabolism.
Materials and Methods
Subjects and Experimental Design.
Seven healthy adult males (age 24 ± 6 years, mean ± SD; body weight 81 ± 14 kg), participated in the main experiment. One of these and an additional adult male subject participated in a preliminary set of experiments aimed to determine a tracer dose of l-[1-13C] cysteine for sufficient labeling of whole blood GSH. They were studied in the Clinical Research Center of the Massachusetts Institute of Technology. All were in good health, as determined by history and physical examination, analysis of cell blood count, biochemical profile, and urine analysis. Their daily energy intake was calculated to maintain body weight, based on a dietary history and an estimate of the subject's usual level of physical activity. The subjects were encouraged to maintain their customary levels of physical activity but did not participate in competitive sports.
The purpose of the study and potential risks involved were fully explained to each subject. Written consent was obtained according to the protocol approved by the Massachusetts Institute of Technology Committee on the use of Humans as Experimental Subjects and the Executive and Policy Committee of the Massachusetts Institute of Technology Clinical Research Center. All subjects received financial compensation for their participation in the experiments and all remained healthy throughout.
Two subjects participated in four pilot experiments, each consisting of a 3-day dietary period during which they received an adequate protein and energy intake, provided as an l-amino acid formula patterned after hen's egg proteins. This was followed by an 8-h, primed constant i.v. infusion of l-[1-13C]cysteine given at different rates of infusion. One of the subjects was studied on three different occasions by using 5, 15, and 30 μmol⋅kg−1⋅h−1 as the tracer doses, and the other subject was studied at a tracer infusion rate of 20 μmol⋅kg−1⋅h−1. All received priming doses equivalent to the 60-min infusion.
The seven subjects who participated in the main study were assigned randomly to each of two 10-day experimental dietary periods providing a complete amino acid formula or a sulfur amino acid (SAA)-free intake. The periods were separated by about 7 days, during which time the subjects consumed a free-choice diet. A 6-h primed, constant, i.v. tracer infusion was conducted on day 10 of each dietary period.
Diet.
Each subject received a diet providing the equivalent of 1 g of protein (N × 6.25) per kilogram of body weight, based on an amino acid formula patterned after hen's egg proteins during one period. The other diet period provided an amino acid mixture devoid of sulfur amino acids (Table 1). The major dietary energy source was provided in the form of protein-free, wheat starch cookies as described (17). The total daily intake before the tracer infusion studies was provided as three separate meals at 0800 h, 1200 h, and 1700 h. Two of these meals were always eaten in the Clinical Research Center under the supervision of the dietary staff. They otherwise continued with their usual everyday activities and slept in their residencies.
Table 1.
Composition of l-amino acid mixtures used for the adequate and sulfur amino acid-free diet (mg⋅kg−1⋅day−1) given to healthy subjects
| Amino acid | Adequate | SAA-free |
|---|---|---|
| Methionine | 35.3 | 0.0 |
| Cysteine | 26.3 | 0.0 |
| Tryptophan | 18.6 | 18.6 |
| Threonine | 56.1 | 56.1 |
| Isoleucine | 74.8 | 74.8 |
| Leucine | 99.1 | 99.1 |
| Lysine⋅HCl | 90.1 | 90.1 |
| Phenylalanine | 65.1 | 65.1 |
| Tyrosine | 48.4 | 48.4 |
| Valine | 83.6 | 83.6 |
| Histidine⋅HCl⋅H2O | 36.5 | 36.5 |
| Arginine⋅HCl | 89.4 | 89.4 |
| Alanine | 73.2 | 73.2 |
| Aspartic acid | 78.9 | 78.9 |
| Glutamic acid | 134.9 | 134.9 |
| Glycine | 39.6 | 75.1 |
| Proline | 49.9 | 49.9 |
| Serine | 99.9 | 99.9 |
| Total Nitrogen | 157.3 | 157.6 |
Tracer Infusion Studies.
The isotope-infusion periods lasted for a total of 6–8 h, and they were carried out when subjects were in the postabsorptive state. Details of the general procedures followed immediately before and during the infusion protocol were similar to those described previously (17–19). The 8-h tracer pilot studies revealed that a [1-13C]-cysteine tracer infusion rate of 5 μmol⋅kg−1⋅h−1 or less would be more than sufficient for our purposes. We, therefore, conducted the main study by using a tracer infusion rate of 3 μmol⋅kg−1⋅h−1.
Thus, on day 10, the subjects entered the infusion room at approximately 7 a.m. and later received a priming dose of 3 μmol⋅kg−1 l-[1-13C]cysteine (obtained from MassTrace, Woburn, MA) (98% 13C), followed by a constant infusion of tracer for 6 h. The bicarbonate pool also was primed at the beginning of the tracer protocol (0.8 μmol⋅kg−1 of [13C] Na bicarbonate; 99% atom) (Cambridge Isotopes Laboratories). Breath samples were collected at intervals during the tracer period 0–360 min and were stored at room temperature until analyzed by isotope ratio mass spectrometry (MAT Delta E, Finnigan, Bremmen, Germany) (19). Total carbon dioxide output was determined by indirect calorimetry (Deltatrack, Sensormedics, Anaheim, CA) for 25 min of every hour during the tracer study period.
Blood samples were drawn at hourly intervals for determination of the plasma (cysteine) and whole blood cysteine isotopic enrichment, as well as the enrichment of [1-13C]cysteinyl-GSH in whole blood. For the plasma isotopic enrichment of l-[1-13C]cysteine, blood samples were placed in tubes containing heparin and were centrifuged immediately, and plasma was stored at −80°C until used for analysis.
Whole blood GSH samples were placed in cold centrifuge tubes (containing 15 μl of 5 M EDTA) and were mixed well. An aliquot (50 μl) of blood was transferred to a second tube (containing 20 mM DTT in 1 ml of 1 M acetic acid). The whole blood samples were stored at −80°C until used.
Analysis.
Samples for isotopic abundance of cysteine in plasma were measured in duplicate as described (18). The cysteine concentrations in plasma and infusates were also measured by this procedure, using [3,3-2H2]cysteine (MassTrace) as an internal standard.
Whole blood total GSH was determined as follows. The red blood cells were lysed by freezing (−80oC) and thawing (room temperature) three times. The samples were then kept at room temperature for 2 h. After centrifuging at 10,000 × g for 5 min, the supernatant solutions were transferred onto an ion exchange column (1.5 ml of the 50w-X8 resin) and were washed with milli-Q water (2.5 ml, three times), and the GSH was eluted into derivatization tubes (10 ml) with NH4OH solution (3 M, 1.5 ml, two times). After drying the samples at 65°C under nitrogen, any dimerized GSH was reduced with a DTT solution (20 mM in 0.5 ml of 0.1 M acetic acid) at room temperature for 2 h. Solvent was removed again at 65°C under nitrogen, and the residue was treated with methanol/acetyl chloride solution (10/1, vol/vol, prepared on 1 ml of ice), at room temperature for 1 h. After removing excess reactants at 65°C under a nitrogen stream, and then cooling down to room temperature, trifluoroacetic anhydride (0.3 ml) was added to each tube. Reaction was allowed to continue at room temperature for 1.5 h. Excess reactants were then removed at 65°C under nitrogen stream, and the sample was reconstituted with acetonitrile (50 μl) for GC/MS analysis.
The GC/MS measurement was carried out with an HP 5890 Series II gas chromatograph coupled to an HP 5988 Mass Spectrometer (Hewlett–Packard). Injections were made with a splitless injection port onto an HP-1 crosslinked methyl siloxane column (30 m, ID 0.25-mm film, thickness 0.25 μm). Column temperature was programmed at 10°C⋅min−1 from 160–290°C, then at 5°C⋅min−1 from 290–320°C; injector and detector were heated at 250°C; ionization source was maintained at 100°C. Helium was used as carrier gas, and methane was used as reactant. The GSH derivative, whose elemental composition was found to be C15 H13 F6 N3 O6 S, is a complex bicycloglutarimide. The nominal structural assignment is shown in Fig. 1. It was determined independently by 13C and 1H nuclear magnetic resonance, two-dimensional nuclear magnetic resonance, fast atom bombardment-mass spectrometry, and negative chemical ionization GC/MS fragmentography. Although an alternative assignment might be proposed, this structure was preferred because (i) we did not detect an intermediate m/z 495, (ii) 13C NMR indicated the presence of the C⩵C bond, and (iii) the mass spectrometry pattern ruled out a free glycyl side chain. A definitive structural statement cannot be made, however, until results of x-ray crystallography become available. The derivative was monitored under negative chemical ionization conditions by selective ion monitoring at nominal masses from m/z 477.1 to 480.1, corresponding to the most abundant and preponderant near-parent ion. The 13C enrichment and concentration of whole blood glutathione were determined by using synthetic [1,2-13C2-glycyl]GSH (20) as an internal standard. The method is sensitive enough to determine tracer enrichments of 13C-GSH with a quantitation limit in the range of 0.3–0.5 mole percent excess.
Figure 1.

The assigned nominal structure of the bicyclo-glutarimide parent ion of derivatized GSH, incorporating [1-13C]cysteine. The glycyl, glutamyl, and cysteinyl moieties within the molecule are indicated by the arrows.
Calculations and Data Evaluation.
Plasma cysteine flux was calculated from isotopic data covering the tracer period 240–360 min, using noncompartmental steady-state isotope dilution equations, derived from a simplified single-pool model (18). Cysteine oxidation was also determined as described (18), assuming a [13C]bicarbonate recovery of 0.77 (19).
The fractional synthesis of whole blood GSH was calculated as follows, using the approach of Jahoor et al. (21):
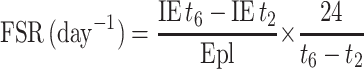 |
where IE t6 − IE t2 is the increase in the 13C isotopic enrichment of whole blood GSH between 2 and 6 h of the tracer infusion period and Epl is the 13C enrichment of the cysteine in the precursor pool. We determined that the ratio of 13C enrichment (net tracer/tracee ratio) of cysteine in plasma to that of cysteine in whole blood was 0.993 ± 0.04 for the adequate and 0.992 ± 0.05 for the SAA-free intakes. We have used the plasma enrichment of cysteine for our calculations.
The absolute synthesis rate (ASR) of whole blood GSH was calculated as the product of the whole blood GSH concentration and the fractional synthesis rate (FSR), as follows:
ASR (μmol⋅liter−1⋅day−1) = whole blood GSH concentration × FSR.
Data were expressed as mean ± SD (±SEM in the figures), for each group. Differences between groups were detected by the paired t test. A probability of P < 0.05 was taken to be statistically significant.
Results
The isotopic abundances for the enrichment of expired CO2 and for the free l-[1-13C]cysteine in plasma and in whole blood [13C]cysteinyl-GSH after the subjects had received the two experimental diets for 10 days are shown in Fig. 2 A, B, and C, respectively. The group averages for isotopic enrichment of plasma l-[13C]cysteine during the period from 2–6 h were 0.073 ± 0.008 and 0.082 ± 0.015 (net tracer/tracee ratio), respectively, for the adequate and SAA-free diets. From these isotopic data, we estimated the kinetics of plasma cysteine and the whole blood GSH-FSR and -ASR for each diet period. Table 2 summarizes the cysteine kinetic data and shows that there was a tendency (P = 0.06) for the postabsorptive cysteine flux to decline with the ingestion of the SAA-free diet. Further, cysteine oxidation was significantly reduced (P < 0.001) in response to this dietary restriction. Oxidation accounted for about 15% of the cysteine disappearance rate when the adequate diet was consumed, falling to about 5% at the end of the SAA-free diet period, reflecting, presumably, a conservation of methionine and cysteine (18). Plasma cysteine concentrations (micromolar values) were 16.41 ± 3.26 for the adequate and 10.61 ± 1.92 for the SAA-free diet periods, respectively (P = 0.07).
Figure 2.

(A) 13C enrichment (atoms percent excess × 10−3) of expired carbon dioxide for the adequate (♦) and the sulfur amino acid-free diet (SAA-free) ( ). (B) Plasma isotopic enrichment (net tracer/tracee ratio) of l-[1-13C]cysteine. (C) Change in the whole blood enrichment (net tracer/tracee ratio) of [13C]cysteinyl-GSH derived from the l-[1-13C] cysteine tracer. Bars are SEM.
Table 2.
Summary of whole body cysteine kinetics in healthy male subjects receiving an adequate or a sulfur amino acid free diet
| Parameter | Adequate | SAA-free |
|---|---|---|
| 13CO2 enrichment (APE × 10−3) | 3.28 ± 0.58*† | 1.19 ± 0.14 |
| VCO2 (mmol⋅kg−1⋅h−1) | 8.48 ± 0.7 | 8.49 ± 0.02 |
| V13CO2 (μmol⋅kg−1⋅h−1) | 0.36 ± 0.07† | 0.13 ± 0.02 |
| Cysteine flux (μmol⋅kg−1⋅h−1) | 38.3 ± 6.4 | 30.9 ± 5.5 |
| Cysteine oxidation (μmol⋅kg−1⋅h−1) | 5.62 ± 0.6† | 1.58 ± 0.16 |
| Percent of cysteine flux oxidized | 14.52 ± 3.15† | 5.16 ± 1.30 |
| Plasma cysteine concentration, μM | 16.41 ± 3.26 | 10.61 ± 1.92 |
APE, atoms percent excess.
*All values are mean ± SD.
†P < 0.01 different from SAA-free.
The rates of whole blood GSH synthesis, expressed as a fraction of the pool and also as an absolute rate, for each diet period, are summarized in Table 3. The fractional synthesis rate was 0.65 ± 0.13 per day when subjects received the adequate diet, and this decreased (P < 0.01) to 0.49 ± 0.13 within 10 days of the SAA-free diet. The ASR was similarly reduced (P < 0.05), by about 23% when the intake of sulfur amino acids was restricted. Whole blood GSH concentrations were not different between the two diet periods, approximating 1.2 mM.
Table 3.
Whole blood fractional (FSR) and absolute (ASR) synthesis rates and concentrations of GSH in young adult men given an adequate or SAA-Free diet for 10 days
| Diet and subjects | GSH
|
Whole blood GSH, μM | |
|---|---|---|---|
| FSR, per day | ASR, μmol⋅liter−1⋅day−1 | ||
| Adequate | |||
| 1 | 0.65 | 557.97 | 858.42 |
| 2 | 0.57 | 600.51 | 1,053.52 |
| 3 | 0.49 | 479.32 | 978.20 |
| 4 | 0.58 | 673.25 | 1,160.77 |
| 5 | 0.60 | 987.00 | 1,645.00 |
| 6 | 0.83 | 829.83 | 999.80 |
| 7 | 0.85 | 1105.30 | 1,300.35 |
| Mean ± SD | 0.65 ± 0.13* | 747 ± 216† | 1,142 ± 243 |
| SAA-free | |||
| 1 | 0.57 | 650.44 | 1,143.13 |
| 2 | 0.56 | 546.53 | 979.45 |
| 3 | 0.34 | 462.16 | 1,359.29 |
| 4 | 0.24 | 356.60 | 1,485.85 |
| 5 | 0.50 | 604.38 | 1,208.75 |
| 6 | 0.58 | 614.12 | 1,058.82 |
| 7 | 0.64 | 815.96 | 1,274.94 |
| Mean ± SD | 0.49 ± 0.13 | 578.6 ± 135 | 1,216 ± 162 |
*P < 0.01 adequate vs. SAA-free diet.
†P < 0.05 adequate vs. SAA-free diet.
Discussion
Using a newly developed gas chromatography/mass spectrometric determination of the isotopic enrichment of intact whole blood glutathione, we have studied cysteine kinetics and oxidation and estimated the rate of whole blood glutathione synthesis in healthy young adults. Cysteine oxidation during the postabsorptive condition when subjects had consumed the adequate diet approximated 5 μmol⋅kg−1⋅h−1. This rate is very similar to that which we have estimated previously in healthy adults consuming diets supplying either adequate methionine or a combination of methionine and cysteine (18). It is also similar to that observed, after subjects have consumed a protein-free or amino acid-free diet for 6 days (18). When our subjects were given a diet free of sulfur amino acids, but otherwise adequate with respect to amino acid and nitrogen content, the rate of cysteine oxidation declined to about 30% of that with the fully adequate diet. This response is reminiscent of that which we have reported previously, when leucine and methionine oxidation rates were compared between those for protein (amino acid)-free diets versus diets otherwise adequate but specifically devoid of leucine or the sulfur amino acids (22). It is evident that there is a more efficient conservation of indispensable or conditionally indispensable amino acids, such as cysteine, when intake of an amino acid is specifically restricted, rather than when its intake is lowered as part of an overall reduction in amino acid intake. Nevertheless, the lowered rate of cysteine oxidation when subjects were given the SAA-free diet reflects a conservation of body cysteine, and this determines in part the quantitative changes in GSH metabolism in response to a diet limiting in methionine and cysteine.
Based on our approach and method, we have estimated that, when receiving an adequate diet, the mean whole blood GSH-FSR was estimated to be 0.65 per day. This is essentially identical to the erythrocyte glutathione FSR reported by Jahoor et al. (11), who used [2H2] glycine as the tracer in a study with five normal healthy British residents. The mean absolute synthesis rate (ASR) that we have determined here is about 748 μmol⋅liter−1⋅day−1 when subjects consumed a fully adequate diet, which compares with a value of about 1.6 mmol⋅liter−1⋅day−1 for the ASR of erythrocyte glutathione obtained by Jahoor et al. (11). The difference between the two studies is attributable to the fact that we have made our measurements on whole blood whereas Jahoor et al. (11) made their studies on the erythrocyte. When corrected for a hematocrit of 41.8 ± 0.8% in our subjects, the ASRs reported here and by Jahoor et al. (11) are virtually the same. This also infers that it was the erythrocyte that was responsible for the bulk of glutathione synthesis in the whole blood of our subjects. Furthermore, this comparison also suggests that glycine and cysteine are equally suitable tracers to study in vivo rates of glutathione synthesis in whole blood and erythrocytes in humans. This point is also supported by Jahoor et al. (21), who found a similar erythrocyte FSR for reduced GSH, in piglets, when using [13C2]glycine or [2H2]cysteine as tracers. Our findings, however, may differ from those of Capitan et al. (23) who used [3,3- 2H2]cysteine and a GC-MS procedure, based on analysis of sonicated whole blood and preparation of the N,S-ethoxycarbonyl methyl ester derivative of the intact peptide with mass spectrometry under electron ionization conditions. These investigators (23) report for one subject a FSR of 0.23 per day. However, it is difficult to draw meaningful comparisons based on just a single subject. Furthermore, it would now be highly desirable to carry out nonprimed tracer studies that would allow the fitting of data to a more formal model for purpose of estimating GSH-FSR (24), particularly because of the tendency for the approach used by us and others (11, 21, 23) to underestimate the FSR. For example, if the data of Fig. 2C is fitted to a minimal two-compartment precursor product model (24) with sequential first order (priming bolus) and zero order (infusion) input, the analytically derived FSR is three to four times higher than presently estimated, suggesting that the enrichment of the “true” intracellular precursor pool is correspondingly lower than that which can be determined from the apparent cysteine plateau (Fig. 2B). In this connection also, the assumption needs to be tested further that the noncompartmental kinetics of plasma (or erythrocyte) cysteine are indeed reflective of glutathione's precursor pool, in comparison to noncompartmental FSR calculations based on the enrichment of glutamyl-cysteine, the immediate intracellular anabolic precursor, or, in cysteinyl-glycine, the immediate intracellular, catabolic product.
It is clear that a change in the dietary availability of cyst(e)ine and its precursor, methionine, can modulate the rate of whole blood GSH synthesis in healthy adults. Although this is the first demonstration in humans that a specific restriction of SAA intake over a relatively short period alters GSH synthesis, the extent to which our findings reflect altered rates of GSH synthesis in other organs and tissues or how acute the response of the GSH machinery is to an altered substrate intake cannot be determined from this study. It might be useful, however, to indicate that an ASR for whole blood glutathione synthesis of 750 μmol⋅liter−1⋅day−1 is roughly equivalent to a rate of 3 μmol⋅kg−1⋅h−1, assuming that blood volume accounts for 8% body weight in a 75-kg subject. This latter rate compares with an endogenous GSH disappearance rate of about 25 μmol⋅kg−1⋅h−1 based either on the plasma pharmacokinetics of an administered bolus of glutathione (25) and indirectly from an evaluation of plasma cysteine kinetics before and after i.v. administration of glutathione (26). Therefore, whole blood, and presumably mainly erythrocyte, glutathione synthesis may account for some 10% of whole body synthesis.
With respect to the rapidity of the response of GSH metabolism to dietary change, we will need to assess this in new studies. However, but in the clinical context, parenteral nutrition in stressed patients frequently involves little cyst(e)ine intake and often under a prolonged time frame. This suggests that the deterioration in GSH status seen in different clinical conditions could be attributable to a primary substrate limitation. At least provision of cysteine as N-acetyl cysteine or as l-2-oxothiazolidine-4-carboxylate (11, 27, 28) can raise GSH concentrations in various clinical situations associated with GSH depletion. The extent to which a nutritional (substrate) modality might be effective in improving GSH levels, where there may also be a reduction in the in vitro activity of γ-glutamyl cysteine synthetase, the rate limiting enzyme (29), and GSH synthetase (30), also needs to be examined. This should involve ideally a detailed study of the relationships between the activity of tissue enzymes when measured in vitro, the in vivo rate of GSH synthesis in various tissues and organs, and a labeled metaprobe of the γ-glutamyl cycle such as [2-13C]l-2-oxothiazolidine-4-carboxylate (31).
The reduced FSR of whole blood GSH was not paralleled by a decline in the whole blood GSH concentration. In patients undergoing abdominal surgery, Lou et al. (30) found that whole blood GSH did not change after surgery whereas it declined in quadriceps femoris muscle, and this was associated with a reduced GSH synthetase activity, suggesting a diminished GSH synthetic capacity in this tissue. Thus, we cannot determine from the present data whether there were changes in the rates of GSH synthesis and in GSH concentrations in tissues such as muscle, intestine, liver, and lung because of the restricted SAA intake, but it seems likely that this would be so, from animal experiments (1, 12). Because the concentration of whole blood glutathione did not differ between the two dietary periods, it would appear that the rate of GSH turnover was reduced when the SAA intake was restricted. This could be attributable to an adaptive decrease in the transport of glutathione in an outward direction (32). In this context, it would be informative to know more about the pharmacokinetics of GSH under the present dietary conditions, possibly by using a suitably labeled GSH tracer such as that which we have synthesized (20). However, to date, we have been unable to prepare efficiently the tracer in sufficient amounts and purity to be able to apply it in human investigations. In disease states, it is possible that synthesis is reduced and breakdown of GSH increased, each requiring possibly different therapeutic strategies to attenuate GSH depletion.
Finally, our findings should be connected with our recent and related studies on an intermediate in the γ-glutamyl cycle of GSH synthesis, or the kinetics of plasma oxoproline and its urinary excretion. After 5 days of a sulfur-free diet in similar healthy adults, we observed a higher oxoproline flux and oxidation rate and an increased urinary output of oxoproline on day 6 (33). Thus, a restricted sulfur amino acid intake in healthy adults appears to alter the activity of a number of steps in the γ-glutamyl cycle, leading to changed GSH synthesis and turnover. To better understand the detailed kinetic and mass balance relationships between and among the components of the γ-glutamyl cycle will require that compartmental models of the metabolism of the cycle intermediates are developed. This is a goal of our research, which is aimed at characterizing fully the complex physiology of glutathione metabolism in vivo in humans (34), under various pathophysiological states. In turn, we would then be in a better position to exploit the wealth of biochemical and cellular data on GSH metabolism and function that now exists. The achievement of an effective and safe manipulation of tissue and organ GSH levels through alternative strategies, including use of GSH precursors (35, 36), would benefit from an integration of this knowledge.
Acknowledgments
We thank the volunteers for their commitment, and we are grateful for the assistance of the nursing, technical, and dietary staff at the Clinical Research Center. This study was supported by National Institutes of Health Grants DK15856, RR88, and GM 02700, and grants-in aid from the Shriners Hospitals for Children and the Anesthesia Research Foundation from Children's Hospital, Harvard Medical School.
Abbreviations
- ASR
absolute synthesis rate
- FSR
fractional synthesis rate
- GSH
glutathione
- SAA
sulfur amino acid
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.090083297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.090083297
References
- 1.Meister A, Anderson M E, Hwang O. J Am Coll Nutr. 1986;5:137–151. doi: 10.1080/07315724.1986.10720121. [DOI] [PubMed] [Google Scholar]
- 2.Beutler E. Annu Rev Nutr. 1989;9:287–302. doi: 10.1146/annurev.nu.09.070189.001443. [DOI] [PubMed] [Google Scholar]
- 3.Herzenberg L A, de Rosa S C, Dubs J G, Roederer M, Anderson M T, Elo S W, Deresinski S C, Herzenberg L A. Proc Natl Acad Sci USA. 1997;94:1967–1972. doi: 10.1073/pnas.94.5.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sido B, Hack V, Hochlehnert A, Lipps H, Herforch C, Dröge W. Gut. 1998;42:485–492. doi: 10.1136/gut.42.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammarqvist F, Luo J-L, Cotgreave J A, Anderson K, Wernerman J. Crit Care Med. 1997;25:783–784. doi: 10.1097/00003246-199701000-00016. [DOI] [PubMed] [Google Scholar]
- 6.Helgling B, Von Overbeck J, Lauterburg B H. Eur J Clin Invest. 1996;26:38–44. doi: 10.1046/j.1365-2362.1996.88237.x. [DOI] [PubMed] [Google Scholar]
- 7.Buhl R, Holroyd D, Mastrangeli A, Cantin A M, Jajfe H A, Wells F B, Saltini C, Crystal R G. Lancet. 1989;ii:1294–1298. doi: 10.1016/s0140-6736(89)91909-0. [DOI] [PubMed] [Google Scholar]
- 8.Thomas S H. Pharmacol Ther. 1993;60:91–120. doi: 10.1016/0163-7258(93)90023-7. [DOI] [PubMed] [Google Scholar]
- 9.McCellan L I, Wolf C R. Drug Resist Updates. 1999;2:153–164. doi: 10.1054/drup.1999.0083. [DOI] [PubMed] [Google Scholar]
- 10.Griffith O W. Free Rad Biol Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 11.Jahoor F, Jackson A, Gazzard B, Philips G, Sharpstone D, Frazer M E, Heird W. Am J Physiol. 1999;276:E205–E211. doi: 10.1152/ajpendo.1999.276.1.E205. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi M, Hiramaya K, Yamaguchi N, Tateishi N, Suzuki M. In: Glutathione: Chemical, Biochemical and Medical Aspects. Dolphin D, Poulson R, Avramovic O, editors. New York: Wiley; 1989. , Part B, pp. 645–727. [Google Scholar]
- 13.Klimberg V S, McCellan J L. Am J Surg. 1996;172:418–424. doi: 10.1016/s0002-9610(96)00217-6. [DOI] [PubMed] [Google Scholar]
- 14.Amores-Sanchez M I, Medina M A. Mol Genet Metab. 1999;67:100–105. doi: 10.1006/mgme.1999.2857. [DOI] [PubMed] [Google Scholar]
- 15.Yu Y-M, Burke J F, Young V R. J Trauma. 1993;35:1–7. doi: 10.1097/00005373-199307000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Marchesini G, Bugianesi E, Bianchi G, Fabbri A, Marchi E, Zoli M, Pisi E. Hepatology. 1992;16:149–155. doi: 10.1002/hep.1840160125. [DOI] [PubMed] [Google Scholar]
- 17.Storch K J, Wagner D A, Burke J F, Young V R. Am J Physiol. 1998;255:E322–E331. doi: 10.1152/ajpendo.1988.255.3.E322. [DOI] [PubMed] [Google Scholar]
- 18.Raguso C, Regan M M, Young V R. Am J Clin Nutr. 2000;71:491–499. doi: 10.1093/ajcn/71.2.491. [DOI] [PubMed] [Google Scholar]
- 19.El-Khoury A E, Fukagawa N K, Sanchez M, Tsay R H, Gleason R E, Chapman T E, Young V R. Am J Clin Nutr. 1994;59:1000–1011. doi: 10.1093/ajcn/59.5.1000. [DOI] [PubMed] [Google Scholar]
- 20.Lu X-M, Fischman A J, Keeneway M, Tompkins R G, Young V R. J Labelled Compds Radiopharm. 1997;39:205–213. [Google Scholar]
- 21.Jahoor R A, Wykes L J, Reeds P J, Henry J F, Del Rosario M P, Frazer M E. J Nutr. 1995;125:1462–1472. doi: 10.1093/jn/125.6.1462. [DOI] [PubMed] [Google Scholar]
- 22.Raguso C, Pereira P, Young V R. Am J Clin Nutr. 1999;70:474–483. doi: 10.1093/ajcn/70.4.474. [DOI] [PubMed] [Google Scholar]
- 23.Capitan P, Malmezat T, Breuille D, Obled C. J Chromatogr B Biomed Sci Appl. 1999;732:127–135. doi: 10.1016/s0378-4347(99)00273-x. [DOI] [PubMed] [Google Scholar]
- 24.Foster D M, Hugh P, Barrett R, Toffolo G, Beltz W F, Cobelli C. J Lipid Res. 1993;34:2193–2205. [PubMed] [Google Scholar]
- 25.Burgunder J M, Lauterburg B H. Eur J Clin Invest. 1987;17:408–414. doi: 10.1111/j.1365-2362.1987.tb01135.x. [DOI] [PubMed] [Google Scholar]
- 26.Fukagawa N K, Ajami A M, Young V R. Am J Physiol. 1996;270:E209–E214. doi: 10.1152/ajpendo.1996.270.2.E209. [DOI] [PubMed] [Google Scholar]
- 27.Anderson M E, Luo J-L. Semin Liver Dis. 1998;18:415–424. doi: 10.1055/s-2007-1007174. [DOI] [PubMed] [Google Scholar]
- 28.Dröge W, Gross A, Hack V, Kinscherf R, Schykowski M, Bockstette M, Mihm S, Galter D. Adv Pharmacol. 1997;38:581–600. doi: 10.1016/s1054-3589(08)61000-5. [DOI] [PubMed] [Google Scholar]
- 29.Lu S C. Semin Liver Dis. 1998;18:321–343. doi: 10.1055/s-2007-1007168. [DOI] [PubMed] [Google Scholar]
- 30.Luo J-L, Hammarqvist F, Anderson K, Wernerman J. Am J Physiol. 1998;275:E359–E365. doi: 10.1152/ajpendo.1998.275.2.E359. [DOI] [PubMed] [Google Scholar]
- 31.Young V R, Ajami A M. J Parenter Enteral Nutr. 1999;23:175–194. doi: 10.1177/0148607199023004175. [DOI] [PubMed] [Google Scholar]
- 32.Lunn G, Dole G L, Beutler E. Blood. 1979;54:238–244. [PubMed] [Google Scholar]
- 33.Metges, C. C., Yu, Y.-M., Cai, W., Lu, X.-M., Wong, S., Reagan, M. M., Ajami, A. & Young, V. R. (2000) Am. J. Physiol., in press.
- 34.Smith C V, Jones D P, Guenther T M, Lash L H, Lauterburg B H. Toxicol Appl Pharmacol. 1996;140:1–12. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 35.Wernerman J, Hammarqvist F. Curr Opin Clin Nutr Metabol Care. 1999;2:487–492. doi: 10.1097/00075197-199911000-00010. [DOI] [PubMed] [Google Scholar]
- 36.Dröge W, Breitkreutz R. Curr Opin Clin Nutr Metabol Care. 1999;2:493–498. doi: 10.1097/00075197-199911000-00011. [DOI] [PubMed] [Google Scholar]