

Abstract
We report a very fast and accurate physics-based method to calculate pH-dependent electrostatic effects in protein molecules and to predict the pK values of individual sites of titration. In addition, a CHARMm-based algorithm is included to construct and refine the spatial coordinates of all hydrogen atoms at a given pH. The present method combines electrostatic energy calculations based on the Generalized Born approximation with an iterative mobile clustering approach to calculate the equilibria of proton binding to multiple titration sites in protein molecules. The use of the GBIM (Generalized Born with Implicit Membrane) CHARMm module makes it possible to model not only water-soluble proteins but membrane proteins as well. The method includes a novel algorithm for preliminary refinement of hydrogen coordinates. Another difference from existing approaches is that, instead of monopeptides, a set of relaxed pentapeptide structures are used as model compounds. Tests on a set of 24 proteins demonstrate the high accuracy of the method. On average, the RMSD between predicted and experimental pK values is close to 0.5 pK units on this data set, and the accuracy is achieved at very low computational cost. The pH-dependent assignment of hydrogen atoms also shows very good agreement with protonation states and hydrogen-bond network observed in neutron-diffraction structures. The method is implemented as a computational protocol in Accelrys Discovery Studio and provides a fast and easy way to study the effect of pH on many important mechanisms such as enzyme catalysis, ligand binding, protein–protein interactions, and protein stability.
Keywords: protein ionization, pK prediction, continuum electrostatics, Generalized Born, CHARMm, hydrogen-bond network
The development of accurate methods to study ionization processes in proteins is important because pH-dependent changes in the protonation state can affect almost all molecular mechanisms related to protein function and stability. The modeling of protein ionization has a very long history. After the pioneering work of Linderstrom-Lang (1924), Tanford and Kirkwood (1957), and Tanford and Roxby (1972), a large number of methods have been proposed to model the titration of acidic and basic residues in protein molecules. The majority of contemporary physical models combine statistical thermodynamics with continuum electrostatics (Bashford and Karplus 1990, 1991). Most of these methods are based on finite-differences techniques to solve the Poisson–Boltzmann equation (FDPB) (Yang et al. 1993; Antosiewicz et al. 1994; Alexov and Gunner 1997; Schaefer et al. 1997). Some FDPB-based models have been extended to protein-membrane systems by including an implicit membrane in the solvation models (Karshikoff et al. 1994; Engels et al. 1995). A detailed description of the physical formalism of the electrostatic approach (Bashford 2004) and reviews of existing methods can be found in the literature (Honig and Nicholls 1995; Juffer 1998; Simonson 2001; Fitch and García-Moreno 2006). However, despite the progress in theoretical understanding of the problem, accurate prediction of ionization properties of proteins remains a challenge in protein modeling (Simonson 2001). The main sources of errors are well known, including the imperfectness in experimental structures used as input data, the use of crystal structures as models of solvated proteins, and the simplification of nonuniform dielectric properties of protein interior. In addition, due to the inherent coupling between the binding of protons and conformational changes (Laskowski Jr. and Sheraga 1954), flexibility of protein structures creates a serious combinatorial problem if the combined states of protonation and conformation are treated explicitly (Spassov and Bashford 1999). The easiest way to reduce the combinatorial problem is to ignore the conformational flexibility completely as in most popular FDPB and semi-empirical models. In contrast, traditional methods of molecular mechanics and molecular dynamics are typically based on the exact opposite approximation, completely ignoring the possible changes of protonation states. However, ignoring protonation changes can be misleading in many interesting cases such as predicting the properties of active sites of enzyme molecules or inside the membrane proteins, because of large effects of desolvation of buried charged groups and stronger interactions inside the protein interior. All of the above has motivated the development of algorithms that couple the treatment of conformational flexibility with the exchange of protons between the acidic and basic groups and the solvent. Examples include mean-field models with side-chain flexibility (You and Bashford 1995; Spassov and Bashford 1999; Barth et al. 2007), Monte Carlo sampling (Beroza and Case 1996; Georgescu et al. 2002), and advanced protocols for molecular-dynamics simulations (for review, see Mongan and Case 2005). Recently an implementation of replica-exchange method in constant pH dynamics (Khandogin and Brooks III 2006) demonstrated an improvement of the accuracy of this class of models that moves the predictions from first principles to a quantitative level. At the same time semi-empirical methods remain popular (Li et al. 2005), filling the existing gap between computational efficiency and the accuracy of compute-intensive physical approaches.
The main goal of the present work was to develop a fast and efficient computational tool to calculate a general set of protein properties related to the protonation of acidic and basic amino acid residues that can be used in diverse applications of protein modeling. Our intention was to replace FDPB with a Generalized Born (GB) model (Still et al. 1990) and to combine it with an iterative mobile clustering (IMC) approach to the binding of protons to molecules with multiple sites of titration (Spassov and Bashford 1999). After implementing a novel algorithm for preliminary optimization of hydrogen coordinates and a revision of the conception of model compounds, even in the simplest variant, without an explicit treatment of the structural relaxation, the computational method achieved high accuracy of pK predictions that was comparable to, or even better than, state-of-the-art methods reported in the literature. The combination of the GB and IMC approach (GB/IMC) provides a new computational protocol for pK calculations and modeling of the complex effects of protein ionization. The program we describe in this work includes modules to calculate the fractional protonation of titration sites and integral titration curves, the electrostatic contribution to the protein free energy, as well as the approximate folding energy as a function of pH. An additional module provides the option to reconstruct and refine the coordinates of all hydrogen atoms at a given pH. The optimization of the hydrogen-bond network includes assignment of hydrogen atoms according to calculated ionization characteristics, as well as a search for optimal tautomeric forms of histidine residues and the flipping of amide groups of Asn, Gln, and protonated carboxyl groups of Asp and Glu residues.
The method has been implemented as the “Protein Ionization and pK prediction” computational protocol in Accelrys Discovery Studio 2.0. In this work we provide validation results on a set of 24 proteins as well as several examples of complex modeling of pH-dependent properties of protein molecules, including membrane proteins.
Theory
The underlying physical formalism of our method is close to the model of protein ionization proposed by Bashford and Karplus (1990, 1991). It combines statistical physics with continuum electrostatics. A similar approach is used in many FDPB models of protein ionization (Bashford and Gerwert 1992; Yang et al. 1993; Antosiewicz et al. 1994; Schaefer et al. 1997; Gunner and Alexov 2000; Nielsen and Vriend 2001; Warwicker 2004) including membrane proteins (Karshikoff et al. 1994; Engels et al. 1995). However, the method proposed here has several novel features and algorithms that contribute to improvement of the accuracy of its predictions. Recently Onufriev et al. (2000) demonstrated that a modification of GB approximation can effectively replace the finite-difference technique to solve the Poisson–Boltzmann equation. We have implemented a similar variant of the GB solvation model (Dominy and Brooks III 1999a) extended to membrane proteins (Spassov et al. 2002) using CHARMm (Brooks et al. 1983). The GB model has been preferred not only because of computational efficiency but, equally important, to avoid the limitations of molecular size and shape specific for grid methods such as FDPB. In addition, the replacement of FDPB with a GB model allows us to substitute monopeptide model compounds with a library of precalculated polypeptide structures. This enables us to take advantage of recently reported accurate pKa values estimated from potentiometric titration of natural acidic and basic residues in blocked alanine pentapeptides (Thurlkill et al. 2006) and to use them as a consistent set of model pK's. In the work of Nielsen and Vriend (2001) and Georgescu at al. (2002) it has been demonstrated that an optimization of the hydrogen-bond network in the input X-ray structures can improve the accuracy of pK predictions of FDPB-based calculations. In our method a novel protocol for preliminary optimization of hydrogen positions has also been implemented. Our algorithm adds a search for optimal proton binding centers to a CHARMm protocol to construct and optimize the hydrogen coordinates.
Basics
A detailed recent description of the continuum electrostatics approach to protein ionization can be found in Bashford (2004). Here we will repeat briefly some of the key points, to avoid any confusion about the exact equations used in our implementation of the method.
A protein is modeled as a molecule with N titration sites that can exchange protons with the solvent. The ionization state l of a system is described by a microstate vector X l. With each component of X l, x i, can take either the value 1 or 0 depending on whether site i is protonated or deprotonated. If the molecule is modeled as a single conformer, i.e., neglecting the coupling between the changes in the ionization state and the changes of protein structure, the probability to find the molecule in a microstate X l at given pH is
 |
where G(X l, pH) is the free energy of the microstate X l. Note that the summation is carried out over all possible N p = 2N microstates, which, even in a single conformer case, creates a serious combinatorial problem if the molecules had more than 15–20 titratable sites.
Almost all pH-dependent characteristics of a protein molecule, such as the titration of the individual ionogenic groups, the integral titration curves, or the pH-dependent contribution to the protein free energy, can be derived from the fractional protonation θi(pH) of individual sites of titration i:
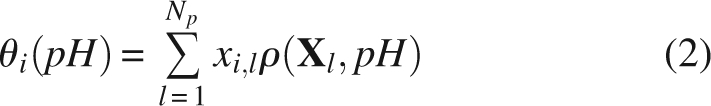 |
In the analysis below we will compare the experimental values of apparent pKa, of individual residues with computed pKhalf values. pKhalf is defined as the pH value at which the fractional protonation of a titration site is equal to 0.5. However, in cases of abnormal shape of titration curves, pKhalf must be used with caution and the pH dependence of fractional protonation should be regarded as the true ionization characteristic of protonation sites. It is convenient for the calculations if the microstate energy in Equation 1 is referenced to the completely deprotonated state (Bashford and Karplus 1991; Schaefer et al. 1997):
 |
where pKintr,iare the calculated intrinsic pK's of the titratable groups assuming that all other residues in the protein are in their neutral state, according to the definition proposed by Tanford and Roxby (1972). In models with distributed formal charges over the multiple atoms of titratable groups, the interaction terms Wij represent the additional energy to protonate site i if site j is protonated (Bashford and Gerwert 1992); qi are the corresponding formal charges equal to −1 for acidic and +1 for basic groups. In the case of flexible molecules Equations 1–3 can be extended by adding pH-independent terms, which depend on conformational state (Spassov and Bashford 1999).
Calculating the electrostatic energy terms
All interaction terms in Equation 3 are calculated using the GB continuum electrostatic model. The use of GB models rather than grid-based techniques to solve the Poisson–Boltzmann equation not only significantly reduces computation time, but also makes the computations less dependent on molecular shape and size. The two main approximations in pure electrostatic approaches to proton binding are first, the assumption that all chemical contributions to the binding energy cancel each other in the thermodynamic cycle used in the definition of intrinsic pK's (see Equation 5 below) and second, that all nonbonded interactions of nonelectrostatic nature are insignificant compared to electrostatic interactions. The latter is not strictly true for short-range, H-bond type interactions. However, it is still a reasonable assumption considering that, in molecular mechanics force fields, such as in CHARMm, the hydrogen-bond interaction energy is often modeled by the sum of a van der Waals and a Coulombic term only and that, at nonoverlapping distances, the van der Waals contribution is negligible compared to the electrostatic interaction term. Similar to most of continuum electrostatic models, electrostatic energy terms in GB models are calculated as a sum of Coulomb term and a Generalized Born solvation energy term (Bashford and Case 2000). For this study, we used the same variant of GB model as in Onufriev et al. (2000):
 |
where εm and εslv are the intramolecular dielectric constant and the dielectric constant of the solvent, respectively, and the αi are effective Born radii. In difference to many other GB implementations, Equation 4 is extended with a correction for the ionic strength (Hawkins at al. 1995); an unchanged value of κ = 0.7 is used in all calculations.
Introducing libraries of model compounds
In most FDPB electrostatic models (Bashford and Gerwert 1992; Yang et al. 1993; Antosiewicz et al. 1994; Schaefer et al. 1997) the pKint r values are derived from calculations on model compounds with known experimental pK data according to a thermodynamic cycle described in Bashford and Karplus (1990):
 |
where ΔΔG terms represent the noncovalent energy differences between the protonated (PH and MH) and deprotonated (P and M) forms of titratable atomic groups situated in the protein and model compound, respectively. Equation 5 is derived from a thermodynamic cycle applied to an “alchemic” process of transferring titratable groups to different structures and environments. The atomic groups of protonation sites are bonded to structures that are similar enough to cancel the “chemical” contributions to binding energy. However, the calculations of energy terms in Equation 5 depend not only on the chemical structure but also on the conformation of model compounds. The structures of model compounds is one of the key differences between our approach and other electrostatic models, where usually the model compound for each particular residue is represented by a monopetide compound with exactly the same atomic coordinates, as of the corresponding residue in the protein structure. In FDPB methods this is a necessary approximation to cancel the effects of numerical singularities due to the grid distribution of atomic charges. However, in the dense space of protein interior, it is not unusual for a titratable residue to obtain a conformation with intra-residue atomic contacts that are quite different from the contacts it would have in a relaxed structure in a water environment. Obviously, this difference is a potential source of error in predicted pK values and it introduces an inconsistency because of use of different model compound structures for residues of the same kind. One possible way to avoid such an inconsistency is to use reduced, nonpeptide reference compounds, for example, acetic acid instead of Glu or Asp monopeptides as in the MCCE method (Georgescu et al. 2002). However, in preliminary tests of the GB approach, our attempts to use the structure of acetic acid failed to reproduce the low pKhalf values of carboxyl group in nonblocked Asp and Glu monopeptides. In a GB-based method it is not needed to use the same conformation of titratable residues in the protein and in the model compound. Based on this, we changed the concept of model compounds: In our method, we calculate the ΔΔG(MH, M) term in Equation 5 for a single, relaxed peptide structure, assumed as representative conformation of the model compound in solvent. In general, the monopeptide models used to calculate reference energies in Equation 5 can be replaced with any structure fulfilling the requirement of chemical similarity, for example, short polypeptides with known titration data. For the present study we took advantage of recently reported accurate NMR data for all natural acidic and basic amino acid residues in blocked pentapeptides of Ala-Ala-X-Ala-Ala type (Thurlkill et al. 2006). For each supported force field we constructed a library of low energy pentapeptide structures in β-strand conformation with the side chain optimized by sampling the side-chain dihedral angles and minimizing the CHARMm energy. The pKmod values of pentapeptide model compounds are taken the same as determined by Thurlkill et al. (2006): 3.67 for X = Asp, 3.67 (a-carboxyl), 4.25 (Glu), 6.54 (His), 8.00 (a-amino), 8.55 (Cys), 9.84 (Tyr), and 10.40 (Lys), and a value of 12.0 is used for arginine.
A more realistic approach would be to construct representative ensembles of model compound conformations to derive the model compound free energy terms. However, during validation tests we achieved sufficient accuracy with the single conformation models and therefore we assumed them an appropriate approximation at the current stage of development.
Electrostatic free energy and pH stability
If the fractional protonation of the titration sites is known, an integration over the binding isotherms (Schellman 1975; Yang and Honig 1994) makes it possible to calculate the pH dependence of electrostatic free energy differences between any two conformers in a convenient way. This approach can be used not only for comparison of different conformational states, but also for comparing different binding states, such as models with bound or unbound ligand. It can also be used in modeling of ensembles of conformers, weighted by the calculated absolute free energy (Spassov and Bashford 1999). For this purpose, the computational method reports the pH-dependent electrostatic contribution to absolute free energy calculated according to a convenient definition (Schaefer et al. 1997):
 |
where G(∞) is the electrostatic energy of the completely deprotonated state. The deprotonated state is constructed by removing the release hydrogen from all titratable groups. Q(pH) can be either the total charge or the average number of bound protons calculated from the fractional protonations of individual sites. In the calculations the infinite pH value at the upper limit in the integral in Equation 6 is substituted with a value, pHinf, estimated to be large enough to exclude the possibility of any numerically significant titration beyond that point. pHinf is derived from the maximum possible contributions of charged groups to the pK shifts.
To provide a rough, but instant estimation of pH stability, the protocol reports the pH-dependent relative folding energy. The reference state used is the simplest, “null” model of unfolded state (Yang et al. 1993; Antosiewicz et al. 1994; Schaefer et al. 1997), assuming all groups in the unfolded state to titrate according to pKa values adopted from the pentapeptide model compounds (Thurlkill et al. 2006) described above. Interestingly, the “pentapeptide” pK values of the acidic residues Asp (pKa = 3.67) and Glu (pKa = 4.25) are very close to the optimal pKa values (Tan et al. 1995) estimated for a protein in the denatured state (3.6 and 4.0 for Asp and Glu, respectively). It implies that the pentapeptide pKa values are better approximations to the unfolded state than the values of the model compounds of 3.9 and 4.3 used in Tan et al. (1995) and other papers.
IMC approach to protein ionization
It has been shown that the use of the iterative mobile clustering as in the IMC approach (Spassov and Bashford 1999) can be an effective way to treat the combinatorial problem arising from the exponential growth of protonation states with the number of titratable groups, not only in the single conformer cases, but also in systems with combined local and global conformational flexibility. The comparison with exact Boltzmann statistic calculations have also shown that IMC is not only much faster, but also more reliable on tested cases than a Monte Carlo sampling (Spassov and Bashford 1999). Therefore, we implemented the IMC algorithm to calculate the titration of acidic and basic groups. The computational program is capable of modeling not only rigid structures but also ensembles of distinct conformations in a way shown before (Spassov and Bashford 1999; Spassov et al. 2001). However, in the current study we report results using only the fast, single-conformer mode of the method.
Computational protocol
The equilibrium of proton binding can be very sensitive to small changes in the structure and model parameters. To minimize noise from possible variations of molecular models, we carried out all calculations with all heavy atoms fixed to the coordinates of the experimental structures and with standard force field values for all parameters, including the atomic radii and charges, with the exception of partial charges of neutral histidine, as explained below. The only physical parameter subject to parameterization in the present work is the internal dielectric constant εm as described in the Results and Discussion section. In principle, our approach allows the use of any CHARMm force field. At the current stage, we have validated the method for the CHARMm and CHARMm polar hydrogen force fields (Momany and Rone 1992).
The present method includes four main steps implemented as separate software components.
Step 1: Preliminary optimization and calculation of the effective Born radii
The first step is implemented as a program module written in CHARMm scripting language and includes a reconstruction and preliminary refinement of hydrogen atom positions followed by calculation of the atomic Born radii. The novel feature is the inclusion of a procedure for determination of the optimal proton binding centers on the carboxyl groups of Asp and Glu residues and C terminus.
The coordinates of hydrogen atoms are not present in most PDB files and, even if present, it is difficult to judge whether the data are objective, rather than the result of modeling. To avoid any possible dependency on the uncertainty in experimental hydrogen positions, initially all existing hydrogen atoms are stripped from the structure. Hydrogens are then reconstructed using the CHARMm HBUILD routine (Brünger and Karplus 1988) and energy optimized at fixed positions of all heavy atoms. In all steps of the algorithm the hydrogen building and optimization are carried out using the standard energy function of the selected force field. The next step invokes a search for optimal proton binding center between the two oxygen atoms of titratable groups of any Asp and Glu residue or C terminus. For this purpose the potential at each atom of interest is calculated and the proton binding center is assigned to the atom embedded in the more negative electrostatic potential. In the following step, all missing hydrogens are constructed for all sites of titration, using the HBUILD routine. Finally, the GBIM CHARMm module (Spassov et al. 2002) is invoked to calculate the effective Born radii. GBIM is an extension of GBorn CHARMm module (Dominy and Brooks III 1999a) including an implicit membrane in the GB solvation model.
To avoid complications due to histidine tautomerism, we considered a simplified model of the neutral deprotonated state of the His side chain, assuming equal partial charges for both proton-binding nitrogens. The inspection of results from validation tests showed that in most cases the approximation of “smeared charge” works reasonably well. However, we believe there is room for improvement, especially to better reproduce cases with very large experimental pK shifts. This work is in progress.
Step 2: Calculating intrinsic pK and interaction energy terms
The Born radii calculated in step 1, along with the atomic coordinates of the molecule and model compounds, are used to evaluate the GB terms in Equation 4. This step is encoded as a separate program, GBPK, which computes all energy terms necessary to evaluate the intrinsic pKint r values and interaction terms included in the microstate energy described by Equation 3. The components of the interaction matrix Wij in Equation 3 are calculated as described by Bashford and Gerwert (1992). In the calculations of pKint r, GBPK uses coordinates of model compounds taken from structural libraries, as already described. The libraries are pre-generated separately for the CHARMm and CHARMm polar hydrogens (Momany and Rone 1992) force fields. The input data to GBPK include also the values of the intramolecular dielectric constant and the ionic strength of the solvent.
Step 3: Calculating the protein ionization
This step includes calculations of the fractional protonation of all sites of titration according to Equations 1–3. It also includes the evaluation of the total charge and the electrostatic contribution to the absolute free energy according to Equation 5 as well as the approximate estimation of the relative folding energy with respect to the “null” model of unfolded state as described above. The calculations are carried out by a program, PHPK, based on the IMC approach.
Step 4: Optimization of hydrogen-bond network
In addition to program modules to calculate the protein ionization, we developed a computational module for the assignment of hydrogen coordinates at a given pH and structural optimization of the entire hydrogen-bond network. Similar to step 1, the optimization algorithm is written as a CHARMm script.
The pKhalf values computed in previous steps are used to define the protonation state of the titratable groups. If the selected pH value is less than the pKhalf for an acidic or basic group, i.e., if the fractional protonation is >0.5, the structure of the group is generated in the protonated state and vice versa. The coordinates of all missing hydrogens are constructed by the CHARMm HBUILD routine. The next step includes a cycle over all sites of titration to optimize the geometry of groups with ambiguous binding center of H atom, such as OD1 or OD2 atoms of aspartate carboxyl groups, OE1 or OE2 of glutamate, C-terminal carboxyl groups or tautomeric forms of deprotonated histidines. The cycle also includes optimization of the ambiguous conformation of side-chain amide groups of Asn and Gln residues. The determination of the histidine tautomeric form is based on the comparison of the computed electrostatic potential at ND1 and NE2 atoms. The proton is assigned to the site with the larger negative potential. Two conformations of each protonated Asp, Glu, and C-terminal carboxyl groups, as well as each Asn and Gln are tested based on 180° rotations of side-chain carboxyl or amide groups, while the rest of the protein atoms are fixed and the low energy conformation is chosen. Note that the described flipping is the only case in all of the computations, which include manipulation of some heavy atoms. At the end of the cycle, the coordinates of all hydrogen atoms are re-optimized by energy minimization.
It has been shown recently that the interactions of side chains with backbone atoms are on average stronger than side-chain-to-side-chain interactions, with a very strong trend for polar Asp, Glu, Asn, Gln, Ser, and Thr residues (Spassov et al. 2007). Assuming the hypothesis of a dominant role of backbone atoms in determining the orientation of side chains of polar residues, we constructed our algorithm without sampling of mutual orientations of protein side chains avoiding a possibly substantial increase in computation time. We validated this approach in tests on several neutron-diffraction structures as discussed in the Results and Discussion section.
Results and Discussion
Lysozyme ionization and parameterization of the method
As discussed above, the only adjustable parameter in this study is the value of the molecular dielectric constant εm in Equation 4. The preliminary parameterization of εm has been carried out on a single structure—the structure of hen-egg white lysozyme (HEWL, PDB code 2lzt), a structure widely used to test the predictive capabilities of computational approaches to protein ionization (Tanford and Roxby 1972; Imoto 1987; Spassov et al. 1989; Bashford and Karplus 1990; Nielsen and McCammon 2003; Mongan et al. 2004; and others). We mainly selected hen-egg lysozyme because of the complete list of existing experimental titration data for all residues of this molecule, excluding arginines. Also, the prediction of lysozyme ionization is challenging because of a number of residues with unusually high or low experimental pKa values.
Figure 1 shows the accuracy of pK calculations as a function of εm. The model achieves the highest prediction accuracy between εm = 10 and 11 with an RMSD value <0.5 compared to recent results reported in the literature (Table 3, see below). The sharp decrease in accuracy at low εm is in agreement with many authors suggesting the use of high values of average dielectric permittivity when the flexibility of permanent dipoles and the mobility of charged groups are not modeled explicitly (Antosiewicz et al. 1994; Pitera et al. 2001; Simonson 2001). Interestingly, the optimal εm value, according to our GB model, is half the value εm = 20 obtained from FDPB calculations (Antosiewicz et al. 1994).
Figure 1.

Average RMSD error of the predicted pK values of the acidic and basic residues as function of the value of internal dielectric constant. Experimental data from Kuramitsu and Hamaguchi (1980) and Bartik et al. (1994). The data are obtained on the example of the 2lzt PDB structure of HEWL.
Table 3.
Comparison of the accuracy of pK predictions with other methods

The preliminary tests on other structures showed a similar dependence on εm, and no systematic difference in accuracy has been observed between using εm = 10 or 11. We therefore used εm = 11 in all calculations in this study, with the exclusion of membrane proteins.
Table 1 shows the comparison between the experimental pKa values and the calculated pKhalf values for all titratable groups in a pH interval from 2 to 12. While both force fields, CHARMm and CHARMm polar H, lead to a correct estimation of the direction of the pK shifts for almost all residues, the all-hydrogen CHARMm models more accurately reproduced the unusually high experimental pK of Glu35 from the active site of the enzyme (pKa = 6.2) (Bartik et al. 1994). On the other hand, the data show a better overall RMSD value for the CHARMm polar H than the CHARMm model. Note that most continuum electrostatic models, if successful in prediction of pKa of buried groups such as Glu35, usually fail to predict the titration of surface groups, or vice versa (Bashford 2004). However, the difference between the two force fields is not drastic and in both cases the direction of pK shifts is predicted correctly for most of the residues. We were unable to find any reported results with better overall RMSD value than the RMSD of the calculations with CHARMm polar hydrogen force field in Table 1.
Table 1.
Comparison of the computed pKhalf values with the experimental pKa values of the acidic and basic groups of HEWL

The tests show that, without the initial optimization of hydrogen positions and using the standard set of pKmod, instead of pentapeptide values, the calculated RMSD error in 2lzt case increases from 0.45 to 0.85. The increased accuracy of 0.27 pK units are from hydrogen optimization and the additional 0.13 pK units are due to the use of pentapeptide pKmod values.
In addition to pK predictions we used the same lysozyme structure to test the ability of the method to predict some integral titration characteristics such as pH dependence of total charge and isoelectric point. Figure 2 shows the comparison between the calculated titration curves and the results of potentiometric titration at two different ionic strengths (Tanford and Roxby 1972). It is seen that at I = 0.1 M the calculated ionization curve is very close to the experimental data with some disagreement at low pH values. Although at I = 1.0 M the “low pH” disagreement increases, it is important that the calculated and the experimental curves show approximately the same pH dependence of the differences between the total charges at low and high ionic strength, suggesting that including the ionic strength dependence in Equation 4 is an appropriate approach. The calculations also give a perfect estimation of the lysozyme isoelectric point, pI = 10.21 versus the experimental value of 10.2 (Tanford and Roxby 1972).
Figure 2.

The pH dependence of lysozyme total charge at different ionic strengths. Open circles and solid line: experiment and computed values at 0.1 M ionic strength; filled circles and broken line: the corresponding curves at 1.0 M ionic strength.
Comparison of calculated pKhalf values with experimental data for different proteins
To investigate the reliability of the method we tested it on a relatively large set of different protein structures. The set contains 24 crystallographic and NMR structures and a total number of 331 titratable groups with known experimental pKa values. For each PDB entry in Table 2, the RMSD between calculated and experimental pK's of titratable groups is shown. Note that the calculations with both CHARMm force fields systematically show a high accuracy of the predictions with an RMSD of ∼0.5 pK units, and only for a small number of outliners (1trs, 1rgg, and 1xnb) the RMSD is elevated, but still below one pK unit. In a recent comparative study by Davies et al. (2006) an empirical method, PROPKA (Li et al. 2005), appears to be more accurate than some of the well known programs based on a continuum electrostatic model—MEAD (Bashford 1997), UHBD (Madura et al. 1995), and MCCE (Alexov and Gunner 1997). Contrary to the findings of the authors implying superiority of the empirical approach, the results in Tables 2 and 3 demonstrate that it is possible to achieve a considerably better accuracy, even better than those reported by the authors of PROPKA (RMSD 0.8–0.9), using a continuum electrostatic model of the same class as cited by the FDPB programs. Of equal importance is the fact that the use of the GB continuum model significantly reduces computational cost, compared to FDPB methods. According to the results of performance tests (Fig. 2) the CPU time used for a middle-size protein usually is about one minute, which is comparable to timings of the fastest empirical methods. Figure 3 also shows that the CPU timescales in an almost linear fashion with the size of proteins. The efficient scale-up is mostly because of the great reduction of sampling in the IMC algorithm, where only the number of mobile clusters increases with protein size, but the number of sites inside the clusters depending on an energy cutoff parameter stays small and not affected by protein size. In all studied cases a cutoff value of 0.5 kcal/mol was enough to achieve the prediction accuracy and to keep the maximum cluster size below 10–15 sites.
Table 2.
The RMSD between calculated and experimental pKa values obtained on a test set of 24 protein structures

Figure 3.

The CPU time used to calculate the ionization of proteins with different chain lengths. The data were generated on an Intel Pentium4 3.0 GHz machine.
A smaller set of seven protein structures with well-characterized dissociation characteristics is widely used for test purposes by many authors. We used the same structures to compare the RMS error of GB/IMC calculations to several of the most accurate methods based on either physical or empirical approaches (Table 3). Note that GB/IMC not only shows better accuracy, as seen in Table 3, but also does that at very low computational cost, if compared to more sophisticated physical methods such as constant pH dynamics or MCCE. The accuracy is even better than the accuracy of the empirical SCP approach, in which results are achieved after intensive fitting of multiple parameters.
Modeling the pH stability
Some examples of the computed pH-dependent contribution to the folding energy comparing to the data from unfolding experiments in urea are given in Figure 4. It is interesting to note that, although the pH dependence of relative folding energy is calculated using the simplest “null” model of unfolded state (Schaefer et al. 1997), i.e., ignoring the possible effects of any resting electrostatic interactions in unfolded state, in most cases the results show a reasonable estimation of the pH stability which is comparable to the results obtained from more realistic models of the electrostatic interactions in the unfolded state, such as the native-like structures modeled by Elcock (1999). In the Rnase T1 example shown in Figure 4, the method is unable to predict sufficiently well the shape of the denaturation curve but gives a close estimation of the pH optimum.
Figure 4.

The comparison of calculated free energy of unfolding compared to the data from urea denaturation. (A) Hen-egg lysozyme; (B) Rnase T1; (C) Rnase A. Experimental data from Pfeil and Privalov (1976) and Pace et al. (1990).
The implementation also allows the curves of pH stability to be obtained using other simple models, for example calculating the difference between the absolute electrostatic energies of native structure and a relaxed structure in β-strand conformation (Schaefer et al. 1997). Here we do not provide such data because during the tests the null model systematically showed a better overall fit to experimental curves. However, at extreme pH, an extended conformation could be a reasonable model because the noncompensated repulsion between the groups of the same charge will be reduced.
Membrane proteins: bacteriorhodopsin ionization
The modeling of ionization characteristics of membrane proteins is important for the understanding of molecular mechanisms, and many of them are of high interest for pharmacology and bioenergetics, such as the regulation of ion channels, the activation of membrane receptors or utilization of light energy. Several studies demonstrate that membrane environment can be successfully approximated by including an implicit low dielectric slab in FDPB ionization models (Karshikoff et al. 1994; Engels et al. 1995) However, despite the availability of several GB solvation models with implicit membrane (Spassov et al. 2002; Im et al. 2003; Tanizaki and Feig 2006), none of the models, to our knowledge, have been used in pK calculations. To test the possibility of replacing grid-based FD calculations with the fast GB code, we used the GBIM CHARMm module to include an implicit membrane and to repeat some of the calculations reported in the MEAD-based study of ionization of bacteriorhodopsin (BR) (Spassov et al. 2001). For the sake of comparison we used the same protein model, based on the 1c3w X-ray structure of ground state. Here we compare the results from the simplest variant of the BR ground state, without including O2H5 ions or larger water clusters as possible sites of proton storage as in the more realistic model proposed by the authors. A more detailed modeling of bacteriorhodopsin ionization and the complex mechanism of proton transfer was beyond the goals of this study and can be found elsewhere (Engels et al. 1995; Sampogna and Honig 1996; Spassov et al. 2001; Song et al. 2003; Ferreira and Bashford 2006). A common problem for all “two dielectric” models with implicit membrane is the choice of the molecular dielectric constant, because of the requirement for this value to be the same in both membrane and protein interior. A low value of εm = 2–4 is suitable for a dielectric slab formed by lipid tails, but inconsistent with the optimal value of 10–11 obtained above on a set of 24 water-soluble proteins. Because a significant fraction of membrane proteins is exposed to the solvent, a compromised value is needed to represent the dielectric properties of both transmembrane region and the molecular interior outside the membrane. We found that a value of εm ∼28–10 reproduces the balance of electrostatic interactions in BR molecule well enough, based on the comparison of calculated pKhalf of the residues in the transmembrane region with the experimental data shown in Table 4. It is seen that GB/IMC results are no less accurate than the results obtained in the previous FDPB calculation on the same model. Note that removing the membrane dramatically changes the proton affinities of some functionally important residues, such as Asp85 and Asp212, as well as the titration behavior of the proton release dyad Glu194–Glu204. We consider the results in Table 4 to be quite encouraging and believe this extension of the GB/IMC method to membrane proteins can be very helpful in modeling the membrane proteins, despite some issues regarding the uniform dielectric environment in the GB implicit solvent model.
Table 4.
Comparison of calculated and experimental pKhalf values of the titratable residues from the Bacteriorhodopsin interior

HIV-1 protease
In an attempt to demonstrate the complete functionality of the method, the GB/IMC implementation was tested on HIV-1 protease using several different experimental structures. This enzyme was selected, not so much because of its biomedical importance, but because numerous titration experiments of different kinds are reported in the literature (Trylska et al. 1999). The calculations were carried out using three X-ray structures, one for apo-enzyme and two complexes with different inhibitors, DMP-232 and KNI-272 (Table 5). KNI-272 is modeled as an ionizable group—it titrates in water with a pKa of 4.9 (Wang et al. 1996).
Table 5.
Computed pKhalf of the acidic residues of HIV-1 protease compared with experimental data for apo-enzyme and two inhibitor complexes

From Table 5, the data show very good agreement between experimental and calculated pKhalf values for apo-enzyme and the complex with the symmetric DMP-232 inhibitor. The single, but important disagreement is the KNI-272 case where our calculations do not predict a low value of the first pKhalf of catalytic dyad formed by Asp25 residues from A and B subunits as suggested by Wang et al. (1996). To study the problem further, we used several models, including models with conformational flexibility and incorporated water molecules, but we have been unable to find any structural or other reasons for the low pK of Asp 25.
In Figure 5 the stability curves computed by using two, “null” and “beta,” reference models of unfolded state (see Materials and Methods) are compared to the experimental data from unfolding in urea.
Figure 5.

The pH-dependent contribution to unfolding energy (kcal/mol) of HIV-1 protease compared to the data from unfolding in urea (Todd et al. 1998). Solid line: data obtained using the null model; broken line: using β model of unfolded state.
In contrast to the FDPB methods, using the GB models makes it technically possible to model the energy of binding between two molecular units in a very easy manner. This can be done simply by calculating the difference of the absolute electrostatic contribution to the free energy, ΔΔG bind, between (Equation 5) the native structure and a structure with the units separated on long distance, say 200 Å. Figure 6 shows the calculated pH dependence of negative energy of binding (−ΔΔG bind) of KNI-272 inhibitor to HIV-1 protease, compared to the corresponding energy curve derived from the experimental pH-dependent association constant. It is seen that the calculation reproduces remarkably well the pH optimum of binding as well as the association of the inhibitor in the low pH region. The observed disagreement at elevated pH could be due to the fact that possible changes in oligomeric state with pH are not taken into account.
Figure 6.

The pH-dependent contribution to the binding energy of KNI-272 inhibitor to HIV-1 protease, compared to the experimental values of association constant K a taken from Velazquez-Campoy et al. (2000). The solid line represents the computed values of −ΔΔG bind. The triangles correspond to the values of 2.303RTlogK a.
Figure 7 shows the hydrogen bonds between residues from the active site and the inhibitor obtained after the final step of the computations on DMP-323 complex. It is notable that the predicted configuration of hydrogen bonds is exactly the same as suggested earlier from an analysis of the experimental data (Yamazaki et al. 1994).
Figure 7.

The predicted hydrogen bonds in the complex of HIV-1 protease with DMP-232 inhibitor.
Structural optimization of hydrogen-bond networks
Most of the protein structures in the PDB are solved without enough resolution to determine the position of the hydrogen atoms. Only a small number of neutron-diffraction and high-resolution X-ray structures contain hydrogen coordinates obtained from the diffraction maps. Because the positions of hydrogen atoms, especially polar hydrogens, can be of significant importance for many atomistic models, numerous algorithms have been developed to construct proton positions from the coordinates of heavy atoms. One of the most widely used tools for building and refinement of a hydrogen-bond network is the CHARMm HBUILD routine (Brünger and Karplus 1988). Recently, several new methods have addressed hydrogen-bond optimization by incorporating some more structural features such as the tautomeric forms of deprotonated histidines or the ambiguous orientation of Asn and Gln side-chain amide groups (Hooft et al. 1996; Word et al. 1999). Two recent methods of this class also include pH dependency in the optimization algorithm (Labute 2007; Li et al. 2007). Interestingly, both of the new methods use a search for a microstate with minimal energy for assignment of protons to titratable groups. In principle, the microstate approach can be a reasonable approximation when the contribution of a single state to the partition sum in Equation 1 is strongly dominant. However, in many interesting cases the use of optimal microstate energy carries the risk of producing misleading results, because in certain conditions, the molecular system can occupy the state with minimal energy with a low probability. Also, a microstate of protonation cannot be observed experimentally, while the calculated fractional protonations are the quantities that correspond directly to both experimental titration data and to the existence of the protons in neutron-diffraction structures of titratable residues. In the present work we used a completely different approach. Instead of the “optimal” microstate, the hydrogen assignment is based on the average fractional protonation of the residues obtained in the calculations of ionization equilibrium, as is described in the Computational protocol/step 4 section.
Table 6 summarizes the results of the refinement of the network of hydrogen interactions obtained on a set of five neutron-diffraction structures crystallized at different pH. In the comparison of the hydrogen structure of individual residues we adopted a criterion used recently for similar purposes (Labute 2007): A prediction is assumed to be correct if the ionization states are the same, the tautomeric form or the orientation of flipping groups is the same, and the hydrogen atoms are rotated by no more than 60°. In rare cases, some groups of hydrogen atoms are completely missing in crystallographic coordinate lists, such as all protons from the lysine NH3 group. In these cases we assume the prediction as correct, because it does not contradict with an experimental estimation.
Table 6.
Comparison between the predicted hydrogen position and the neutron-diffraction structures

While comparison to neutron-diffraction structures is the appropriate method to validate the positions of hydrogen atoms (Brünger and Karplus 1988), these structures have not been used in recent studies, and high resolution X-ray structures (Labute 2007) or quite indirect testing (Li et al. 2007) are used instead. In a previous study on the same subject (Hooft et al. 1996) the authors discard the neutron-diffraction structures as inappropriate for validation of the optimized hydrogen positions, suggesting experimental problems as a reason for too many cases of poor predictions. In contrast, our results demonstrate a good agreement between the predicted and neutron-diffraction position not only for hydrogen atom positions but also for the calculated and observed protonation states of titratable residues as can be seen in Tables 6 and 7. A closer inspection of structures shows an extremely good fit of nonpolar hydrogen atoms with very rare cases of differences >0.2–0.3 Å. Those differences are observed in the hydrogen positions of some methyl groups rotated occasionally by ∼50°–60°, such as in Leu49 and Leu89 in 2l2k and Val196 in 2gve structures. Note, however, that the rotations of the symmetric CH3 or NH3 groups are usually insignificant from the modeling point of view. As could be expected, the positions of polar atoms show more differences, especially on the surface, but even in these cases the agreement to neutron-diffraction structures is quite good. The inspection of the 2gve structure of xylose isomerase, the largest of the studied proteins, shows that, out of 114 groups, the position of the polar protons are predicted correctly for 93 (82%), with differences coming mostly from the Ser and Thr residues situated on the surface of the protein.
Table 7.
Comparison between calculated and experimental protonation states in neutron-diffraction structures

In Table 7 the comparison between the calculated protonation states and the protonation states determined from the neutron-diffraction structures can be seen. The data provide two possible ways to make the comparison—from a strict prediction based on the calculated pKhalf values, and from an analysis based on the calculated fractional protonation corresponding to the experimental pH value. An experimental state is defined as protonated if the corresponding hydrogen atom is present in the PDB coordinate list, and vice versa. For each entry only the nontrivial cases are listed, discarding the residues that are unlikely to titrate because of large differences between experimental pH and model pK's.
When using neutron-diffraction structures for comparison, it must be noted that, for a proton to be observed, it is not absolutely necessary that the occupancy be more than one-half. If we assume that states with reasonable fractional protonation, say no less than 0.25, can be seen on neutron-diffraction maps, then the predicted states of 31 from 36 residues, or 86%, can be assumed as consistent with neutron-diffraction structure. Even using the stricter criterion of 50% occupancy, a high percentage (75%) of the states is predicted correctly. It is notable that, in lysozyme, not only is the protonation state of Glu35 from active site predicted correctly, but the hydrogen, HE2, is bound to the same oxygen atom as the deuterium atom, DE2 in the experimental structures (Fig. 8), and has a similar orientation. The only difference observed in the 1lzn structure is in the protonation of Glu7. The observed protonated state in 1lzn structure does not agree with the predicted state, and also with the low pKa of 2.9 observed in NMR experiments in water (see Table 2). It is tempting to speculate that the difference is related to the effect of the crystal field, but we will leave such a hypothesis for further investigation.
Figure 8.

The differences between predicted and neutron-diffraction hydrogen positions in the active site of HEWL (1lzn). In boldface, the structure of Glu35. In light-blue are the hydrogen and deuterium atoms in neutron-diffraction structure.
The most striking result of hydrogen optimization is the 100% prediction of the tautomeric states of all 10 deprotonated forms of histidine residues in the test set as shown in Table 7. It is not trivial to predict the only one from 210 or 1024 possible combinations. We regard this result as valuable, because the right histidine tautomers can be critical for important implementations of method such as ligand docking. Figure 9 illustrates a prediction of histidine protonation on the example of myoglobine, 1l2k structure.
Figure 9.

The protonation and tautomeric states at pH 6.8 of the histidine residues in 1l2k structure of met-myoglobin. (A) Predicted. (B) Neutron-diffraction structure.
Conclusion
The results of several studies show that, without an explicit treatment of conformational flexibility, the traditional methods based on continuum electrostatic models have accuracy problems in the prediction of the dissociation characteristics of titratable residues in proteins (Simonson 2001; Bashford 2004; Khandogin and Brooks III 2006). The existing gap between the accuracy and computational efficiency of direct physical approaches motivates the development of empirical methods (Li et al. 2005) recently suggested as more accurate (Davies et al. 2006) than most well known FDPB programs. In contrast, the results of this study demonstrate that it is possible to create an accurate and computationally effective approach to protein ionization based entirely on the traditional continuum electrostatic model. The proposed GB/IMC approach systematically yields results that appear to be superior not only to the fast empirical methods, but also to the results of all other methods we found in literature, some of them based on much more detailed physical models. Several factors incrementally contributed to the improvement of the method, such as the replacement of monopeptide model compounds with structural libraries of polypeptides and the novel algorithm for preliminary optimization of the hydrogen structure. Among the other factors contributing to high accuracy, we like to mention the efficiency of the IMC approach, as well as the use of CHARMm GB models with improved accuracy (Dominy and Brooks III 1999a,b), and the well-balanced CHARMm (Momany and Rone 1992) force field atomic parameters. The implementation of GBIM (Spassov et al. 2002) CHARMm module in the calculations of the effective Born radii makes GB/IMC, to our knowledge, the first GB-based approach to protein ionization that is applicable to membrane proteins.
The proposed algorithm for structural optimization of hydrogen-bond networks also shows a reasonable agreement with the crystallographic protonation states and position of hydrogen atoms in neutron-diffraction structures. It is notable that the high percentage of correct predictions is achieved using input structures that are completely stripped of all hydrogen atoms. We believe this method will be valuable for future implementation in molecular dynamics protocols, ligand docking, and protein docking algorithms, as well as in hybrid protocols including molecular dynamics or quantum mechanics calculations.
The integration of the GB/IMC program modules within Discovery Studio provides a convenient graphical tool that saves a lot of effort usually needed in the preparation of input data. The Protein Ionization and pK prediction protocol is already in the hands of many investigators and the authors are hopeful that it will be useful in many areas of protein modeling.
Materials and Methods
Data sets
A set of 24 proteins has been constructed for the purpose of accuracy tests of pK predictions (Table 2). The entries include X-ray structures representing proteins with pKa values of a relatively large number of residues determined by NMR experiments. The set contains the proteins from the collection of Forsyth et al. (2002) extended with some proteins taken from MCCE and PROPKA studies (Georgescu et al. 2002; Li et al. 2005). A small number of PDB entries (1hic, 1mhi, 1a91, and 2ci2) are discarded because of an incomplete list of atomic coordinates or too many residues with ambiguous titration. Most of references to the sources of experimental pK values can be found in the literature (Forsyth et al. 2002; Georgescu et al. 2002; Li et al. 2005). Additionally, the experimental pK list is updated with data for more residues, found after a search in pKa database PPD (Toseland et al. 2006).
A second set of five neutron-diffraction structures has been selected for testing the optimization of the hydrogen position. The selection (see Table 6) includes representatives of all neutron-diffraction structures deposited in PDB with resolution <2.0 Å and with reported pH of the solvent during the crystallization.
Acknowledgments
We thank Drs. Paul Flook, Dipesh Risal, Marc Fasnacht, Yi-Shiou Chen, and Eric Yan for their help and useful discussions.
Footnotes
Reprint requests to: Velin Z. Spassov, Accelrys Inc., 10188 Telesis Court, Suite 100, San Diego, CA 92121, USA; e-mail: vss@accelrys.com; fax: (858) 799-5100.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.036335.108.
References
- Alexov, E.G., Gunner, M.R. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys. J. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antosiewicz, J., McCammon, J.A., Gilson, M.K. Prediction of pH-dependent properties of proteins. J. Mol. Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- Barth, P., Alber, T., Harbury, P.B. Accurate, conformation-dependent predictions of solvent effects on protein ionization constants. Proc. Natl. Acad. Sci. 2007;104:4898–4903. doi: 10.1073/pnas.0700188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartik, K., Redfield, C., Dobson, C.M. Measurement of the individual pKa values of acidic residues of hen and turkey lysozymes by two-dimensional 1H NMR. Biophys. J. 1994;66:1180–1184. doi: 10.1016/S0006-3495(94)80900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashford, D. An object-oriented programming suite for electrostatic effects in biological molecules. In: Ishikawa Y., et al., editors. Lecture notes in computer science. Vol. 1343. Springer; Berlin: 1997. pp. 233–240. [Google Scholar]
- Bashford, D. Macroscopic electrostatic models for protonation states in proteins. Front. Biosci. 2004;9:1082–1099. doi: 10.2741/1187. [DOI] [PubMed] [Google Scholar]
- Bashford, D., Case, D. Generalized Born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- Bashford, D., Gerwert, K. Electrostatic calculations of the pK values of ionizable groups in Bacteriorhodopsin. J. Mol. Biol. 1992;224:473–486. doi: 10.1016/0022-2836(92)91009-e. [DOI] [PubMed] [Google Scholar]
- Bashford, D., Karplus, M. pKa's of ionizable groups in proteins: Atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- Bashford, D., Karplus, M. Multiple-site titration curves of proteins: An analysis of exact and approximate methods for their calculations. J. Phys. Chem. 1991;95:9556–9561. [Google Scholar]
- Beroza, P., Case, D.A. Including side chain flexibility in continuum electrostatic calculations of protein titration. J. Phys. Chem. 1996;100:20156–20163. [Google Scholar]
- Brooks, B., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- Brünger, A.T., Karplus, M. Polar hydrogen positions in proteins: Empirical energy placement and neutron diffraction comparison. Proteins. 1988;4:148–156. doi: 10.1002/prot.340040208. [DOI] [PubMed] [Google Scholar]
- Davies, M.N., Toseland, C.P., Moss, D.S., Flower, D.R. Benchmarking pKa prediction. BMC Biochem. 2006;7:18. doi: 10.1186/1471-2091-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy, B.N., Brooks C.L., III Development of a Generalized Born model parametrization for proteins and nucleic acids. J. Phys. Chem. B. 1999a;103:3765–3773. [Google Scholar]
- Dominy, B.N., Brooks C.L., III . Parameterization of a Generalized Born model for the MSI CHARMm Momany and Rone force field. Accelrys Document; San Diego, CA: 1999b. [Google Scholar]
- Elcock, A.H. Realistic modeling of the denatured states of proteins allows accurate calculations of the pH dependence of protein stability. J. Mol. Biol. 1999;294:1051–1062. doi: 10.1006/jmbi.1999.3305. [DOI] [PubMed] [Google Scholar]
- Engels, M., Gerwert, K., Bashford, D. Computational studies of the early intermediates of the bacteriorhodopsin photocycle. Biophys. Chem. 1995;56:95–104. doi: 10.1016/0301-4622(95)00020-x. [DOI] [PubMed] [Google Scholar]
- Ferreira, A.M., Bashford, D. Model for proton transport coupled to protein conformational change: Application to proton pumping in the bacteriorhodopsin photocycle. J. Am. Chem. Soc. 2006;128:16778–16790. doi: 10.1021/ja060742d. [DOI] [PubMed] [Google Scholar]
- Fitch, C.A., García-Moreno, E. B. Structure-based pKa calculations using continuum electrostatics methods. Curr. Protoc. Bioinformatics. 2006;8:8.11. doi: 10.1002/0471250953.bi0811s16. [DOI] [PubMed] [Google Scholar]
- Forsyth, W.R., Jan, M., Antosiewicz, J.M., Robertson, A.D. Empirical relationships between protein structure and carboxyl pKa values in proteins. Proteins. 2002;48:388–403. doi: 10.1002/prot.10174. [DOI] [PubMed] [Google Scholar]
- Georgescu, R.E., Alexov, E.G., Gunner, M.R. Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys. J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunner, M.R., Alexov, E. A pragmatic approach to structure based calculation of coupled proton and electron transfer in proteins. Biochim. Biophys. Acta. 2000;1458:63–87. doi: 10.1016/s0005-2728(00)00060-8. [DOI] [PubMed] [Google Scholar]
- Hawkins, G.D., Cramer, C.J., Truhlar, D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995;246:122–129. [Google Scholar]
- Honig, B., Nicholls, A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Hooft, R.W., Sander, C., Vriend, G. Positioning hydrogen atoms by optimizing hydrogen-bond networks in protein structures. Proteins. 1996;26:363–376. doi: 10.1002/(SICI)1097-0134(199612)26:4<363::AID-PROT1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Im, W., Feig, M., Brooks C.L., III An implicit membrane Generalized Born theory for the study of structure, stability, and interactions of membrane proteins. Biophys. J. 2003;85:2900–2918. doi: 10.1016/S0006-3495(03)74712-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto, T. Electrostatic free energy of lysozyme. Biophys. J. 1987;44:293–298. doi: 10.1016/S0006-3495(83)84302-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juffer, A.H. Theoretical calculations of acid dissociation constants of proteins. Biochem. Cell Biol. 1998;76:198–209. doi: 10.1139/bcb-76-2-3-198. [DOI] [PubMed] [Google Scholar]
- Karshikoff, A., Spassov, V., Cowan, S.W., Ladenstein, R., Schirmer, T. Electrostatic properties of two porin channels from Escherichia coli . J. Mol. Biol. 1994;240:372–384. doi: 10.1006/jmbi.1994.1451. [DOI] [PubMed] [Google Scholar]
- Khandogin, J., Brooks C.L., III Toward the accurate first-principles prediction of ionization equilibria in proteins. Biochemistry. 2006;45:9363–9373. doi: 10.1021/bi060706r. [DOI] [PubMed] [Google Scholar]
- Kuramitsu, S., Hamaguchi, K. Analysis of the acid-base titration curve of hen lysozyme. Biochemistry. 1980;87:1215–1219. [PubMed] [Google Scholar]
- Labute, P. 2007 Protonate 3D: Assignment of macromolecular protonation state and geometry. Chemical Computing Group. http://www.chemcomp.com/journal/proton.htm
- Laskowski M., Jr, Sheraga, H.A. Thermodynamics considerations of protein reactions. I. Modified reactivity of polar groups. J. Am. Chem. Soc. 1954;76:6305–6319. [Google Scholar]
- Li, H., Robertson, A.D., Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- Li, X., Jacobson, M.P., Zhu, K., Zhao, S., Friesner, R.A. Assignment of polar states for protein amino acid residues using an interaction cluster decomposition algorithm and its application to high resolution protein structure modeling. Proteins. 2007;66:824–837. doi: 10.1002/prot.21125. [DOI] [PubMed] [Google Scholar]
- Linderstrom-Lang, K. On the ionization of proteins. C.R. Trav. Lab. Carlsberg. 1924;15:1–29. [Google Scholar]
- Madura, J.D., Briggs, J.M., Wade, R.C., Davis, M.E., Luty, B.A., Ilin, A., Antosiewicz, J., Gilson, M.K., Bagheri, B., Scott, L.R., et al. Electrostatics and diffusion of molecules in solution: Simulations with the University of Houston Brownian dynamics program. Comput. Phys. Commun. 1995;91:57–95. [Google Scholar]
- Mehler, E.L., Guarnieri, F. A self-consistent, microenvironment modulate descreened coulomb potential approximation to calculate pH-dependent electrostatic effects in proteins. Biophys. J. 1999;77:3–22. doi: 10.1016/S0006-3495(99)76868-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momany, F., Rone, R. Validation of the general purpose QUANTA 3.2/CHARMm force field. J. Comput. Chem. 1992;13:888–900. [Google Scholar]
- Mongan, J., Case, D.A. Biomolecular simulations at constant pH. Curr. Opin. Struct. Biol. 2005;15:157–163. doi: 10.1016/j.sbi.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Mongan, J., Case, D.A., McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004;25:2038–2048. doi: 10.1002/jcc.20139. [DOI] [PubMed] [Google Scholar]
- Nielsen, J.E., McCammon, J.A. On the evaluation and optimization of protein X-ray structures for pKa calculations. Protein Sci. 2003;12:313–326. doi: 10.1110/ps.0229903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, J.E., Vriend, G. Optimizing the hydrogen-bond network in Poisson–Boltzmann equation-based pKa calculations. Proteins. 2001;43:403–412. doi: 10.1002/prot.1053. [DOI] [PubMed] [Google Scholar]
- Onufriev, A., Bashford, D., Case, D.A. Modification of the Generalized Born model suitable for macromolecules. J. Phys. Chem. B. 2000;104:3712–3720. [Google Scholar]
- Pace, C.N., Douglas, V., Laurents, D.V., Thomson, J.A. pH dependence of the urea and guanidine hydrochloride denaturation of ribonuclease A and ribonuclease T1. Biochemistry. 1990;29:2564–2572. doi: 10.1021/bi00462a019. [DOI] [PubMed] [Google Scholar]
- Pfeil, W., Privalov, P.L. Thermodynamic investigations of proteins. III. Thermodynamic description of lysozyme. Biophys. Chem. 1976;4:41–50. doi: 10.1016/0301-4622(76)80005-1. [DOI] [PubMed] [Google Scholar]
- Pitera, J., Falta, M., van Gunsteren, W. Dielectric properties of proteins from simulation: The effects of solvent, ligands, pH, and temperature. Biophys. J. 2001;80:2546–2555. doi: 10.1016/S0006-3495(01)76226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampogna, R., Honig, B. Electrostatic coupling between retinal isomerization and the ionization state of Glu204: A general mechanism for proton release in bacteriorhodopsin. Biophys. J. 1996;71:1165–1171. doi: 10.1016/S0006-3495(96)79320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, M., Sommer, M., Karplus, M. pH-Dependence of protein stability: Absolute electrostatic free energy differences between conformations. J. Phys. Chem. B. 1997;101:1663–1683. [Google Scholar]
- Schellman, J.A. Macromolecular binding. Biopolymers. 1975;14:999–1018. [Google Scholar]
- Simonson, T. Macromolecular electrostatics: Continuum models and their growing pains. Curr. Opin. Struct. Biol. 2001;11:243–252. doi: 10.1016/s0959-440x(00)00197-4. [DOI] [PubMed] [Google Scholar]
- Smith, R., Breton, I.M., Chai, R.Y., Kent, S.B.H. Ionization states of the catalytic residues in HIV-1 protease. Nat. Struct. Biol. 1997;3:946–950. doi: 10.1038/nsb1196-946. [DOI] [PubMed] [Google Scholar]
- Song, Y., Mao, J., Gunner, M.R. Calculation of proton transfers in bacteriorhodopsin bR and M intermediates. Biochemistry. 2003;42:9875–9898. doi: 10.1021/bi034482d. [DOI] [PubMed] [Google Scholar]
- Spassov, V.Z., Bashford, D. Multiple-site ligand binding to flexible macromolecules: Separation of global and local conformational change and an iterative mobile clustering approach. J. Comput. Chem. 1999;20:1091–1111. [Google Scholar]
- Spassov, V.Z., Karshikoff, A.D., Atanasov, A.P. Electrostatic interactions in proteins. A theoretical analysis of lysozyme ionization. Biochim. Biophys. Acta. 1989;999:1–6. [Google Scholar]
- Spassov, V.Z., Luecke, H., Gerwert, K., Bashford, D. pK calculations suggest storage of an excess proton in a hydrogen-bonded water network in bacteriorhodopsin. J. Mol. Biol. 2001;312:203–219. doi: 10.1006/jmbi.2001.4902. [DOI] [PubMed] [Google Scholar]
- Spassov, V.Z., Yan, L., Szalma, S.Z. Introducing an implicit membrane in Generalized Born/solvent accessibility continuum solvent models. J. Phys. Chem. B. 2002;106:8726–8738. [Google Scholar]
- Spassov, V.Z., Yan, L., Flook, P.K. The dominant role of side-chain backbone interactions in structural realization of amino acid code. ChiRotor: A side-chain prediction algorithm based on side-chain backbone interactions. Protein Sci. 2007;16:494–506. doi: 10.1110/ps.062447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Still, W.C., Tempczyk, A., Hawley, R.C., Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990;112:6127–6129. [Google Scholar]
- Tan, Y.-J., Oliveberg, M., Davis, B., Fersht, A.R. Perturbed pKA-values in the denatured states of proteins. J. Mol. Biol. 1995;254:980–992. doi: 10.1006/jmbi.1995.0670. [DOI] [PubMed] [Google Scholar]
- Tanford, C., Kirkwood, J.G. Theory of protein titration curves. I. General equations for impenetrable spheres. J. Am. Chem. Soc. 1957;76:3331. [Google Scholar]
- Tanford, C., Roxby, R. Interpretation of protein titration curves. Application to lysozyme. Biochemistry. 1972;11:2192–2198. doi: 10.1021/bi00761a029. [DOI] [PubMed] [Google Scholar]
- Tanizaki, S., Feig, M. Molecular dynamics simulations of large integral membrane proteins with an implicit membrane model. J. Phys. Chem. B. 2006;110:548–556. doi: 10.1021/jp054694f. [DOI] [PubMed] [Google Scholar]
- Thurlkill, R.L., Grimsley, G.R., Scholtz, J.M., Pace, C.N. pK values of the ionizable groups of proteins. Protein Sci. 2006;15:1214–1218. doi: 10.1110/ps.051840806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd, M.J., Semo, N., Freire, E. The structural stability of the HIV-1 protease. J. Mol. Biol. 1998;283:475–488. doi: 10.1006/jmbi.1998.2090. [DOI] [PubMed] [Google Scholar]
- Toseland, C.P., McSparron, H., Davies, M.N., Flower, D.R. PPD v1.0—an integrated, web-accessible database of experimentally determined protein pK a values. Nucleic Acids Res. 2006;34:D199–D203. doi: 10.1093/nar/gkj035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trylska, J., Antosiewicz, J., Geller, M., Hodge, C.N., Klabe, R.M., Head, M.S., Gilson, M.K. Thermodynamic linkage between the binding of protons and inhibitors to HIV-1 protease. Protein Sci. 1999;8:180–195. doi: 10.1110/ps.8.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez-Campoy, A., Luque, I., Todd, M.J., Milutinovich, M., Kiso, Y., Freire, E. Thermodynamic dissection of the binding energetics of KNI-272, a potent HIV-1 protease inhibitor. Protein Sci. 2000;9:1801–1809. doi: 10.1110/ps.9.9.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y.X., Freedberg, D.I., Yamazaki, T., Wingfield, P.T., Stahl, S.J., Kaufman, J.D., Kiso, Y., Torchia, D.A. Solution NMR evidence that the HIV-1 protease catalytic aspartyl groups have different ionization states in the complex formed with the asymmetric drug KNI-272. Biochemistry. 1996;35:9945–9950. doi: 10.1021/bi961268z. [DOI] [PubMed] [Google Scholar]
- Warwicker, J. Improved pKa calculations through flexibility based sampling of a water-dominated interaction scheme. Protein Sci. 2004;13:2793–2805. doi: 10.1110/ps.04785604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Word, J., Lovell, S., Richardson, J., Richardson, D. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999;285:1735–1747. doi: 10.1006/jmbi.1998.2401. [DOI] [PubMed] [Google Scholar]
- Yamazaki, T., Nicholson, L.K., Torchia, D.A., Wingfield, P., Stahl, S.J., Kaufman, J.D., Charles, J., Eyermann, C.J., Nicholas Hedge, C., Lam, P.Y.S., et al. Catalytic aspartyl groups are protonated in the complex formed by the protease and a non-peptide cyclic urea-based inhibitor. J. Am. Chem. Soc. 1994;116:10791–10792. [Google Scholar]
- Yang, A.S., Honig, B. Structural origins of pH and ionic strength effects on protein stability. Acid denaturation of sperm whale apomyoglobin. J. Mol. Biol. 1994;237:602–614. doi: 10.1006/jmbi.1994.1258. [DOI] [PubMed] [Google Scholar]
- Yang, A.S., Gunner, M.R., Sampogna, R., Sharp, K., Honig, B. On the calculations of pK's in proteins. Proteins. 1993;15:252–265. doi: 10.1002/prot.340150304. [DOI] [PubMed] [Google Scholar]
- You, T.J., Bashford, D. Conformation and hydrogen ion titration of proteins: A continuum electrostatic model with conformational flexibility. Biophys. J. 1995;69:1721–1733. doi: 10.1016/S0006-3495(95)80042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]