

Abstract
We have used mouse embryonic fibroblasts (MEFs) derived from VDR(+/-) and VDR(-/-) mice to determine whether the nuclear vitamin D receptor (VDR) is directly involved in the regulation of NF-κB activation. We found that the basal IκBα protein level was markedly decreased in VDR(-/-) MEFs compared to VDR(+/-) MEFs; however, degradation of IκBα and its phosphorylation were not altered in VDR(-/-) cells, neither were the levels of IKKα and IKKβ proteins. Consistently, p65 nuclear translocation was increased in unstimulated VDR(-/-) cells. The physical interaction between VDR and p65 was absent in VDR(-/-) MEFs, which may free p65 and increase its activity. Consequently, these alterations combined led to a marked increase in NF-κB transcriptional activity. Consistently, induction of IL-6 by TNFα or IL-1β was much more robust in VDR(-/-) than in VDR(+/-) cells, indicating that VDR(-/-) cells are more susceptible to inflammatory stimulation. Therefore, fibroblasts lacking VDR appear to be more pro-inflammatory due to the intrinsic high NF-κB activity. The reduction of IκBα in VDR(-/-) MEFs may be partially explained by the lack of VDR-mediated stabilization of IκBα by 1,25(OH)2D3. These data suggest that VDR plays an inhibitory role in the regulation of NF-κB activation.
Keywords: Vitamin D receptor, inflammation, NF-κB, mouse embryonic fibroblasts
Introduction
Nuclear factor-κB (NF-κB) is a family of transcription factors that play an essential role in innate and adaptive immune responses [1]. The NF-κB transcription factors are homo- or heterodimers formed by five proteins including NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB and c-Rel. Different NF-κB dimers bind to specific DNA sequence in gene promoters to regulate transcription of a wide range of genes, including those involved in immune and inflammatory responses. NF-κB is active in the nucleus and its activity is inhibited by the inhibitor of κB (IκB), which binds to NF-κB to block its nuclear translocation. Phosphorylation of IκB by IκB kinase (IKK) initiates the ubiquitylation and degradation of IκB by proteasome, leading to nuclear translocation and activation of NF-κB [1, 2]. The IKK complex is activated by growth factors, pro-inflammatory cytokines and hormones through TNF receptor and Toll-like receptor superfamily [1].
Previous works have suggested that 1,25(OH)2D3 directly modulates NF-κB activity. In dendritic cells, 1,25(OH)2D3 inhibits IL-12 expression through targeting the NF-κB pathway [3]; 1,25(OH)2D3 directly suppresses RelB transcription [4]. In activated lymphocytes, 1,25(OH)2D3 suppresses the increase in NF-κB p50 and c-rel proteins [5]. 1,25(OH)2D3 has also been shown to decrease the DNA binding capacity of NF-κB in human fibroblasts and keratinocytes [6, 7]. In pancreatic islet cells, a vitamin D analog is reported to significantly down-regulate pro-inflammatory chemokine, which is associated with up-regulation of IκBα transcription and arrest of NF-κB p65 nuclear translocation [8]. However, it remains to be determined whether VDR is directly involved in the regulation of the NF-κB pathway.
In the present study we investigated the effect of VDR ablation on NF-κB activation using mouse embryonic fibroblasts (MEFs) derived from VDR-null mice. We found that cells lacking the VDR exhibit increased NF-κB activity due to the reduction in IκBα levels and the lack of VDR-p65 interaction. Our data suggest that VDR is directly involved in suppression of NF-κB activation, which may partially explain the VDR-mediated anti-inflammatory mechanism of vitamin D.
Materials and Methods
Embryonic fibroblast isolation and culture
Mouse embryonic fibroblasts (MEFs) were isolated from E13.5 embryos generated from VDR(+/-) × VDR(+/-) mouse breeding [9]. The cells were cultured in DMEM containing 10% FBS. Cells from each embryo were genotyped by PCR. VDR(+/-) and VDR(-/-) MEFs were used in experiments after more than 15 passages when they had been immortalized as shown previously [10].
Northern and Western Blot
Northern blot and Western blot analyses were performed as described previously [11].
Immunofluorescence analysis
MEFs were cultured in the presence or absence of Salmonella or TNFα for one hour. The permeabilized cells were incubated with anti-p65 antibody, followed by incubation with Alexafluor594 secondary antibodies, Alexafluor488 FITC-conjugated secondary antibodies or DAPI. The cells were examined with a Leica DMIRE2 scanning laser confocal microscope.
Luciferase reporter assay
MEFs were transfected with pNF-κB-Luc, pRL-TK (control) and pcDNA3.1 or pcDNA-hVDR using LipofectAMINE 2000 (Invitrogen). Luciferase activity was determined using Dual Luciferase Assay Kit (Promega).
NF-κB p65 DNA binding activity
NF-κB activation was determined using TransAM NF-κB p65 Transcription Factor Assay Kit (Active Motif), which specifically measures the amount of NF-κB p65 bound to its consensus binding site (5’GGGACTTTCC3’) with ELISA.
IL-6 assay
The level of IL-6 was determined using mouse IL-6 Enzyme Immunometric Assay (EIA) kit (Assay Designs).
Results
To investigate the effect of VDR inactivation on NF-κB activation, the protein level of the major components of the NF-κB pathway in VDR(+/-) and VDR(-/-) MEFs was compared. We found that VDR ablation led to a marked reduction of IκBα protein (by more than 50%) in VDR(-/-) cells (Fig. 1A).However, IκBα degradation induced by TNFα treatment or Salmonella infection followed a similar time-course pattern in VDR(+/-) and VDR(-/-) MEFs, so is the pattern of IκBα phosphorylation following TNFα treatment. The same levels of IKKα and IKKβ were seen in VDR(+/-) and VDR(-/-) MEFs under unstimulated or TNFα-treated condition. Therefore, despite the reduction, the pathway involved in IκBα degradation appears unaltered in VDR(-/-) cells.
Figure 1. Comparison of NF-κB pathway between VDR(+/-) and VDR(-/-) MEFs.
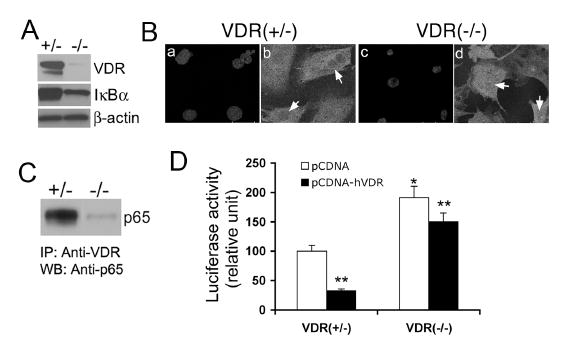
(A) Basal levels of IκBα protein in VDR(+/-) and VDR(-/-) cells determined by Western blotting. Also shown is the VDR levels determined using anti-VDR antibody. (B) Confocal microscopy showing increased p65 nuclear translocation in VDR(-/-) cells. VDR(+/-) and VDR(-/-) MEFs were stained with 4-diamidino-2-phenylindole (DAPI) to visualize the nucleus (a and c) or with anti-p65 antibody (b and d). (C) Protein-protein interaction between VDR and p65. VDR(+/-) and VDR(-/-) cell lysates were immunoprecipitated with anti-VDR antibody (IP), and then the precipitated complex was probed with anti-p65 antibody by Western blotting (WB). (D) Suppression of NF-κB transcriptional activity by hVDR over-expression. VDR(+/-) and VDR(-/-) MEFs were co-transfected with pNF-κB-Luc reporter, pRL-TK and pcDNA3.1 or pcDNA-hVDR, and luciferase activity was determined after 24 hours. *, P<0.05 vs. VDR(+/-) cells; **P<0.05 vs. corresponding pcDNA control.
By confocal microscopy we found that, due to the reduced level of IκBα, more NF-κB p65 was translocated into the nucleus in VDR(-/-) cells than in VDR(+/-) cells in unstimulated state (Fig. 1B), and this immunostaining observation was confirmed by Western blot analysis of p65 levels in the cytosolic and nuclear compartments of VDR(+/-) and VDR(-/-) MEFs. Consistently, the basal p65 DNA binding activity and NF-κB transcriptional activity were significantly increased in VDR(-/-) cells; when the cells were stimulated with TNFα or Salmonella, the induction of both p65 DNA binding and NF-κB activity was much greater in VDR(-/-) cells than in VDR(+/-) cells.
By co-immunoprecipitation assays we were able to pull down p65 protein in VDR(+/-) MEFs, but not in VDR(-/-) cells, using anti-VDR antibody (Fig. 1C), confirming the existence of VDR-p65 interaction in mouse fibroblasts. Interestingly, the basal NF-κB transcriptional activity was markedly increased in VDR(-/-) cells, and transfection of hVDR significantly reduced NF-κB activity in both VDR(+/-) and VDR(-/-) cells (Fig. 1D), suggesting VDR-p65 interaction affects NF-κB transcriptional activity. Therefore, it appears that increased nuclear accumulation of p65 and lack of VDR binding to p65 in the nucleus lead to higher NF-κB activity in VDR(-/-) cells.
Since NF-κB is a key regulator involved in the synthesis of inflammatory cytokines, we measured the production of IL-6, a well-known NF-κB target gene, as a read-out of the downstream biological effect of NF-κB. In the basal state, IL-6 secretion by VDR(+/-) cells was undetectable, but it was easily detected in VDR(-/-) cells; under the stimulation of TNFα or Salmonella, the secretion of IL-6 was much more robust in VDR(-/-) cells than in VDR(+/-) cells (Fig. 2A). Similarly, Northern blot analyses showed that IL-6 mRNA expression induced by TNFα or IL-1β was more robust in VDR(-/-) cells than in VDR(+/-) cells (Fig. 2B and C ), confirming that VDR(-/-) cells are more susceptible to inflammatory stimuli.
Figure 2. IL-6 synthesis in MEFs.
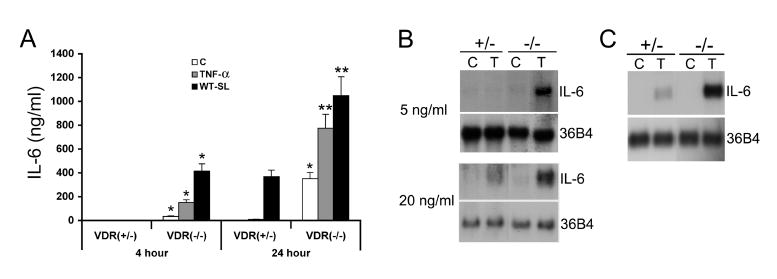
(A) IL-6 secretion. VDR(+/-) and VDR(-/-) MEFs were untreated (C) or treated with TNFα (20 ng/ml) or Salmonella (WT-SL) for one hour, and incubated in media containing Gentamicin for 4 or 24 hours. IL-6 secretion into the media was determined using a mouse IL-6 EIA kit. Note the basal IL-6 production in unstimulated VDR(+/-) cells was below the detectable limit. *P<0.05, **P<0.001 vs. corresponding VDR(+/-) value. (B and C) IL-6 mRNA expression. (B) VDR(+/-) and VDR(-/-) MEFs were untreated (C) or treated with 5 ng/ml (upper panel) or 20 ng/ml (lower panel) TNFα (T) for 6 hours. (C) VDR(+/-) and VDR(-/-) MEFs were untreated (C) or treated with 1 ng/ml IL-1β (T) for 6 hours. IL-6 mRNA levels were determined by Northern blotting. 36B4 is the internal loading control.
The increase in NF-κB activity in VDR(-/-) cells is attributed in part to the reduction in IκBα. To explain the IκBα reduction in VDR(-/-) MEFs, we determined the effect of vitamin D on IκBα. We found that 1,25(OH)2D3 was able to inhibit IκBα protein degradation induced by TNFα or IL-1β treatment in VDER(+/-) MEFs, but not in VDR(-/-) cells, and the inhibition was apparent at a dose as low as 10-10 M of 1,25(OH)2D3. Therefore, VDR is required for vitamin D-mediated stabilization of IκBα protein in MEFs.
Discussion
In the present study we set to address the role of VDR in NF-κB activation by examining the effect of VDR inactivation on the NF-κB pathway. Our data suggest that VDR is directly involved in the regulation of NF-κB activation. VDR(-/-) cells exhibit reduced IκBα levels, leading to increased nuclear translocation of p65, which is consistent with a previous observation that a vitamin D analog can block p65 nuclear translocation [8]; on the other the hand, because of the lack of VDR, the physical interaction between VDR and p65 is absent in VDR(-/-) cells, which may free p65 and increase its activity. Consequently, these changes combined lead to a marked increase in NF-κB activity in VDR(-/-) cells. Therefore, VDR appears to regulate the NF-κB activation pathway by targeting IκBα and p65, thus cells lacking VDR appear to be more susceptible to inflammatory stimuli. It will be interesting to further investigate the inflammatory status of VDR-null mice.
How VDR regulates IκBα remains unclear. In VDR(-/-) cells, the levels of IKKα and IKKβ and IκBα degradation appears normal, suggesting that the events involved in IκBα phosphorylation and the following proteasome-mediated IκBα degradation are unaltered in the absence of VDR. Since VDR is not involved in these events, the reduction of IκBα is unlikely due to altered IKK protein levels or impaired proteasome-mediated degradation; however, whether VDR inactivation alters IKK phosphorylation or IKK activity remains to be determined. Since 1,25(OH)2D3 can prevent or at least reduce the degradation of IκBα protein, and the inhibition of IκBα degradation is dependent on VDR, liganded VDR may help stabilize IκBα. This may partially explain why VDR ablation leads to a decrease in IκBα levels, but exactly how VDR affects IκBα stability needs to be further investigated.
The exact functional significance of VDR-p65 protein interaction remains to be determined. We speculate that the elevated basal NF-κB activity in VDR(-/-) cells results not only from the reduction in IκBα, which increases the nuclear accumulation of p65, but also from the disruption of the VDR-p65 interaction, which releases the restraint on p65 and thus increases its activity. In other words, under normal conditions VDR-p65 interaction helps suppress p65 activity. This speculation seems to be supported by the co-transfection experiment that shows a reduction of NF-κB transcriptional activity in the presence of hVDR over-expression.
The direct involvement of VDR in the regulation of NF-κB activation suggests an intrinsic inhibitory role of VDR in inflammation process, and this inhibitory action may likely be enhanced in the presence of vitamin D ligands. Given the crucial role of NF-κB in inflammatory response, and the therapeutic and pharmacologic potentials of vitamin D and vitamin D analogs in treatment of autoimmune and inflammatory diseases, understanding the relationship and interaction between vitamin D, VDR and NF-κB pathway has broad implications.
Acknowledgments
This work was supported in part by NIH grant DK59327 (Y.C.L), DK47662 and DK35932 (J.L.M) and a Pilot and Feasibility Award DK42086 (J.S).
Footnotes
This work was supported in part by NIH grant DK59327 (Y.C.L), DK47662 and DK35932 (J.L.M) and a Pilot and Feasibility Award DK42086 (J.S).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Nakanishi C, Toi M. Nuclear factor-kappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 3.D’Ambrosio D, Cippitelli M, Cocciolo MG, Mazzeo D, Di Lucia P, Lang R, Sinigaglia F, Panina-Bordignon P. Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J Clin Invest. 1998;101:252–262. doi: 10.1172/JCI1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong X, Craig T, Xing N, Bachman LA, Paya CV, Weih F, McKean DJ, Kumar R, Griffin MD. Direct transcriptional regulation of RelB by 1alpha,25-dihydroxyvitamin D3 and its analogs: physiologic and therapeutic implications for dendritic cell function. J Biol Chem. 2003;278:49378–49385. doi: 10.1074/jbc.M308448200. [DOI] [PubMed] [Google Scholar]
- 5.Yu XP, Bellido T, Manolagas SC. Down-regulation of NF-kappa B protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3 . Proc Natl Acad Sci U S A. 1995;92:10990–10994. doi: 10.1073/pnas.92.24.10990. [published erratum appears in Proc Natl Acad Sci U S A 1996 Jan 9;93(1):524] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harant H, Wolff B, Lindley IJ. 1Alpha,25-dihydroxyvitamin D3 decreases DNA binding of nuclear factor-kappaB in human fibroblasts. FEBS Lett. 1998;436:329–334. doi: 10.1016/s0014-5793(98)01153-3. [DOI] [PubMed] [Google Scholar]
- 7.Riis JL, Johansen C, Gesser B, Moller K, Larsen CG, Kragballe K, Iversen L. 1alpha,25(OH)(2)D(3) regulates NF-kappaB DNA binding activity in cultured normal human keratinocytes through an increase in IkappaBalpha expression. Arch Dermatol Res. 2004;296:195–202. doi: 10.1007/s00403-004-0509-9. [DOI] [PubMed] [Google Scholar]
- 8.Giarratana N, Penna G, Amuchastegui S, Mariani R, Daniel KC, Adorini L. A vitamin D analog down-regulates proinflammatory chemokine production by pancreatic islets inhibiting T cell recruitment and type 1 diabetes development. J Immunol. 2004;173:2280–2287. doi: 10.4049/jimmunol.173.4.2280. [DOI] [PubMed] [Google Scholar]
- 9.Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94:9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li YC, Bolt MJG, Cao LP, Sitrin MD. Effects of vitamin D receptor inactivation on the expression of calbindins and calcium metabolism. Am J Physiol Endocrinol Metab. 2001;281:E558–E564. doi: 10.1152/ajpendo.2001.281.3.E558. [DOI] [PubMed] [Google Scholar]