

Abstract
Recent progress in understanding the Q-cycle mechanism of the bc1 complex is reviewed. The data strongly support a mechanism in which the Qo-site operates through a reaction in which the first electron transfer from ubiquinol to the oxidized iron-sulfur protein is the rate determining step for the overall process. The reaction involves a proton-coupled electron transfer down a hydrogen bond between the ubiquinol and a histidine ligand of the [2Fe-2S] cluster, in which the unfavorable protonic configuration contributes a substantial part of the activation barrier. The reaction is endergonic, and the products are an unstable ubisemiquinone at the Qo-site, and the reduced iron-sulfur protein, the extrinsic mobile domain of which is now free to dissociate and move away from the site to deliver an electron to cyt c1 and liberate the H+. When oxidation of the semiquinone is prevented, it participates in bypass reactions, including superoxide generation if O2 is available. When the b-heme chain is available as acceptor, the semiquinone is oxidized in a process in which the proton is passed to the glutamate of the conserved –PEWY- sequence, and the semiquinone anion passes its electron to heme bL to form the product ubiquinone. The rate is rapid compared to the limiting reaction, and would require movement of the semiquinone closer to heme bL to enhance the rate constant. The acceptor reactions at the Qi-site are still controversial, but likely involve a “two-electron gate” in which a stable semiquinone stores an electron. Possible mechanisms to explain the cytb150 phenomenon are discussed, and the information from pulsed EPR studies about the structure of the intermediate state is reviewed.
The mechanism discussed is applicable to a monomeric bc1 complex. We discuss evidence in the literature that has been interpreted as shown that the dimeric structure participates in a more complicated mechanism involving electron transfer across the dimer interface. We show from myxothiazol titrations and mutational analysis of Tyr-199, which is at the interface between monomers, that no such inter-monomer electron transfer is detected at the level of the bL hemes. We show from analysis of strains with mutations at Asn-221 that there are coulombic interactions between the b-hemes in a monomer. The data can also be interpreted as showing similar coulombic interaction across the dimer interface, and we discuss mechanistic implications.
Introduction
The ubiquinol:cytochrome c oxidoreductase (bc1 complex) family of redox-linked proton pumps form the core of all major electron transfer chains, with an ancestry dating back to before the great divide. Their importance needs no further emphasis beyond their role in carrying the energy flux of the biosphere. It is generally agreed that a Q-cycle mechanism accounts for the activity of the enzyme, but there are many variants, and which particular variant best explains the finer points is still under debate [1–11]. In the simpler bacterial systems, the kinetic and thermodynamic behavior is well described by the modified Q-cycle [12, 13]. This has been discussed extensively in recent reviews [2–5, 11, 13], and is shown schematically in Fig. 1. The bc1 complex has a dimeric structure incorporating 4 metal centers in each monomer (hemes bH and bL in cytochrome (cyt) b, heme c1 in cyt c1, and a [2Fe-2S] cluster in the iron-sulfur protein (ISP)), which provide electron transfer chains connecting two quinone processing sites, and three additional catalytic interfaces. Despite this complexity, the mechanism is better understood than that for other respiratory complexes. The oxidation of ubiquinol (QH2) occurs at the Qo-site (also called the QP-site), where the two electrons are diverted down two different chains. The first electron goes down the high-potential chain of ISP, heme c1, and cyt c (or c2 in bacteria), to an oxidant (a cytochrome oxidase in respiratory chains, or the oxidized reaction center in photosynthetic systems). Removal of one electron leaves a semiquinone (SQ) at the Qo-site. The second electron is delivered to a low-potential chain of hemes bL and bH, which passes the electron on to the Qi-site (QN-site), where it reduces either ubiquinone (Q) to SQ, or SQ to QH2. Because reduction of Q at the Qi-site requires two electrons overall, the bifurcated reaction at the Qo-site has to occur twice to provide these, and to complete a full turnover of the enzyme. The location of the two Q-sites on opposite sides of the membrane gives rise to an electrogenic flux through the low-potential chain, which provides the electrogenic arm, while diffusion of Q and QH2 across the membrane with oxidation of 2QH2 (with release of 4H+), or reduction of Q (with uptake of 2H+) provide the neutral arms of a Mitchellian proton-pumping loop.
Fig. 1. The modified Q-cycle mechanism.
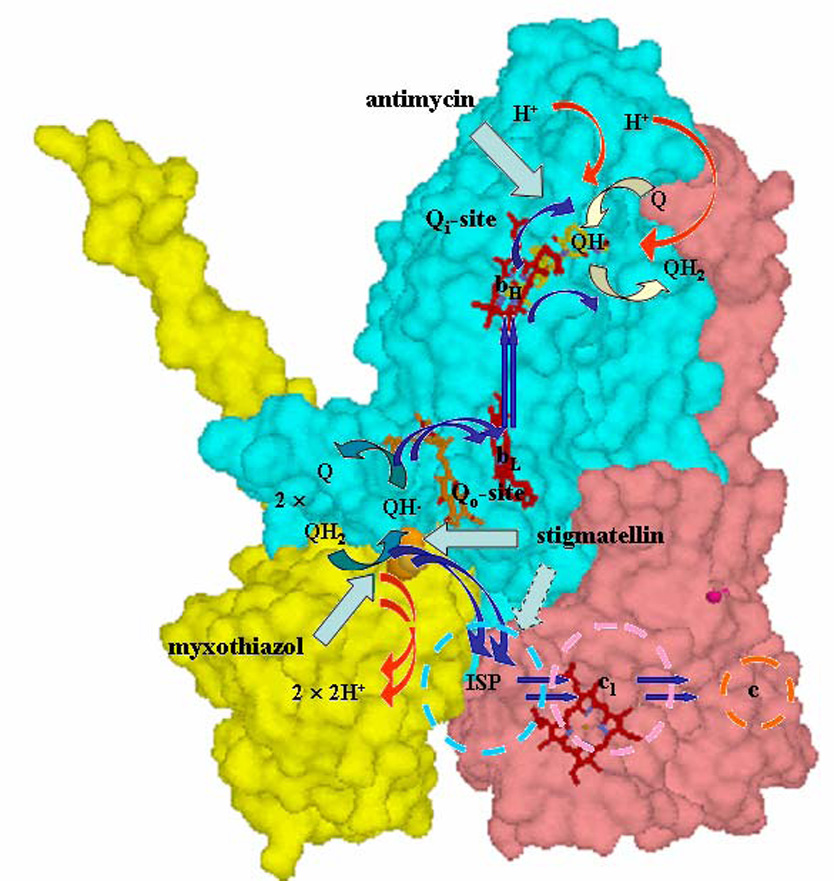
The mechanism demonstrated in the 1980’s, superimposed on the Rb. sphaeroides structure (PDB file 2QJK). Electron transfers are shown by blue arrows; H+ release or uptake by red arrows, exchange of Q or QH2 by open blue-green (Qo-site) or yellow (Qi-site) arrows; inhibition sites by cyan arrows. See text for description of operation.
The ancestral history of the bc1 complex has left an unfortunate mechanistic legacy [14–18]. Oxidation of ubihydroquinone (ubiquinol, QH2) at the Qo-site of the complex operates through an intermediate semiquinone (SQ) that has a high reactivity with O2. This leads to production of superoxide anion, the precursor of reactive oxygen species (ROS) that play havoc with the cellular machinery, and ultimately lead to cellular suffocation through damage to mitochondrial DNA [18, 19]. There is some controversy over what fractional contribution this reaction makes to the overall cellular ROS production in humans, but a substantial literature suggests that it may be the major source under some physiological conditions and in some genetic pathologies [20–23]. This design-defect likely reflects an “invention” when the biosphere was anaerobic, and one of the interests in studying the complex has been to see how evolution has subsequently honed the architecture to minimize the effect.
Much of the critical work has investigated the bc1 complex in Rhodobacter species of photosynthetic bacteria. The major advantage of working in photosynthetic systems lies in the fact that the bc1 complex is tightly integrated into the photosynthetic machinery [24], so that excitation with a saturating flash generates the substrates stoichiometrically, allowing exploration of the kinetics of a single turnover of the intact apparatus on the microsecond time scale. From this work, a detailed description of the mechanism has been proposed, in which the partial reactions have been dissected out, and characterized through kinetic and thermodynamic parameters sufficient to account for the bulk of available experimental data [4, 5, 25, 26]. With the availability of structures [27–33], this physicochemical description has been fleshed out in molecular detail. Although the structures provided strong support for the modified Q-cycle, they also introduced some novel features [34–36]. The structures have also put much detailed work using mutagenesis on a firmer basis, providing insights that have allowed an atomic-level analysis of mechanism.
In this paper, we briefly review recent developments that have provided us with an economical model for turnover in a monomeric complex, and then explore the question whether this is sufficient to explain turnover of the dimer.
Mobility of the ISP ectodomain
Recognition of the mobility of the extrinsic domain[28] launched a period of controversy about the role of movement of the ISP in control of function. Much of the speculation has focused on a possible controlling role of such movement. From our own work [36, 37], and from Millet’s results[38, 39] it is clear that movement is always more rapid than the rate determining step, supporting the interpretation derived from examination of the early structures, which led us to focus instead on specific interactions between the ISP and its reaction partners that may restrict mobility by binding at the reaction interfaces. As we have shown, some of these can be measured directly through thermodynamic effects, and have provided the basis for our models for formation of ES- and EP-complexes[4, 34–36, 40]. Some can be observed either through crystallographic solution [28, 33, 36, 41, 42], or through spectroscopic studies [42, 43]. For many of these complexes, the dominant interaction, either observed directly or proposed, is through H-bonding between His-161 (bovine numbering is indicated by italics) of ISP and the occupant of the Qo-site. The nature of these interactions is strongly determined by the protolytic properties of this group, which are also reflected in the redox dependences of the half-cell reactions, and the pH dependence of redox titration data [40, 44, 45]. We have therefore invested much effort in characterizing these properties, and investigating how they are determined at the atomic level [43, 46, 47]. Our general approach in this latter investigation has been through specific mutagenesis, thermodynamic measurement (including protein film voltammetry), crystallographic solution at high resolution, and detailed spectroscopic investigation of the local protein and solvent environment using pulsed-EPR, Resonance Raman (Fig.2), and FTIR. Structures at high resolution (<1.5 Å) have been published for wild-type (2NUK) and 5 mutants (2NUM (Y156F), 2NWF (Y156W), 2NVE (S154T), 2NVF (S154C), and 2NVG (S154A) [47]. An additional structure for G133S has been described briefly in the context of spectroscopic data [46]. Five additional structures are at different stages of refinement for mutant strains modified at Leu-132 and Gly-133 (in collaboration with Satish Nair). Our spectroscopic studies have been published in four papers characterizing the WT protein in situ in the complex, and as the purified extrinsic domain [43, 46, 48, 49]. Additional work is nearing completion, in which we have dissected the proton environment of the [2Fe-2S] cluster, and identified sites undergoing deuterium exchange, likely involved in H-bonds, and we have similar data from two mutant strains, Y156F and S154A, in each of which we have identified loss of at least one exchangeable proton, with different properties in the two mutants. From the two structures for mutant strains at these different sites already published, we can say with confidence that the structures are essentially unchanged except for the loss of a single H-bond resulting from the mutation. It follows that the changes in thermodynamic and kinetic properties must arise from this change. For both mutations (Y156F, S154A), DFT/electrostatic calculations have recently been performed [50], and the results were in good agreement with our experimental data (Table 1), and support previous speculation [51–53] as to the importance of these interactions. From this extensive work we now have a detailed picture of the structural underpinnings of the thermodynamic properties of the ISP, and a much clearer understanding of how these properties are modified by mutation. At the resolution provided by our structures [46, 47], crystallography provides true atomic-level positions for all non-H atoms, and in some cases also shows electron density for H-atoms. For some H-bonds, this makes it possible to identify angular parameters. This information is also in principle available from pulsed-EPR data. The special interest of our studies lies in the fact that we are accumulating such data from the same set of proteins, which will allow a cross-validation of methodologies. Our recent observation that the backbone -NH of Leu-132 provides a H-bond to S2 of the cluster, and that the geometry leads to significant spin density on the N-atom (and by extension, on the S-atom of the cluster) is a nice demonstration of the synergy between these approaches [46].
Fig. 2. Synergy of spectroscopy, crystallography, and thermodynamic analysis.

(Left) Structures of wild-type, S154A and Y156F mutants (modified residues shown with electron densities); (middle) 2D ESEEM plots showing proton profiles in wild-type and S154A, and difference (bottom), showing loss of protons a, b (similar data have been obtained for the Y156F mutant); (right) RR spectra showing differences in ISP mutants S154A and Y156F, compared to SDX mutants and wild-type, and (bottom) PFV titrations, showing voltammogram (left, WT, pH 7), and Em vs. pH curves from titration data for wild-type and five mutant strains (right).
Table 1.
Electrochemical parameters measured by CD spectrum, protein film volatammetry, and kinetic titration
| Strains | Em values from CD spectra (mV) | Protein Film Voltammetry | Kinetic titration at 83% reduced quinine pool 4) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Eacid (mV) | Ebase (mV) | pKox1 | pKox2 | pKred1,2 | pKA | pKB | Vopt | ||
| wild-type/BH6 1) | 303 ± 4 2) / 312 ± 5 3) | 308 ± 3 | −134 ± 6 | 7.6 ± 0.1 | 9.6 ± 0.1 | 12.4 ± 0.4 | 6.66 ± 0.10 | 9.14 ± 0.26 | 973.3 |
| S154T | 260 ± 5 2) | 288 ± 3 | −151 ± 7 | 7.71 ± 0.14 | 9.32 ± 0.15 | 12.2 ± 0.1 | 6.83 ± 0.12 | 8.48 ± 0.14 | 826.1 |
| S154C | 313 ± 5 2) | 324 ± 4 | −97 ± 6 | 7.17 ± 0.11 | 9.77 ± 0.13 | 12.0 ± 0.1 | 6.29 ± 0.05 | 8.46 ± 0.06 | 804.0 |
| S154A | 167 ± 6 2) | 178 ± 4 | −239 ± 10 | 8.06 ± 0.15 | 9.89 ± 0.17 | 12.5 ± 0.1 | 7.52 ± 0.20 | 10.2 ± 0.28 | 199.2 |
| Y156F | 256 ± 4 3) | 252 ± 4 | −165 ± 6 | 7.76 ± 0.15 | 8.83 ± 0.21 | 11.8 ± 0.1 | 6.90 ± 0.06 | 8.54 ± 0.08 | 714.6 |
| Y156W | 198 ± 3 3) | 208 ± 3 | −228 ± 4 | 7.87 ± 0.09 | 9.25 ± 0.11 | 12.2 ± 0.1 | 6.97 ± 0.08 | 9.69 ± 0.12 | 242.7 |
The wild-type strain in this work is a complementary mutant strain of Rb. sphaeroides BC17, which contains the pRK415 with a modified fbc operon which has a histidine-tag at the C-terminus of the cyt b gene. The protein film voltammetric data of the wild-type was reported [44].
The values are measured at pH 7.4.
The data represent the limiting value for Em at pH << pK1, as described in [59].
Vopt is the optimal reaction rate achieved when the first protonable group is deprotonated (pKA) and the second group is protonated (pKB), according to the following equation from the reference [148].
Qo-site reaction
From our earlier studies relating Berry’s first complete structures to function, we had suggested a scenario in which movement of the extrinsic domain of the ISP was a spontaneous diffusional process with an expected timescale in the sub-microsecond range [28, 35, 36]. We demonstrated the feasibility of this scenario through molecular dynamic simulations. We pointed out that binding processes at the catalytic interfaces were likely relatively weak, based on the previously measured kinetic constants, and expected collision-limited rates, and on the occupancy of ISP in structures. In a more recent extended series of kinetic measurements [37], we were able to confirm these expectations, and demonstrate limits on reaction rates which were in line with these earlier speculations. These measurements provided a number of useful constraints on mechanism.
a) We demonstrated that the oxidation of QH2 in the absence of antimycin, determined from the electrogenic events associated with turnover, occurred at the same rate as that in the presence of antimycin, determined from the rate of reduction of heme bH [37]. This result was important because it precluded any mechanisms in which physicochemical properties affecting turnover at the Qo-site were modified by binding of antimycin at the Qi-site (cf. [9, 10, 53]).
b) We analyzed the rates of partial processes in the high-potential chain, and compare these with the turnover of the Qo-site as determined by the kinetics of reduction of heme bH in the presence of antimycin. We demonstrated that the 120 µs lag before onset of heme bH reduction was almost all accounted for by the time needed to get an oxidizing equivalent to the Qo-site. This result was important because it placed constraints on movement of ISP, and on the stoichiometry of intermediate states, including that of the SQ. Since the rate limiting step was identified as the first electron transfer, with a rate-constant of ~770 µs, the unaccounted lag of <20 µs would limit any occupancy to <0.026 for any intermediate between QH2 and heme bH [37]. This constraint on occupancy can be lowered to <0.015 on the basis of work from Millett’s lab in which the reaction was initiated in the isolated Rb. sphaeroides complex by flash activation via a bound ruthenium complex [39, 54]. This result eliminates all reaction mechanisms in which a stable (K >0.015) SQ-ISPH intermediate is required [55, 56].
c) For the Qo-site reaction itself, the first electron transfer (to ISPox) had been identified as the rate determining step in QH2 oxidation on the basis of dependence on driving force [25, 26, 57–59]. We had proposed a specific role for the H-bond between His-161of ISP, and either QH2 as H-bond donor in formation of the ES-complex, or Q as H-bond acceptor in formation of the EP-complex [25, 40, 45, 59, 60]. The difference between these roles depended on the protolytic properties of the histidine. We suggested that this group was responsible for the pK at 7.6 measured on the oxidized ISP. This pK was shifted to >12 in the reduced form. From this hypothesis, several predictions could be made about the properties of the first electron transfer reaction. The predictions were framed in the context of a mechanism in which the transfer of an electron (together with a proton) from QH2 to ISPox occurred in a strongly endergonic process to generate the intermediate SQ at low occupancy. From the structures, and our proposal for the nature of the ES-complex, several paradoxical features had to be explained. These related to the high activation barrier, the slow rate of the overall reaction, and the very short distance for electron transfer (~7 Å). Since, after reduction, the ISP would be protonated, the reaction effectively involves H-transfer in a proton-coupled electron transfer. In the treatment proposed[4, 40, 42], the pK of His-161contributed two separate effects. The first of these involved the role of the dissociated form of ISPox in formation of the ES-complex, in which QH2 was H-bond donor in the complex with ISPox. This meant that the concentration of ISPox as a substrate was determined by the pK on His-161. We demonstrated that this was the case by measurement of the pH dependence of electron transfer, which showed Michaelis-Menten saturation for ISPox [25]. Support for this interpretation was provided by use of a mutant ISP (Y156W) with a shifted pK, which also showed a corresponding shift in pH dependence [59]. The second effect of the pK on ISPox came from its role in determining the energy level of an intermediate proton configuration [61]. We suggested that electron transfer proceeded from an unfavorable configuration of the H+ in the H-bond, determined by a Brønsted relationship and the pK values for QH2 and ISPox. The reaction interface was anhydrous, so this necessitated a proton-coupled electron transfer along the H-bond. We pointed out that the unfavorable intermediate state would contribute part of the energy barrier determining the rate of electron transfer, and that this could explain the observed properties. A detailed Marcus-type analysis showed that the behavior of WT could be accounted for economically by this mechanism, and the same treatment also explained the behavior of a set of mutant strains with modified Em and pK values for ISP [4].
Because of the important role of the pK on His-161, and the coupling between redox and protolytic properties, we have invested much effort in determining the structural basis of these properties (see above). The attribution of the pK to a liganding histidine has now been confirmed by spectroscopy [62–64] and computational modeling [50] (see above). The mechanism proposed successfully accounts for the properties of the first electron transfer. Since this is the rate determining step, our detailed mechanism in the context of the modified Q-cycle also explains much of the subsequent behavior of the complex.
d) We had proposed that the second electron transfer at the Qo-site might require a movement of the SQ from a location in the site distal from heme bL where it was generated on oxidation of QH2 to a proximal location, allowing ~1000-fold enhancement of rate constant and rapid reduction of the heme at low occupancy [25, 60]. We had suggested a role for the conserved glutamate (Glu-295 of the -PEWY- span), which acted as a second H-bonding residue involved in formation of the ES-complex [34]. In this mechanism, oxidation of QH2 by H-transfer to ISPox would generate the QH. form of SQ. Glu-295 would facilitated H+ released by accepting a H+ from QH., and rotating to deliver the H+ to a water chain leading to the exterior, leaving Q.− as the mobile form. Hunte and colleagues [8, 65] have adopted a similar mechanism based on their yeast bc1 complex structure, which was the first to show the water chain we had suggested from MD simulations [36]. More recently, Osyczka et al. [1, 7] discussed possible mechanisms for prevention of bypass reactions. They concluded that none of those previously proposed would work, because any configuration of the site allowing occupancy by SQ under circumstances in which heme bL was reduced would give bypass rates similar to forward rates. We demonstrated that the alternatives discussed by them where physicochemically unrealistic, but that our earlier mechanism could function both to allow rapid forward electron transfer, and to limit bypass reactions by a coulombic gating [3, 5]. We showed that the behavior of strains in which Glu-295 was mutated were consistent with the mechanism proposed. However, we should note that several other groups have constructed similar mutations in other systems, and although the results are for the most part consistent between groups, the interpretations have provided a different gloss on mechanistic aspects [66–68]. This residue is not absolutely conserved, and none of the mutants is completely inhibited, and some (for example E295D) show rates as high as 20% wild-type, and these facets have been interpreted as showing that Glu-295 is not mechanistically important. Our own analysis of bypass reactions clarified the arguments of Osyczka et al. [1, 7], rejected their main conclusions, and provided severe constraints on possible mechanisms. The coulombic gating mechanism proposed involving Glu-295 offered a novel and successful resolution of the paradox they uncovered. A different gating mechanism was suggested by Mulkijanian [9], with alternative proton exit pathways, but a similar role for the glutamate. A role for glutamate in proton exit has been supported by the results of electrostatic calculations of different conformers at the Qo-site [69]. However, the site involves a metastable ES-complex, and because transitory states were not included in the calculations (which considered only the fully reduced or oxidized complex), the results can only be taken as providing an indication of potential protonation changes coupled to thermodynamically accessible states.
The question of the role of SQ in the Qo-site reaction has been controversial for many years. Suggested mechanisms have ranged widely, from expectations for a major fraction of centers occupied by SQ in the intermediate state [31, 55], through low occupancy models [25, 26], to scenarios in which no SQ was expected [1, 7]. In addition to the properties that led us to favor a low occupancy model [4, 5, 25], two other lines of evidence have strongly supported our suggestion that the SQ intermediate is formed at low occupancy by an endergonic proton-coupled electron transfer. Forquer et al. [70], in studies of by-pass reactions and super oxide (SO) formation, have confirmed that in yeast, as in the Rb. sphaeroides complex, the rate of the first electron transfer is determined by driving force (and is therefore the limiting process), and have shown that the normal forward reaction, and the formation of SO both have the same activation energy. Secondly, Cape et al.[71] have demonstrated that SQ is formed at the site at low occupancy (~0.01) under anaerobic conditions in the presence of antimycin. Zhang et al. [72] have also demonstrated a SQ signal with different line shape, also under anaerobic conditions, in a mutant truncated so as to remove heme bH, so that in each case electron transder out of the b-heme chain was prevented. Each of these observations is consistent only with the second of these models. Cape et al. also showed that the SQ is not in H-bonding contact with any N-atoms (excluding a complex with ISP), but was in a constrained environment (assumed to be the Qo-site volume), as expected from the Hong et al. [25] mechanism. The occupancy of SQ was at the high end of the range expected from the rate of bypass reactions, but the occupancy expected was based on an assumed reaction distance similar to that in the ES-complex. The appearance of SQ was not time resolved in either of these studies. However, it is clear from the very low rate of bypass reactions under normal forward electron transfer that the re-oxidation of SQ keeps the level much lower than this [5], and it seems probable that SQ appearance in these experiments required reduction of the b-heme chain by one or more turnovers of the Qo-site before SQ could accumulate. This is likely also the explanation for the discrepancy between the results above [71, 72] and the observation of Zhu et al. [73] in which no SQ was observed on initiation of turnover of the oxidized bovine complex by QH2 addition. The authors make no mention of redox conditions, but the complex was initially fully oxidized and no antimycin was added, and it seems likely that the conditions were aerobic. Under aerobic conditions Cape et al. did not see SQ, in line with previous reports [26, 74], and attributed this to reaction of SQ with O2. The failure of Zhu et al. [73] to observe SQ could therefore be attributed either its oxidation by O2, or rapid reoxidation by heme bL, and is as expected from all these prior reports. Zhu et al. [73] reported the kinetics of reduction of heme bL and ISP, and also showed raw data indicating the reduction of heme bH and the appearance of SQ at the Qi-site. For ISP and heme bL, 10% of the signal showed matched rates of reduction with t½ ~ 200 µs. This half-time would represent a rate ~4-fold faster than the fastest rates previous reported in native photosynthetic membranes [25], or in the isolated complex [54], from Rb. sphaeroides, and ~25-fold faster than rates of Qo-site turnover measured by stopped flow in the isolated bovine complex in the presence of antimycin. Taken at face value, the rapid and matched rates of reduction of ISP and heme bL might be thought to place more severe constraints on the possible occupancy of SQ at the Qo-site than those previously suggested [5], but the noise level apparent in the EPR data shown would preclude more quantitative estimation. One possible explanation for the initial kinetics seen by Zhu et al. [73] is that the initial reaction represents a reduction kinetics truncated by re-oxidation, since the complex was initially fully oxidized. Indeed, extrapolation of the initial rate shows 50% completion at 1.1 ms, suggesting that the initial rate was in line with that measured previously for the rate limiting first electron transfer reaction. If this is the case, and if a mechanism limited by the first electron transfer is assumed, the new data provide no tighter constraints than those previously estimated (see discussion above and [5, 26, 39, 54]). The lower range of plausible occupancies calculated was ~ 10−8 SQ/site [3–5, 25], and such levels are certainly not precluded by the Zhu et al. [73] results. Any estimate of occupancy less than the previously calculated upper limit would make it the more imperative to account for the rapid overall rate observed in terms of a rate constant for the second electron transfer faster than that possible if SQ had to react from the distal domain of the Qo-site [5]. No kinetic data in the presence of antimycin were reported by Zhu et al. [73], although effects of antimycin addition were discussed. De Vries et al. [75] had shown a faster rate of cyt bH reduction in the absence than in the presence of antimycin, which was insensitive to myxothiazol, but the data reported by Zhu et al. suggest a delayed reduction of heme bH and a simultaneous appearance of SQ in the absence of antimycin.
Mechanism of the Qi-site
Any mechanism must account for the fact that the Qi-site operates as an interface between the 1-electron chemistry of the b-heme chain, and the 2-electron chemistry of the quinone pool, and involves intermediate states tuned to binding of the three components of quinone redox chemistry, Q, SQ and QH2. Since the liganding requirements for these differ markedly, the site must be reconfigured during the catalytic cycle to accommodate these different requirements. In addition, pathways for uptake of two protons and for exchange of substrate and product need to be identified. It is generally supposed that the site operates through a two-electron gate in which heme bH reduces Q to SQ on one turnover of the Qo-site, and SQ to QH2 on a second turnover [12, 24].
The two-electron gate at the Qi-site
The reaction at the Qi-site has been treated as a simple two-electron gate [24]. However, it has long been recognized that even such a simple treatment has built-in complexities relating to the involvement of three different components, Q, SQ and QH2, as binding partners. The site has to accommodate each of these in any catalytic cycle, and each has its own requirements in terms of H-bonding [76]. The paradigm for discussion favored in the literature is in terms of the disproportionation reaction [77, 78]. Values for the differential binding of Q and QH2 and SQ stability are based on analysis of titration data from EPR measurement of SQ. However, a disproportionation is clearly not appropriate as a description of the catalytic cycle, because, on the time-scale of turnover, the site can only accommodate one species at a time, and the SQ is not exchangeable. Alternative approaches have been suggested, which attempt to reconcile the SQ data with the properties of the high-potential form of heme bH associated with the cyt b150 phenomenon [79–81]. Our recent efforts on the Qi-site have been directed at construction and characterization of mutant strains, and exploration of the local protein and solvent environment of the SQ form, since this information is accessible though pulsed-EPR [76, 82–85]. These have provided a detailed picture of local structure [76, 84], which, together with the available crystallographic data [41, 65, 86, 87], has allowed new insights to mechanism. The pattern of H-bonding interpreted from crystallographic data has varied between labs. Berry’s structures of the bovine and chicken mitochondrial complexes have shown a conserved His-201and Asp-228 as ligands to carbonyl O-atoms of a bound quinone species, and the most recent structures of the Rb. sphaeroides complex from Xia’s group, have also shown a direct ligand from the equivalent His-217. In structures of the yeast mitochondrial complex from the Hunte group, the H-bond to the histidine has been through a water molecule. In structures of mitochondrial complexes from Xia and collaborators, both H-bonds are modeled as through waters. Several of these structures are at close to atomic resolution, but because the quinone present can become reduced to SQ by X-ray exposure[88, 89], the nature of the occupant in these structures is ambiguous. Our own EPR results are interpreted as showing ligands equivalent to those seen in Berry’s structures (His-217 and Asp-252). However, MacMillan (personal communication) has reported that the Nε of histidine seen in our ESEEM spectra is not visible in his using the yeast complex. The difference may come from the protocol used for generation of the SQ, - reduction by ascorbate at pH 7.8–9.0 in our experiments, and by dithionite at pH 9.5 in MacMillan’s. The discrepancies between labs might also provide clues about variability in liganding of the three species occupying the site during catalytic turnover, and variability with pH on dissociation of liganding sidechains [76, 84], but we have not yet explored all these possibilities. Our conclusions are summarized in a scheme in which conformational plasticity allows the site to reconfigure to adapt to the binding requirements of the bound Q, SQ, and QH2 forms that participate in the reaction [76]. It is likely that the primary ligands, His-217 and Asp-252, act as proton exchange sites; His-217 lies at the surface, so provides a direct pathway, but Asp-252 is quite buried. Electrostatic calculations on the yeast complex suggest a strong coupling between residues equivalent to Asp-252 and Lys-251, other nearby lysines in cyt c1, and possibly bound cardiolipin. These are suggested to provide a channel for proton exchange with the aqueous phase [69].
Our more recent efforts have involved experiments to explore the N-environment, and the possibility of a third H-bond from Asn-221 (equivalent to Ser-205)[90]. In our previous work, no spin density on any additional N-atom apart from the histidine Nε could be detected using 14N X-band. From the 14N S-band and 15N X-band spectra, we have now determine the complete nuclear quadrupole tensor of the 14N, and the isotropic and anisotropic couplings, with the Nε of His-217. However, no significant spin-density on other N-atoms was detected. DFT calculations showed that the energies expected from the Asn-221 H-bond would be masked by the dipole-dipole signals from other N-atoms, leaving the question open. Several mutant strains at Asn-221 were constructed, were able to grow photosynthetically, and showed turnover of the Qi-site reactions, but with inhibition in rate, and modified properties for heme bH. We therefore concluded that the potential H-bond from Asn-221 is not essential, but is important for function. The characteristics of the weak H-bond observed through exchange with deuterium suggest that it involves a bound H2O, but some of the diagonal peak in 15N HYSCORE spectra could have been contributed by a H-bond from Asn-221. The most recent structures [87] include one showing a quinone occupant (PDB ID 2qjy), in which Nε of His-217 serves as a direct H-bonding ligand, as seen by EPR [90], no H-bond from Asn-221, and a distance to Asp-252 too great for a direct H-bond, but compatible with a water-mediated H-bond. The structure most likely reflects the occupancy by the oxidized Q.
The role of cyt b150 in the mechanism of the Qi-site
The component known as “cyt b150” is due to a perturbation of the properties of a fraction of heme bH on interaction with SQ at the Qi-site. It is seen in redox titrations of chromatophores [91], mitochondria [92] or isolated complexes [93], as a component titrating with Em,7 150 mV only in the absence of antimycin[81]. From the properties of cyt b150 we suggested that the Qi-site catalyzes the following reaction, in which the cyt b150 is represented by the state cyt bHH Q·− [2, 94]. The reaction (eq. 1) can be considered as a reversal of the normal forward reaction by which ferroheme bL reduces SQ to QH2, and, after release of the product, binds Q to re-establish the starting state for the Qi-site.
| (1) |
This overall reaction can be split into partial processes:
allowing definition at any defined pH, of an equilibrium constant for formation of the cyt b150 form:
| (2) |
where KA values are association (binding) constants for QH2 and Q, and Em(cytbH) and Em(QH·/QH2) refer to the mid-point potentials of cyt bH, and of the SQ/quinol couple bound at the site, respectively.
The affinities for Q and QH2 can be subsumed under terms for the potential of the quinone pool and the Q/SQ couple at the site, since the ratio of binding constants for Q and QH2 contributes to the value for Em(QH·/QH2) so as to cancel the ratio of binding constants. This gives:
| (3) |
(where Em(Q/QH·) refers to the bound couple), which can also be derived directly from eq. 1. A computer model based on this set of equations allows exploration of the variation of redox states of all components as a function of redox potential, pH, etc. In this treatment, no account is taken of different protonation states, local coulombic effects, etc., and these effects are subsumed under existing terms. However, the observed pH dependence of both the b150 and SQ titrations are handled by a single pK value, currently assigned to SQ (see below).
The mechanism proposed here is in contrast to the mainstream of discussion which starts from consideration of the disproportionation reaction as a basis for mechanism [77, 82, 95, 96]. Since structures became available, there has been little discussion of how this might occur at a site that appears too small to contain more than one species. The Qi-site catalyzes a quinol-quinone transhydrogenase reaction that allows transfer from exogenous quinol to bound quinone, or between exogenous quinones [97, 98]. A plausible mechanism would be for two electrons from a donor QdH2 to be stored in the two b-hemes, allowing dissociation of the Qd, and its replacement by an acceptor Qa, which would then leave the site as QaH2 [98]. The rate of such a reaction would depend strongly on the probability of populating the bH −bL− state. The unfavorable equilibrium constant for reduction of heme bL through bH [12, 99] would account for the relatively slow rate observed. Nevertheless, this would likely be sufficient to allow equilibration on the time scale of a redox titration. Equilibration through 1-electron transfer processes might also occur through direct or indirect interaction with mediators. Since the conditions used to assay semiquinone by EPR (high concentrations of redox mediators, and a long equilibration time) would allow the reaction to reach equilibrium, it is reasonable to use the data in discussion of mechanism, but it is highly unlikely that it reflects the kinetically important intermediate states.
Since the hemes are 1-electron acceptors, it is necessary to take account of the equilibration of the Q/QH2 couple with heme bH through 1-electron transfer reactions, - an attractive feature of the cyt b150 mechanism proposed. Reduction of heme bH through the Qi-site by exogenous quinones, with generation of a semiquinone, occurs on addition of quinol substrate to the oxidized complex in the presence of myxothiazol. In a definitive study by de Vries [75], reduction by duroquinol led to generation of ferroheme bH and SQ simultaneously, with the reaction ~90% complete at the first point of measurement at 5 ms (using 300 µM QH2). From this, a value of τ < 3 ms seems appropriate. In chromatophores, where the reactions using the native substrate can be followed in situ, the myxothiazol insensitive reduction of heme bH on generation of 1 QH2 in the oxidized pool also occurred with τ ~ 3 ms [80]. Since this is ~two orders of magnitude faster than the transhydrogenase reaction, the disproportion reaction is excluded. It seems likely that duroquinol, and presumably other quinols, can react directly and rapidly at the Qi-site by exchange with the initial occupant.
Rich et al. [96] have discussed the cyt b150 phenomena in terms of differential affects of occupancy by Q, SQ or QH2 on the Em of heme bH (see also [82]). A set of 7 different Em values and 2 pKs was required to account for the properties of both the SQ and cyt b150, but these parameters did not include the possibility for differential binding of the different components. Furthermore, in deriving this set, Rich at al. [96] had to assume that SQ generated in the presence of oxidized heme bH was EPR silent due to magnetic quenching (see also [100]). If the semiquinone signal at the Qi-site is modulated by magnetic interaction with the spin of oxidized b-heme [100, 101], this would lead not only to loss of amplitude, but also to some displacement of the maximum of the bell-shaped titration curve [96], and hence to misleading values for derived thermodynamic parameters. There is no doubt that occupancy by inhibitors can modify properties of heme bH, and a substantial literature showing changes in both Em and λmax, but the complexity of the Rich et al. treatment makes it somewhat intractable to testing. In contrast, the model proposed here has only a single equilibrium constant, and singular Em values for heme bH and the quinone system taken directly from experiment. Computational modeling shows that the properties of the cyt b150 phenomena are reproduced quite well using measured values for Em for heme bH, values for binding constants and Em values for the Q/SQ/QH2 system derived from redox titrations and kinetic experiments. The properties of the SQ are also reproduced quite well, but are mechanistically linked to formation of SQ through reaction 1 above, so that the SQ observed is in the form cyt bHH.Q·−. An interesting feature of these simulations was that, because of the importance of Em of heme bH and the Q/SQ couple in eq. 3, the pH dependence of both b150 and the SQ were well explained, but are determined by a single pK. Previously we have used the published pK at ~7.8 on the heme bH. However, we have recently revisited these titrations (see below) and now prefer a pK in this range on the semiquinone, or associated with interaction with the protein (cf. [69]).
The forward reaction
The mechanism for formation of the cyt b150 state provides the main elements of a mechanism for the forward reaction [2]. Additional parameters needed are those for reduction of Q to SQ by electron transfer from heme bH. The fact that the observed rate is that of the limiting step at the Qo-site restricts use of the kinetic data to estimation of lower limits for the forward rate constants. However, the equilibrium constant can be estimated from the kinetics with the quinone pool initially oxidized (and the site presumably occupied by Q). After flash activation to populate the ferroheme bH (generated as the one QH2 in the pool produced by excitation of the RC is oxidized through the Qo-site), the heme remains partly reduced in the 100 ms range, suggesting an equilibrium constant for reduction of Q by heme bHH with Keq ~ 3. This relatively stable level of heme bH reduction titrates away as the pool starts to become reduced over the range below Eh,7 150 mV. This is the range over which the cyt b150 state is populated, so the acceptor at the site will change from Q to SQ over this range. A mechanism for the forward reaction can then be formulated, as discussed more extensively elsewhere [2].
Can a monomeric mechanism account for the observed functional data?
The structures all show a dimer, which raises the main question to be discussed in the remainder of this paper. What do we know of the consequences of this dimeric nature? Are there mechanistic parameters dependent on interactions across the dimer interface?
The structures now available (including several structures from two Rhodobacter species [31, 32, 87]) have demonstrated the high degree of conservation of catalytic subunits across the bacterial/mitochondrial divide, highlighted the dimeric nature of the complex, and provided a framework for extensive discussion of the role of the dimer in mechanism [6, 7, 53, 102–106]. The attractiveness of the modified Q-cycle treatment rests on the fact that the properties can be related to a few constants, measured directly from thermodynamic or kinetic experiments on the intact membrane systems, or calculated from structural data. It is appropriate to emphasize that the kinetic data for native and mutant strains accumulated over the past 25 years have allowed us to subject the modified Q-cycle model to detailed analysis in terms of stoichiometric ratios of reactants, the equilibrium constants determining their distribution, and the kinetic constants determining the time course of changes on introduction of substrates [12, 37, 107–110]. Because the system can be flash-activated, no mixing of substrates is needed, and as a consequence, the initial state of the system can be manipulated over a wide range of Eh and pH to change substrate concentrations, thermodynamic poise, etc. These data are satisfactorily and economically explained by the monomeric modified Q-cycle. No ad hoc intrusions spoil the simplicity; - there is no need to introduce secondary interactions between sites, or actions-at-a-distance, beyond those introduced by simple coulombic considerations. The mechanism is completely natural, - a conclusion reinforced by more recent data in which kinetic contributions were more accurately resolved by deconvolution, allowing finer points to be explored [111–113]. This is in contrast to schemes introduced by other groups, in which mechanistically occult interactions between catalytic centers, or between reaction intermediates and redox states, have been proposed, which have been given prominent controlling roles [10, 53, 103, 104]. These speculative interactions are based on interpretation of kinetic experiments, usually using an isolated mitochondrial complex in stopped-flow mixing protocol. Although this approach has been applied with skill, intrinsic difficulties limit the utility of some experiments. These arise from the nature of the system, - the experiments require the detergent solubilized complex, and use of more soluble quinol substrates than the native ubihydroquinone-10. As a result, the binding of QH2 leading to formation of the ES-complex is always unnatural. Even with decylUQH2 or analogs with truncated isoprenyl sidechains, binding involves partition from a mixed aqueous and micellar phase in which the quinol has a limited aqueous solubility [114] (CMC ~30 µM). Often, quinones without the lipophilic side chain, and with very different redox properties (menaquinone or duroquinone, for example) have been used, and these differences change both the kinetics of binding and the equilibrium constants for partial processes, with sometimes dramatic kinetic consequences ([115] and Kokhan, O. and Wraight, C.A. personal communication ), so that the observed effects often differ from those seen in situ. Because of the limitations of stopped-flow, one has to be careful in interpreting the results, - in particular when using them to justify hypotheses about the physiological mechanism. There is a strong temptation to presume that any kinetic effect that cannot be understood in terms of the simple model must reflect some new twist to the Q-cycle theme, and this has encouraged much speculation centered on “hidden” interactions. Unfortunately, the scope for speculation increases dramatically, with a power-law dependence on the number, n, of such interactions proposed, further compounded by a square function if interactions between monomers in the dimer are considered.
We will take as a starting point for discussion the mechanism summarized in Fig. 1, and examine the hypothesis that this minimal model of a monomeric mechanism [2–5] is sufficient to account for the metabolic role of the bc1 complex. Although the structure is dimeric, the model can accommodate those features from the structural evidence that involve the dimer in a scaffolding role. The iron-sulfur subunit of one monomer is structurally constrained by specific interactions at the membrane level, involving a clamp between cyt b subunits, and interactions with other subunits mainly in one monomer, which leave the extrinsic mobile head free to interact with catalytic interfaces in the other monomer. This dimeric involvement could in principle account for the dimeric nature without any extraneous functional role. However, several labs have claimed to demonstrate inter-monomer electron transfer [6, 53, 103], and others have assumed on the basis of strong theoretical grounds that this must occur [7]. Furthermore, a substantial body of evidence has shown that binding of antimycin modifies the interaction of ISP so as to change the susceptibility to proteolytic cleavage [116], and recent results from Daldal’s group have shown conditions in which the g-tensor of the [2Fe-2S] cluster appeared to change with respect to the orientation of chromatophore films on binding of antimycin [102]. Both these latter effects are discussed in the context of a modified interaction with the Qo-site occupant, reflecting a coupled conformational change induced by antimycin binding at the Qi-site.
As several commentaries have noted [7, 27, 53, 117], the structures show distances between the bL hemes across the dimer interface that are expected to facilitate rapid electron transfer. These are Fe-Fe at 21.58 Å, nearest conjugate atoms at 10.54 Å (vinyl sidechains), and pyrrole rings at 14.45 Å (in 1pp9) (Fig. 3A). Values for rate constant depend on which distances is used, and on whether a classical Marcus [118, 119] or Moser-Dutton [120–124] treatment is used for the exponential term. Some representative values for the competing pathways are shown in Table 2. If the appropriate distance is that between conjugate systems, it would likely include the vinyl sidechains, the path assumed in the Table. The values for ΔG chosen depend on the redox status, and hence on any effect of coulombic interactions between hemes. For example, if heme bH is reduced in one monomer, the coulombic effect on bL will be about −60 to −80 mV, generating an exergonic pathway for electron transfer to the neighboring heme bL, if the heme bH in that monomer is oxidized (bottom line). Note however that all bL-bL rate constants would allow rates more rapid than the rate-determining step (~1.35 × 103 s−1) [25, 26]. A succinct review of this problem by Shinkarev and Wraight [125] is couched in terms of competition between the heme bL → heme bH, and heme bL1 → heme bL2 paths. In assigning rate constants, they assumed the appropriate distance was that between conjugate rings, (14.45 Å), but since the vinyl groups are part of the conjugate system, it does not seem natural to exclude them, and this increases rate constants by a factor of ~200. Then, although competitive effects could lower expected rates as they suggest, under conditions in which both hemes bH are oxidized, no combination of effects within the conventions of the Moser-Dutton treatment of the intrinsic rate constant would prevent electron transfer across the dimer interface at rates in the range of the limiting rate once the “active side” bH became reduced. The expectation is therefore that electrons could be shared between monomers at this level, and that this pathway would be functional, and easily observable on the timescale of ~100 ms used in typical experiments.
Fig. 3. Schemes to show potential electron transfer pathways involved in electron equilibration between monomers at the level of heme bL.

A. The redox centers of the dimer are shown, and the Qo- and Qi-sites indicated by stigmatellin and antimycin, respectively. The scheme shows the dimer with the Qi-site blocked in both monomers by antimycin (blue crosses), and the Qo-site of one monomer blocked by myxothiazol (red cross). Under these conditions, if electron transfer between hemes bL could occur, both hemes bH could be reduced by turnover of the uninhibited Qo-site (broken red arrows). B. The dimer interface, showing residues referred to in the text. The dotted line shows the 10.21 Å distance between heme bL 4-vinyl groups (taken from PDB file 2qjy, chains A–F).
Table 2.
| Reaction pairs and conditions | Moser-Dutton k/s−1 | Marcus k/s−1 |
|---|---|---|
| bL-bH 2-vinyl (assuming ΔG = −60 mV, λ = 0.75, R = 7.02 Å) | 8.97 × 108 | 1.73 × 108 |
| bL-bH 2-vinyl (assuming ΔG = −130 mV, λ = 0.75, R = 7.02 Å) | 2.15 × 109 | 5.68 × 108 |
| bL-bL 4-vinyl (assuming ΔG = −0 mV, λ = 0.75, R = 10.54 Å) | 2.86 × 106 | 4.08 × 105 |
| bL-bL 4-vinyl (assuming ΔG = −60 mV, λ = 0.75, R = 10.54 Å) | 6.51 × 106 | 1.25 × 106 |
Experimental evidence in support of dimeric operation has come from several studies. Covian et al. [126] interpreted stopped-flow kinetic experiments as showing that the stoichiometry of cyt c1 reduction on addition of decylUQH2 was half that expected from the total complement, and that only half of the complex present participated in electron transfer. Under these conditions, two cyt b hemes became reduced per active enzyme in the presence of antimycin (although no effort was made to distinguish between hemes bH and bL). These properties were similar to previously observed kinetic anomalies, which were explained by the modified Q-cycle. For example, on flash activation in the presence of antimycin, a discrepancy between amplitudes of heme b and heme c reduction is observed, and had been seen as contrary to the expectations of a Q-cycle [127]. However, the modified Q-cycle requires two turnovers of the Qo-site for a full turnover of the complex, and detailed analysis in the early 1980s showed the equilibrium constant for these depends on the acceptor. The value depends on Em for heme bH on the first turnover (Keq~480), and that of bL on the second turnover (Keq~3), and the degree of reduction of the high potential chain is simply a reflection of this change in the nature of the acceptor [12]. However, Covian et al. [126] explained their results in terms of an anti-cooperative mechanism in which the Qo-site of only one monomer was functional, but fed into both sides of the dimer through electron exchange at the heme bL level. In support of this, they showed titration curves with antimycin of the initial rate of cyt c reduction in a steady-state assay of activity that gave a convex curve, with a stimulation of rate in the sub-stoichiometric range. In a later paper [6], the same group reported titrations by antimycin of cyt b reduction on addition of decylUQH2 to the myxothiazol inhibited complex, in which the titration curves showed a convex shape. They again interpreted the results as showing electron transfer between the bL hemes.
Although such convex curves might be expected from electron sharing, a substantial literature from the 1970s, following the observation of Kröger and Klingenberg [128, 129] of hyperbolic titration curves with antimycin resulting from a “pool” effect of ubiquinone, demonstrates that alternative explanations are possible. In general, a Kröger-Klingenberg effect will be found whenever the turnover of the process being titrated is greater than that of the rate-limiting reaction, since a fraction of centers can be eliminated without a full effect on the overall process. We have been able to generate convex titration curves under a variety of such conditions with chromatophores ([130], and in collaboration with Vlad Shinkarev), or with isolated bc1 complex. However, when care was taken to ensure that the rate-limiting step was the process titrated, linear curves were always seen. Indeed, such linear titrations are the norm in the literature. Several other mechanisms can give non-linear titration curves, as shown on titration of the amplitude of cyt c1 + c2 oxidation following flash activation in chromatophores [130], where the degree of convexity is dependent on which time-point in the kinetic curve is chosen. This is because mobility of the substrates QH2 and cyt c2 allows one active complex per chromatophore to re-reduce all cyt c2, given time. This effect has an n-value from Hill plots of ~ 9, likely reflecting the number of bc1 complexes per chromatophore. Similarly, titration by antimycin of the kinetics of cyt bH reduction and re-oxidation shows a similar effect, possibly, as suggested by Bechmann et al. [131], because antimycin can leave one Qi-site and find another, so long as one remains open. In the isolated complex this gave an n-value of 2, but we found a higher value in chromatophores (>4.9) (Fernandez-Velasco and Crofts, unpublished), which suggests that the inhibitor can redistribute as long as an open site is available in a chromatophore. In both cases, linear titrations are seen under conditions in which the rate limiting step was titrated.
| (4) |
| (5) |
In the work from Covian and colleagues, the case for an interpretation in terms of electron exchange between monomers depended on a kinetic model in which a questionable value for the equilibrium constant between hemes bH and bL was used [6, 126]. The treatment involved a reaction equation, bH− + bL ⇆ bH + bL−, appropriate for the reaction in solution. Then a condition [bH−] = [bL], and [bH] = [bL−] was imposed because the actual reaction was intramolecular (eq. 4), and this changed the equilibrium constant determining occupancy of the bL− state by 18.6-fold (compare eq. 4 and eq. 5). The value derived allowed sufficient occupancy of the bL− state to provide an inter-monomer rate that explained the observed rate. Without the larger value for Keq their model fails to match the kinetic data. The treatment is in error on both mechanistic and thermodynamic counts; the reaction is not 2nd order, and by assigning separate identities to four reactants, they are trying to have their entropic cake and eat it too. Intramolecular electron transfer is an exchange between two states in which the electron is on either one or the other center, and treating the system as 1st order gives the conventional equilibrium constant expected from a one-electron transfer. For the transition bH−bL ⇆ bHbL−, Keq is determined as in eq. 5. Any alternative path with a different equilibrium constant would make possible a perpetual motion machine. Because the experiments of Covian et al. [6, 104, 126] did not discriminate between several possible mechanisms, and because their model is suspect, it can hardly be taken as established from these results that the mechanism is dimeric, or that electron exchange must occur between monomers on the timescale of turnover.
Gong et al. [104] have constructed mutants in Rb. sphaeroides with changes at three aromatic residues, Phe-195, Tyr-199, and Phe-203, which, on the basis of a computer model, were expected to line the dimer interface between the bL hemes. They studied steady-state electron transfer and SO production in bc1 complex isolated from variants in which each residue was changed to alanine, with some additional changes at F195. The electron transfer rates reported (2.5 µmol cyt c reduced /nmol heme b/min or 21 mol/mol/s for the wild-type) were only ~1.5% physiological rates (1,350 mol/mol/s at saturating substrate), indicating that the turnover was substantially inhibited in all these preparations. None of these mutations had a strong inhibitory effect. The greatest effect was seen when Phe-195 was changed; F195A had a lower rate (80% of WT) than F195W, F195H or F195Y, and a higher rate of SO production. On this basis, the authors concluded that an aromatic residue was required, and that the critical function was electron transfer between the bL hemes. However, this electron transfer path was conjectural; no evidence was presented showing that any such reaction occurred, and the effects could as well have been attributed to a weak inhibition of the Qo-site, which (on the basis of myxothiazol occupancy) is 14–19 Å from these residues. In structures now available, only Tyr-199 is in the most direct path, whether interpreted as through bond or through protein. We discuss below our own results based on a series of mutations at this residue, Y199T, S, C, W and L, and show that these are clearly against an inter monomer exchange.
In another challenge to the modified Q-cycle, Mulkidjanian [9, 10] has suggested an “activated” Q-cycle with a number of novel features, at least some of which seem untenable. He attributed the acceleration of the rate of QH2 oxidation in the range ~Eh 150 mV to a priming process involving formation of SQ at the Qi-site (the cyt b150 effect). However, the same acceleration over this Eh range occurs in the presence or absence of antimycin, which specifically eliminates the Qi-site SQ, so this must be wrong. It had previously been demonstrated that this acceleration was due to an increase in [QH2] on reduction of the Q-pool, and preferential binding of QH2 at the Qo-site [12]. Another odd experimental result under special conditions was a marked difference in rates of turnover of the Qo-site in the presence or absence of antimycin. However, this contradicts studies from a number of labs [12, 37, 132–135] showing the rates to be the same under these two conditions. It seems likely that these aberrant observations reflect a lack of technical finesse rather than real functional differences.
Other groups have suggested control functions based on conformational coupling linked to structural changes on occupation of the Qo-site by different redox species [39]. However, the causal relationship proposed could be as well explained by dynamic flexibility to accommodate different reaction partners [40].
Myxothiazol titrations to test for electron transfer between monomers
In the presence of antimycin, oxidation of heme bH is prevented, so electrons passing out of the Qo-site accumulate in heme bH. In chromatophores, the stoichiometry of reaction centers to bc1 complex is about 2:1, ensuring that sufficient substrate (2 cyt c2 + plus 1QH2) is generated on a saturating flash to allow one full turnover of the complex. In the presence of antimycin, the two oxidizing equivalents in the high-potential chain could in principle oxidize 2 QH2, with reduction of hemes bH and bL. However, the equilibrium constant for the second turnover, resulting from the low Em (~−90 mV) of heme bL, prevents this, restricting the complex to 1 turnover of the Qo-site [24, 109]. If inter-monomer transfer could occur [27, 53, 104, 117], electrons from either Qo-site in the dimer could reduce both hemes bH. Even with one monomer per dimer inhibited at the Qo-site by myxothiazol, full reduction of heme bH should be observed, because both bH centers would operate at the same higher Em (~40 mV), so that the equilibrium constant would strongly favor reduction. In practice, statistical effects would lead to a bowed titration curve (for example, at 50% occupancy, the distribution of dimers expected is 25% each of 0,0; 0,1; 1,0; and 1,1, where 0 and 1 are vacant and myxothiazol inhibited sites, so 75% of the full reduction might be expected). This convex titration curve is not seen. This could possibly be attributed to some special role for the intervening protein. We have constructed mutant strains in which the residue at the contact point between monomers at the level of heme bL, Tyr-199, was modified to give Y199W, T, S, L, and C. Since this residue is next to His-298, a ligand to heme bL, and lies on the most direct path of electron transfer between hemes bL (Fig. 3B), we anticipated substantial effects when inter monomer electron transfer was probed by titration with myxothiazol (Fig. 4). Fig. 4 shows the results observed. All strains showed similar rates for Qo-site turnover in the absence of myxothiazol. None showed any marked change in redox properties of the cytochromes (Table 3), although all showed small changes in the α-band spectrum of heme bL. Titrations for all strains (including WT) were strictly linear, whether measured through initial rate, or amplitude of reduction, or after 1 flash or two (Fig. 4B). Since the second flash leads to reoxidation of the high potential chain, the driving force for inter-monomer electron transfer would be substantially increased, and an “extra” reduction of heme bH would have been expected if electron transfer between the bL hemes could occur, but this was not observed. Instead, heme bL went reduced after the second flash, as expected in the monomeric Q-cycle [12]. Furthermore, no indication of the biphasic character expected from a branched pathway was apparent in the kinetic curves. We also tested these properties in a strain (H198-T199-Y200) constructed so as to mimic the extra residue in the sequence at this point found in the cytochrome b6f complex of the cyanobacterial/chloroplast photosynthetic chain (with sequence –HTF-) [136]. Again, myxothiazol titrations were linear, although the structures now available show a distinctive change in configuration of the interface in the b6f complex compared to the bc1 complex, which might also have been induced by the mutation. The simplest interpretation of these results is that no inter-monomer electron transfer occurred, despite the short distance between the hemes and the rapid rate constant expected.
Fig. 4. Titrations of Qo-site with myxothiazol showing linear curves in WT and mutant strains.


a) Typical curves showing the kinetics of heme bH reduction in WT in the presence of antimycin. Similar sets of data were taken for each strain, all of which had similar maximal rates. Myxothiazol concentrations are indicated to the right of each curve. b) Fraction of heme bH reduced at 20 ms (expressed as %) in the presence of Qi-site inhibitor antimycin A, as a function of inhibitor concentration. The system was excited with a xenon flash (~5 µs at half-height) and the reduction of bH monitored from the difference kinetics measured at 561–569 nm. Ambient redox potential was poised at ~100 mV (QH2 oxidation close to maximal). Strains are indicated by labels; bc1 complex in the range 70–220 nM.
Table 3.
| Strain | Redox Potentiometry Em, (V) | Nernst Equation Solution Fitted at Each Wavelength of the Spectrum, Em, (V) | ||||||
|---|---|---|---|---|---|---|---|---|
| bL | bH | b150 | cyt c1 | bL | bH | b150 | cyt c1 | |
| WT + antimycin | −0.113 | 0.049 | --- | 0.250 | −0.090 | 0.040 | --- | 0.250 |
| WT | −0.120 | 0.054 | 0.169 | 0.250 | −0.090 | 0.040 | 0.150 | 0.250 |
| Y199L | −0.123 | 0.038 | 0.149 | 0.259 | −0.100 | 0.030 | 0.148 | 0.250 |
| Y199S | −0.077 | 0.057 | 0.172 | 0.265 | −0.094 | 0.053 | 0.170 | 0.260 |
| Y199T | −0.108 | 0.045 | 0.160 | 0.262 | −0.092 | 0.027 | 0.152 | 0.250 |
| 199-insert | −0.096 | 0.035 | 0.155 | 0.251 | −0.092 | 0.033 | 0.135 | 0.250 |
| Y199W | −0.080 | 0.075 | 0.170 | 0.237 | −0.091 | 0.050 | 0.150 | 0.250 |
The above conclusion presents an interesting paradox. From treatments using the Moser-Dutton approximation for intrinsic rate constant at this distance, electron transfer should occur, but the experiments show that it does not. If unequivocal evidence becomes available showing that electron-sharing occurs, then the data interpreted as showing that it does not will have to be re-evaluated. If on the other hand, no such evidence is found, then it will be necessary to explore the possibility that other parameters must be important in determining electron transfer rate, over and above those in the Moser-Dutton treatment. Either result would be of interest, and advance the field.
Several possibilities look interesting. The Moser-Dutton treatment uses the Hopfield approximation [119] to allow for quantum mechanical effects, but this effectively modifies kB, giving rise to thermodynamic inconsistency in applying the Marcus term to the Arrhenius expression [137]. From the Marcus curve generated using Moser-Dutton value of 3.1, values for log10k at any chosen ΔG appropriate for a forward reaction, and for a reverse reaction at a value taken symmetrically with respect to ΔG = 0, do not give the ratio expected from the relation of ΔG to Keq. In the Moser-Dutton treatment of the intrinsic rate constant, the approach is implicitly classical, since fluxes through all pathways are additive, and distance and density are taken to provide all information pertinent to the probability of electron transfer through the protein matrix. Alternative approaches have treated the intrinsic rate constant quantum mechanically [119, 138]; then a standard path integral treatment opens the possibility of destructive interference, so that, in this scenario, interference in the inter-heme bL path could limit rate. Indeed, several recent papers [139–143] have considered more sophisticated treatments that include interference terms. In a few cases, especially those involving pathways including heme and the liganding histidine, destructive interference was found to play an important role. Additional complications arise in systems involving electron transfer between subunits [142]. An more conventional approach is in terms of the electron distribution on the hemes. If the electron distribution at the 2-vinyl position is more favorable than at the 4-vinyl, this would favor electron transfer from heme bL to bH. Walker [144, 145] has kindly provided useful insights into the electronic structure as determined by calculation and EPR, and has provided data showing that the β-carbons of the 2-vinyls of the ferrihemes should both have substantially higher spin occupancies than those of the 4-vinyls (Ann Walker, personal communication). Since rate depends on k[occupancy], the bL→bH pathway (in which the 2-vinyls are in the shortest path) should be favored.
Coulombic interactions in the dimer
When the bc1 complex was mutated to replace one of the histidine ligands to heme bH, the complex was still assembled with a full complement of redox centers, except for that heme. The complex retained a fully functional Qo-site reaction, but turnover was restricted by the availability of heme bL as the only low-potential chain acceptor [146]. Potentiometric titration of the hemes showed that the Em, 7 values for heme bL were changed from ~ −90 mV in wild type to −8 mV (in H111N) or −14 mV (in H212D), an increase of ~80 mV. We suggested that this change could be explained by loss of a coulombic interaction on loss of the negative charge of heme bH, which, with Em ~50 mV, would be reduced over the range in which heme bL titrates. If this speculation is accepted, the value and the distances shown by structures would allow calibration of other coulombic interactions in the dimeric complex, and construction of the curves like those shown in Fig. 5. From this, it is evident that substantial coulombic effects might be expected both within the monomer, and across the dimer interface. Such interactions have been extensively investigated in other systems. In order to explore these possibilities, we have re-evaluated the thermodynamic properties of the b-hemes by analysis assuming such effects.
Fig. 5. Coulombic free-energy change expected between hemes.


The change in Em expected from coulombic effects between hemes calculated from distance apart, for different intervening dielectric values.
Thermodynamic properties of the b-hemes
Titrations were performed using full spectral analysis over the α- and β-band range, long equilibration times between measurements, and relatively high concentrations of mediators. Typical curves are shown in Figs. 6 (wild type) and 8 (mutant N221I) [147]. Space does not permit a full discussion, but several interesting features emerge from this new analysis.
Fig. 6. Thermodynamic behavior of the b-type hemes in H6B strain (his-tagged cyt b with wild-type sequence) in situ.

(Top left) typical titration data in the absence and presence of antimycin (pH 7); (bottom left) spectra from redox cuts showing cyt b150, heme bH and heme bL; the dashed line shows the curve expected for an n=1 component, with amplitude for degree of reduction plotted as (Bottom, right) spectra derived by fitting titrations at each wavelength using fixed values for Em, and n, for each component; (top, right) the pH-dependence of Em values for individual components, assuming single species for each component.
i) The pK at 7.8 previously observed is not seen [91, 92]. It seems likely that in previous analysis, titration curves for heme bH were convoluted with cyt b150 and the pH dependence of its amplitude (Fig. 6). Two brief reports suggests that the same is true in Rb. capsulatus [7, 66].
ii) A second new observation is the apparent independence of pH for the Em values for heme bH in the presence of antimycin.
iii) Finally, in fitting the curves, especially those for bH in the presence of antimycin, and for bL, we found that n-values less than 1 often provided better fits (Fig. 6, top left). When n=1 was imposed, analysis of the curves into two components was required for a good fit (Fig. 7).
Fig. 7. Reanalysis of the thermodynamic behavior assuming the coulombic effects are in play.

Titration data for heme bH in the absence or presence of antimycin were assumed to reflect two components split by coulombic interaction.
Because small changes in the extremes of the fitted curves can give quite large differences in fitted values, the data presently available are not accurate enough in themselves to provide unambiguous values. Furthermore, analysis was complicated by the presence of additional components from other redox complexes, most notably a heme with a spectrum similar to heme bH titrating in the range of the cyt b150 component, which was not observed in titrations of the isolated complex, but which contributed 10–15 % of the measured change in chromatophores. With these necessary caveats, when an analysis of heme bH was performed, assuming that each component of the bc1 complex was split, some additional features became apparent (Fig. 7). The titration was resolved into two components in the presence or absence of antimycin, each showing the same pH dependence. The lack of pH dependence for heme bH in the presence of antimycin disappeared, and was seen to reflect a pH dependence of the amplitude of a higher potential component. The higher potential component in the presence of antimycin had a lower Em than the cyt b150 component, and so merged with the low Em component to give what appeared to be a single component. We hesitate to put too much emphasis on this reanalysis, but it suggests two populations with the same spectrum titrating with different Em values, which is the behavior expected from two hemes in identical environments interacting through coulombic repulsion. Clearly, a more detailed analysis with full electrostatic calculations (cf. [69]) is needed, since the local fields and dielectric properties are not well represented by mean values assumed in Fig. 5.
Asn-221 mutants
Similar experiments with strains N221T, H, I, S, P and D have shown dramatic effects on the thermodynamic parameters measured in the presence of antimycin. Effects of mutation are also reflected in major changes in kinetics. These experiments are still in progress, but example data have been reported elsewhere [90, 147], and are summarized in Table 4.
Table 4.
| Kinetic characteristics of strains with Asn-221 of cytochrome b modified | ||||||||
|---|---|---|---|---|---|---|---|---|
| Strain | Photosynthetic growth | Reduction of bHa | Fraction of bH reduced b | Em,7, pH dependence d | Reoxid. (t½/ms) e | |||
| cyt b150 | bH, no inhib. | with AA | ||||||
| Wild type | +++ | 455 | 0.05 | 148, −59 | 32, −59 | 46, 0 | <1.6 | |
| N221D | +++ | (as wt) | 0.8 | 150 −59 | 35, −54 | −47, −59 | 15 | |
| N221H | +++ | 520 | 0.68 | 150, −59 | 35, −59 | 13, −50 | 12 | |
| N221P | +++ | 442 | 0.167 | 154, −59 | 38, −59 | −3, −59 | 2.0 | |
| N221T | +++ | (as wt) | 0.37 | 140, −59 | 29, −50 | 13, 59 | 8.0 | |
| N221I | +++ | (as wt) | 0.75 | 104, −59 | 36, −59 | −95, −59 | 14 | |
Notes: Rate of reduction of heme bH measured following a 5 µs flash at Eh ~120 mV in the presence of antimycin (units – mol cyt bH /mol bc1 complex / sec).
Fraction of heme bH reduced at the maximal amplitude (at ~3–5 ms) of the kinetic trace in the absence of antimycin, compared to the fraction reduced in the presence of antimycin.
Rate of reoxidation of heme bH in the absence of antimycin, measured by subtraction of the traces in the presence and absence of antimycin (units – mol cyt bH /mol bc1 complex / sec). The rate in the wild-type reflects the rate-limiting reduction of the low potential chain through the Qo-site reaction. Since v = k(occupancy), and the fraction of heme bH in the reduced form (column 4) is <<1, the rate constant must be much greater than that for the limiting step.
Em,7 in mV, pH dependence of Em in mV/pH unit, assuming a single component for each heme.
Half-times for reoxidation of heme bH measured from kinetic traces.
Several interesting effects can be noted in these data. In all Asn-221 mutant strains, the redox properties of both b-hemes (and the cyt b150 component) were relatively unaffected in the absence of antimycin, but Em values for heme bH were shifted to lower values in the presence of antimycin, quite dramatically in some strains. However, in the presence of antimycin, the electron transfer rates to the bH heme were not markedly changed, indicating that turnover of the Qo-site was unaffected. All mutations showed some inhibition of electron transfer from heme bH to the acceptor at the Qi-site in the absence of antimycin. Inhibition was apparent both at Eh ~200 mV, where the acceptor would be Q, and 100 mV, where SQ is likely the acceptor in WT. Titrations of SQ have shown similar values to wild type for amplitude and peak of the bell-shaped titration curve in strains so far tested (N221P and S), so we have no reason to believe that the SQ stability is markedly changed. For several strains, rates were substantially slower than wild-type. Surprisingly, N221P, in which H-bonding potential is lost, showed the least kinetic effect.
In the presence of antimycin, all strains showed changes in thermodynamic properties of heme bH (but not bL). When analyzed according to conventional single component titrations, all mutants showed a dependence of ~−59 mV/pH. For several strains, the changes in Em in the presence of antimycin were quite dramatic, as seen in N221I (Fig. 8). This can be seen most easily through the difference spectra in Fig. 8c. In the absence of antimycin, the difference ΔA for the redox cut between Eh,8.7 values −380 and −40 mV shows a spectrum of heme bL similar to that over the same range in wild type. On addition of antimycin, an additional component appears in this difference spectrum, shown as the antimycin induced change, which has the spectrum of heme bH, but with an Em shifted to the same range as heme bL. As with wild type, the titration data in the mutant strains were not well fit by single components of n=1 (Fig. 8b), and this was also obvious in titrations of heme bL (not shown). Because in some mutants, the two hemes showed similar Em values, unambiguous resolution of the different components was difficult. The effects of the lowered Em were apparent in kinetic data, so that in the presence of antimycin at ambient potentials at which the quinone pool was oxidized enough to sustain turnover (Eh,7 >40 mV), and where both hemes were expected to be substantially oxidized, reduction of components with both spectra was observed on flash activation. This is in contrast to wild type, where reduction of heme bL was observed only when heme bH had been pre-reduced before the flash [109]. The interpretation of these results is again ambiguous, but we favor a picture in which coulombic interactions can occur both within the monomer, and across the dimer interface, as shown schematically in Fig. 9.
Fig. 8. Titration of heme bH in chromatophores from N221I in the presence and absence of antimycin.

A (top), titration curves showing shift in Em on addition of antimycin; B (middle), titration with antimycin fit by 2-component, n=1; C (bottom), absorbance spectra showing the difference (−380)-(−40) mV from pH 8.7 titration. The difference with and without antimycin shows the contribution of heme bH with its low potential in the presence of antimycin.
Fig. 9. Scheme to show some of the coulombic interactions suggested.

The dimer is represented by four Q-sites (circles) with antimycin blocking Qi (blue cross). The hemes are shown as horizontal bars, either empty, or occupied by an electron (red dot). Top, in wild-type, heme bH has Em,7 ~50 mV, heme bL has in intrinsic Em,7 ~−30 mV, but titrates with Em ~−100 mV because of coulombic interaction with heme bH.
Bottom, in N221I, both hemes have the same intrinsic Em,7 ~−30. The first electron can go to either, and change the Em of the other.
For clarity only the coulombic interactions within a monomer are shown However, the titrations suggest that additional interactions across the dimer interface may also occur.
It will be obvious that detailed characterization of these strains, especially with respect to the SQ properties, will provide parameters suitable for testing the hypothesis proposed for cyt b150 formation. The dramatic effects of antimycin in the heme bH properties will also help in understanding how binding can modify these parameters. A comprehensive review of structural aspects of antimycin binding by Huang et al.[86], the electrostatic calculations for the yeast complex [69], and the recent higher resolution structures of the Rb. sphaeroides complex [87] have provided a firm basis for discussion. Since the N221P strain showed minimal effects on function in the absence of antimycin, it is clear that the H-bonding possibilities of the asparagine in wild type are not essential to function [90]. Despite the minimal effect of mutation on function, on addition of antimycin this strain showed a marked shift in Em for heme bH. The pattern of this effect among the neutral residue changes tested was for a larger shift in Em with increasing hydrophobicity. The N221D mutant falls outside this pattern, but this may reflect its acidic nature. There is no obvious feature of these characteristics that allows us at present to suggest a mechanistic explanation. Examination of the protein environment with respect to heme bH shows a highly polar region, likely related to the need for equilibration of the heme with external pH. If such equilibration were through protonation of the D-propionate of heme bH on redox change, this volume would be expected to support proton equilibration through H+ conducting pathways. We have noted previously the extensive H-bonding in this volume, and the presence of a bound water that forms a H-bonding link between the propionate and Ser-203 in the bovine structure. Since there is considerable variability in conformation within this volume in the six different monomers of the Rb. sphaeroides bc1 complex that are included in PDB file 2qjy, it seems likely that this part of the protein is structurally plastic. It may be involved in equilibration of the heme propionate with the N-phase pH. However, this propionate was not included in the set of residues shown to undergo protonation change in response to reduction in electrostatic studies [69].
In summary, it seems likely that coulombic interactions are “felt” between all centers in the dimeric structure that carry charge. The mechanistic consequences for interactions within a monomer are subsumed under the monomeric Q-cycle, since the Em values derived from redox titration are those that reflect the interactions. As noted elsewhere, coulombic interactions within the Qo-site provide a possible mechanism for prevention of bypass reactions [5]. Interactions across the dimer interface have not been analyzed in detail. However, it seems reasonable to look to an interaction between Q.− species in the Qi-site as a possible explanation for the otherwise anomalous observation that the maximal SQ population observed is 1/dimer.
Conclusions
The modified Q-cycle continues to provide the most economical model for the mechanism of the bc1 complex. The evidence for inter-monomer electron traffic does not seem compelling, and our own experiments suggest that no such reactions occur on the time scale expected. Coulombic effects are expected, and can be demonstrated for the intra-monomer case. Analysis of titration curves suggests that coulombic interactions also occur across the dimer interface, as would be expected from simple physicochemical considerations.
Acknowledgements
We gratefully acknowledge support for the work reported here from grants NIH RO1 GM35438 (to ARC) and NIH RO1 GM62954 (to SAD).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Osyczka A, Moser CC, Dutton PL. Fixing the Q-cycle. Trends in Biochemical Science. 2005;30:176–182. doi: 10.1016/j.tibs.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Crofts AR. The cytochrome bc1 complex – function in the context of structure. Annu. Rev. Physiol. 2004;vol. 66:689–733. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]
- 3.Crofts AR. The bc1 complex: what is there left to argue about? In: Wikström M, editor. Biophysical and Structural Aspects of Bioenergetics. Cambridge: Royal Society of Chemistry Publishing; 2005. pp. 123–155. [Google Scholar]
- 4.Crofts AR. Proton-coupled electron transfer at the Qo-site of the bc1 complex controls the rate of ubihydroquinone oxidation. Biochim. Biophys. Acta. 2004;1655:77–92. doi: 10.1016/j.bbabio.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Crofts AR, Lhee S, Crofts SB, Cheng J, Rose S. Proton pumping in the bc1 complex: A new gating mechanism that prevents short circuits. Biochim. Biophys. Acta. 2006;1757:1019–1034. doi: 10.1016/j.bbabio.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 6.Covian R, Trumpower BL. Rapid electron transfer between monomers when the cytochrome bc1 complex dimer is reduced through center N. J. Biol. Chem. 2005;280:22732–22740. doi: 10.1074/jbc.M413592200. [DOI] [PubMed] [Google Scholar]
- 7.Osyczka A, Moser CC, Daldal F, Dutton PL. Reversible redox energy coupling in electron transfer chains. Nature. 2004;427:607–612. doi: 10.1038/nature02242. [DOI] [PubMed] [Google Scholar]
- 8.Hunte C, Palsdottir H, Trumpower BL. Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett. 2003;545:39–46. doi: 10.1016/s0014-5793(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 9.Mulkidjanian AY. Ubiquinol oxidation in the cytochrome b1 complex: Reaction mechanism and prevention of short-circuiting. Biochim. Biophys. Acta. 2005;1709:5–34. doi: 10.1016/j.bbabio.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 10.Mulkidjanian AY. Proton translocation by the cytochrome bc1 complexes of phototrophic bacteria: introducing the activated Q-cycle. Photochem. Photobiol. Sci. 2007;6:19–34. doi: 10.1039/b517522d. [DOI] [PubMed] [Google Scholar]
- 11.Rich PR. The quinone chemistry of bc complexes. Biochim. Biophys. Acta. 2004;1658:165–171. doi: 10.1016/j.bbabio.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 12.Crofts AR, Meinhardt SW, Jones KR, Snozzi M. The role of the quinone pool in the cyclic electron-transfer chain of Rhodopseudomonas sphaeroides: A modified Q-cycle mechanism. Biochim. Biophys. Acta. 1983;723:202–218. doi: 10.1016/0005-2728(83)90120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crofts AR. The Q-cycle, - a personal perspective. Photosynth. Res. 2003;80:223–243. doi: 10.1023/B:PRES.0000030444.52579.10. [DOI] [PubMed] [Google Scholar]
- 14.Skulachev VP. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996;29:169–202. doi: 10.1017/s0033583500005795. [DOI] [PubMed] [Google Scholar]
- 15.Boveris A. Determination of the production of superoxide radicals and hydrogen-peroxide in mitochondria. Methods Enzymol. 1984;105:429–435. doi: 10.1016/s0076-6879(84)05060-6. [DOI] [PubMed] [Google Scholar]
- 16.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 17.Muller F. The nature and mechanism of superoxide production by the electron transport chain: Its relevance to aging. J. Am. Aging Assoc. 2000;23:227–253. doi: 10.1007/s11357-000-0022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun J, Trumpower BL. Superoxide anion generation by the cytochrome bc1 complex. Archives of Biochemistry and Biophysics. 2003;419:198–206. doi: 10.1016/j.abb.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 19.Raha S, Robinson BH. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genetics. 2001;106:62–70. doi: 10.1002/ajmg.1398. [DOI] [PubMed] [Google Scholar]
- 20.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 21.Kristal BS, Jackson CT, Chung HY, Matsuda M, Nguyen HD, Yu BP. Defects at Center P underlie diabetes-associated mitochondrial dysfunction. Free Radical Biol. & Medicine. 1997;22:823–833. doi: 10.1016/s0891-5849(96)00428-5. [DOI] [PubMed] [Google Scholar]
- 22.Fisher N, Meunier B. The bc1 complex: structure, function and dysfunction. In: Garcia-Trejo J, editor. Recent Research Developments in Human Mitochondrial Myopathies. India: Research Signpost; 2002. pp. 97–112. [Google Scholar]
- 23.Fisher N, Meunier B. Effects of mutations in mitochondrial cytochrome b in yeast and man - Deficiency, compensation and disease. Europ. J. Biochem. 2001;268:1155–1162. doi: 10.1046/j.1432-1327.2001.02010.x. [DOI] [PubMed] [Google Scholar]
- 24.Crofts AR, Wraight CA. The electrochemical domain of photosynthesis. Biochim. Biophys. Acta. 1983;726:149–186. [Google Scholar]
- 25.Hong SJ, Ugulava N, Guergova-Kuras M, Crofts AR. The energy landscape for ubihydroquinone oxidation at the Qo-site of the bc1 complex in Rhodobacter phaeroides. J. Biol. Chem. 1999;274:33931–33944. doi: 10.1074/jbc.274.48.33931. [DOI] [PubMed] [Google Scholar]
- 26.Crofts AR, Wang Z. How rapid are the internal reactions of the ubiquinol:cytochrome c2 oxidoreductase? Photosynth. Res. 1989;22:69–87. doi: 10.1007/BF00114768. [DOI] [PubMed] [Google Scholar]
- 27.Xia D, Yu C-A, Kim H, Xia J-Z, Kachurin AM, Zhang L, Yu L, Deisenhofer J. Crystal Structure of the Cytochrome bc1 Complex from Bovine Heart Mitochondria. Science. 1997;277:60–66. doi: 10.1126/science.277.5322.60. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Huang L-S, Shulmeister VM, Chi Y-I, Kim K-K, Hung L-W, Crofts AR, Berry EA, Kim S-H. Electron transfer by domain movement in cytochrome bc1. Nature (Lond.) 1998;392:677–684. doi: 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- 29.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete Structure of the 11-Subunit Bovine Mitochondrial Cytochrome bc1 Complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 30.Hunte C. Insights from the structure of the yeast cytochrome bc1 complex: crystallization of membrane proteins with antibody fragments. FEBS Lett. 2001;504:126–132. doi: 10.1016/s0014-5793(01)02744-2. [DOI] [PubMed] [Google Scholar]
- 31.Berry EA, Huang L-S, Saechao LK, Pon NG, Valkova-Valchanova M, Daldal F. X-ray structure of Rhodobacter capsulatus cytochrome bc1: comparison with its mitochondrial and chloroplast counterparts. Photosynth. Resc. 2004;81:251–275. doi: 10.1023/B:PRES.0000036888.18223.0e. [DOI] [PubMed] [Google Scholar]
- 32.Esser L, Gong X, Yang S, Yu L, Yu C-A, Xia D. Surface-modulated motion switch: Capture and release of iron–sulfur protein in the cytochrome bc1 complex. Proc. Natl. Acad. Sci. (U.S.A.) 2006;103:13045–13050. doi: 10.1073/pnas.0601149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palsdottir H, Lojero CG, Trumpower BL, Hunte C. Structure of the Yeast Cytochrome bc1 Complex with a Hydroxyquinone Anion Qo Site Inhibitor Bound. J. Biol. Chem. 2003;278:31303–31311. doi: 10.1074/jbc.M302195200. [DOI] [PubMed] [Google Scholar]
- 34.Crofts AR, Barquera B, Gennis RB, Kuras R, Guergova-Kuras M, Berry EA. Mechanism of ubiquinol oxidation by the bc1 complex: the different domains of the quinol binding pocket, and their role in mechanism, and the binding of inhibitors. Biochemistry. 1999;38:15807–15826. doi: 10.1021/bi990962m. [DOI] [PubMed] [Google Scholar]
- 35.Crofts AR, Hong S, Zhang Z, Berry EA. Physicochemical aspects of the movement of the Rieske iron sulfur protein during quinol oxidation by the bc1 complex. Biochemistry. 1999;38:15827–15839. doi: 10.1021/bi990963e. [DOI] [PubMed] [Google Scholar]
- 36.Izrailev S, Crofts AR, Berry EA, Schulten K. Steered Molecular Dynamics Simulation of the Rieske Subunit Motion in the Cytochrome bc1 Complex. Biophys. J. 1999;77:1753–1768. doi: 10.1016/S0006-3495(99)77022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crofts AR, Shinkarev VP, Kolling DRJ, Hong S. The modified Q-cycle explains the apparent mismatch between the kinetics of reduction of cytochromes c1 and bH in the bc1 complex. J. Biol. Chem. 2003;278:36191–36201. doi: 10.1074/jbc.M305461200. [DOI] [PubMed] [Google Scholar]
- 38.Engstrom G, Xiao K, Yu C-A, Yu L, Durham B, Millett F. Photoinduced electron transfer between the Rieske iron-sulfur protein and cytochrome c1 in the Rhodobacter sphaeroides cytochrome bc1 complex: effects of pH, temperature, and driving force. J. Biol.Chem. 2002;277:31072–31078. doi: 10.1074/jbc.M202594200. [DOI] [PubMed] [Google Scholar]
- 39.Rajagukguk S, Yang S, Yu C-A, Yu L, Durham B, Millett F. Effect of Mutations in the Cytochrome b ef Loop on the Electron-Transfer Reactions of the Rieske Iron-Sulfur Protein in the Cytochrome bc1 Complex. Biochemistry. 2007;46:1791–1798. doi: 10.1021/bi062094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crofts AR, Guergova-Kuras M, Kuras R, Ugulava N, Li J, Hong S. Proton-coupled electron transfer at the Qo-site: what type of mechanism can account for the high activation barrier? Biochim. Biophys. Acta. 2000;1459:456–466. doi: 10.1016/s0005-2728(00)00184-5. [DOI] [PubMed] [Google Scholar]
- 41.Gao X, Wen X, Esser L, Quinn B, Yu L, Yu C-A, Xia D. Structural Basis for the Quinone Reduction in the bc1 Complex: A Comparative Analysis of Crystal Structures of Mitochondrial Cytochrome bc1 with Bound Substrate and Inhibitors at the Qi Site. Biochemistry. 2003;42:9067–9080. doi: 10.1021/bi0341814. [DOI] [PubMed] [Google Scholar]
- 42.Crofts AR, Shinkarev VP, Dikanov SA, Samoilova RI, Kolling D. Interactions of quinone with the iron-sulfur protein of the bc(1) complex: is the mechanism spring-loaded? Biochim. Biophys. Acta. 2002;1555:48–53. doi: 10.1016/s0005-2728(02)00253-0. [DOI] [PubMed] [Google Scholar]
- 43.Samoilova RI, Kolling D, Uzawa T, Iwasaki T, Crofts AR, Dikanov SA. The interaction of the Rieske iron sulfur protein with occupants of the Qo-site of the bc1 complex, probed by 1D and 2D Electron Spin Echo Envelope Modulation. J. Biol. Chem. 2002;277:4605–4608. doi: 10.1074/jbc.C100664200. [DOI] [PubMed] [Google Scholar]
- 44.Zu Y, Manon M-J, Couture MM-J, Kolling DRJ, Crofts AR, Eltis LD, Fee JA, Hirst J. The reduction potentials of Rieske clusters: the importance of the coupling between oxidation state and histidine protonation state. Biochemistry. 2003;42:12400–12408. doi: 10.1021/bi0350957. [DOI] [PubMed] [Google Scholar]
- 45.Ugulava NB, Crofts AR. CD-monitored redox titration of the Rieske Fe-S rotein of Rhodobacter sphaeroides: pH dependence of the mid-point potential in isolated bc1 complex and in membranes. FEBS Lett. 1998;440:409–413. doi: 10.1016/s0014-5793(98)01493-8. [DOI] [PubMed] [Google Scholar]
- 46.Dikanov SA, Kolling DRJ, Endeward B, Samoilova RI, Prisner TF, Nair SK, Crofts AR. Identification of Hydrogen Bonds to the Rieske Cluster through the Weakly Coupled Nitrogens Detected by Electron Spin Echo Envelope Modulation Spectroscopy. J. Biol. Chem. 2006;281:27416–27425. doi: 10.1074/jbc.M604103200. [DOI] [PubMed] [Google Scholar]
- 47.Kolling DRJ, Brunzelle JS, Lhee S, Crofts AR, Nair SK. Atomic resolution crystal structures of Rieske iron-sulfur protein of cytochrome bc1 complex: Exploring the roles of hydrogen bonding in tuning the redox potential of iron-sulfur clusters. Structure. 2007 doi: 10.1016/j.str.2006.11.012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwasaki T, Kounosu A, Kolling DRJ, Crofts AR, Dikanov SA, Jin A, Imai T, Urushiyama A. Characterization of the pH-Dependent Resonance Raman Transitions of Archaeal and Bacterial Rieske [2Fe-2S] Proteins. J. Am. Chem. Soc. 2004;126:4788–4789. doi: 10.1021/ja031976p. [DOI] [PubMed] [Google Scholar]
- 49.Iwasaki T, Kounosu A, Kolling DRJ, Lhee S, Crofts AR, Dikanov SA, Uchiyama T, Kumasaka T, Ishikawa H, Kono M, Imai T, Urushiyama A. Resonance Raman characterization of archaeal and bacterial Rieske protein variants with modified hydrogen bond network around the [2Fe-2S] center. Protein Science. 2006;15:2019–2024. doi: 10.1110/ps.052035406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ullmann GM, Noodleman L, Case DA. Density functional calculation of pKa values and redox potentials in the bovine Rieske iron-sulfur protein. J. Biol. Inorg. Chem. 2002;7:632–639. doi: 10.1007/s00775-002-0342-6. [DOI] [PubMed] [Google Scholar]
- 51.Colbert C, Couture MM-J, Eltis LD, Bolin JT. A cluster exposed: structure of the Rieske ferredoxin from biphenyl dioxygenase and the redox properties of Rieske Fe-S proteins. Structure. 2000;8:1267–1278. doi: 10.1016/s0969-2126(00)00536-0. [DOI] [PubMed] [Google Scholar]
- 52.Hunsicker-Wang LM, Heine A, Chen Y, Luna EP, Todaro T, Zhang YM, Williams PA, McRee DE, Hirst J, Stout CD, Fee JA. High-Resolution Structure of the Soluble, Respiratory-Type Rieske Protein from Thermus thermophilus: Analysis and Comparison. Biochemistry. 2003;42:7303–7317. doi: 10.1021/bi0342719. [DOI] [PubMed] [Google Scholar]
- 53.Trumpower BL. A concerted, alternating sites mechanism of ubiquinol oxidation by the dimeric cytochrome bc1 complex. Biochim. Biophys. Acta. 2002;1555:166–173. doi: 10.1016/s0005-2728(02)00273-6. [DOI] [PubMed] [Google Scholar]
- 54.Sadoski RC, Engstrom G, Tian H, Zhang L, Yu CA, Yu L, Durham B, Millett F. Use of a photoactivated ruthenium dimer complex to measure electron transfer between the Rieske iron-sulfur protein and cytochrome c1 in the cytochrome bc1 complex. Biochemistry. 2000;39:4231–4236. doi: 10.1021/bi000003o. [DOI] [PubMed] [Google Scholar]
- 55.Link TA. The role of the "Rieske" iron sulfur protein in the hydroquinone oxidation (Qp−) site of the cytochrome bc1 complex: The "proton-gated affinity change" mechanism. FEBS Lett. 1997;412:257–264. doi: 10.1016/s0014-5793(97)00772-2. [DOI] [PubMed] [Google Scholar]
- 56.Berry EA, Huang LS. Observations concerning the quinol oxidation site of the cytochrome bc1 complex. FEBS Lett. 2003;555:13–20. doi: 10.1016/s0014-5793(03)01099-8. [DOI] [PubMed] [Google Scholar]
- 57.Schröter T, Hatzfeld OM, Gemeinhardt S, Korn M, Friedrich T, Ludwig B, Link T. Mutational analysis of residues forming hydrogen bonds in the Rieske [2Fe2S] cluster of the cytochrome bc1 complex of Paracoccus denitrificans. Eur. J. Biochem. 1998;255:100–106. doi: 10.1046/j.1432-1327.1998.2550100.x. [DOI] [PubMed] [Google Scholar]
- 58.Denke E, Merbitzzahradnik T, Hatzfeld OM, Snyder CH, Link TA, Trumpower BL. Alteration of the midpoint potential of the Rieske iron-sulfur protein by changes of amino acids forming H-bonds to the iron-sulfur cluster. J. Biol. Chem. 1998;273:9085–9093. doi: 10.1074/jbc.273.15.9085. [DOI] [PubMed] [Google Scholar]
- 59.Guergova-Kuras M, Kuras R, Ugulava N, Hadad I, Crofts AR. Specific mutagenesis of the Rieske iron sulfur protein in Rhodobacter sphaeroides shows that both thermodynamic gradient and the pK of the oxidized form determine the rate of quinol oxidation by the bc1 complex. Biochemistry. 2000;39:7436–7444. doi: 10.1021/bi992491+. [DOI] [PubMed] [Google Scholar]
- 60.Crofts AR, Hong SJ, Ugulava N, Barquera B, Gennis RB, Guergova-Kuras M, Berry E. Pathways for proton release during ubihydroquinone oxidation by the bc1 complex. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10021–10026. doi: 10.1073/pnas.96.18.10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Crofts A, Guergova-Kuras M, Ugulava N, Kuras R, Hong S. Proc. XIIth Congress of Photosynthesis Research. Brisbane; Australia: 2002. Proton processing at the Qo-site of the bc1 complex of Rhodobacter sphaeroides; p. 6. [Google Scholar]
- 62.Lin I-J, Chen Y, Fee JA, Song J, Westler WM, Markley JL. Rieske Protein from Thermus thermophilus: 15N NMR Titration Study Demonstrates the Role of Iron-Ligated Histidines in the pH Dependence of the Reduction Potential. J. Am. Chem. Soc. 2006;128:10672–10673. doi: 10.1021/ja0627388. [DOI] [PubMed] [Google Scholar]
- 63.Iwaki M, Yakovlev G, Hirst J, Osyczka A, Dutton PL, Marshall D, Rich PR. Direct observation of redox-linked histidine protonation changes in the iron-sulfur protein of the cytochrome bc(1) complex by ATR-FTIR spectroscopy. Biochemistry. 2005;44:4230–4237. doi: 10.1021/bi047533v. [DOI] [PubMed] [Google Scholar]
- 64.Iwaki M, Osyczka A, Moser CC, Dutton PL, Rich PR. ATR-FTIR spectroscopy studies of iron-sulfur protein and cytochrome c1 in the Rhodobacter capsulatus cytochrome bc1 complex. Biochemistry. 2004;43:9477–9486. doi: 10.1021/bi049211x. [DOI] [PubMed] [Google Scholar]
- 65.Hunte C, Koepke J, Lange C, Roβmanith T, Michel H. Structure at 2.3 Å resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure. 2000;8:669–684. doi: 10.1016/s0969-2126(00)00152-0. [DOI] [PubMed] [Google Scholar]
- 66.Osyczka A, Zhang H, Mathé C, Rich PR, Moser CC, Dutton PL. Role of the PEWY Glutamate in Hydroquinone-Quinone Oxidation-Reduction Catalysis in the Qo Site of Cytochrome bc1. Biochemistry. 2006;45:10492–10503. doi: 10.1021/bi060013a. [DOI] [PubMed] [Google Scholar]
- 67.Wenz T, Covian R, Hellwig P, MacMillan F, Meunier B, Trumpower BL, Hunte C. Mutational analysis of cytochrome b at the ubiquinol oxidation site of yeast complex III. J. Biol. Chem. 2007;282:3977–3988. doi: 10.1074/jbc.M606482200. [DOI] [PubMed] [Google Scholar]
- 68.Seddiki N, Meunier B, Lemesle-Meunier D, Brasseur Gl. Is Cytochrome b Glutamic Acid 272 a Quinol Binding Residue in the bc1 Complex of Saccharomyces cerevisiae? Biochemistry. 2008;47:2357–2368. doi: 10.1021/bi701905a. [DOI] [PubMed] [Google Scholar]
- 69.Klingen AR, Palsdottir H, Hunte C, Ullmann GM. Redox-linked protonation state changes in cytochrome bc1 identified by Poisson–Boltzmann electrostatics calculations. Biochim. Biophys. Acata. 2007;1767:204–221. doi: 10.1016/j.bbabio.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 70.Forquer I, Covian R, Bowman MK, Trumpower BL, Kramer DM. Similar Transition States Mediate the Q-cycle and Superoxide Production by the Cytochrome bc1 Complex. J. Biol. Chem. 2006;281:38459–38465. doi: 10.1074/jbc.M605119200. [DOI] [PubMed] [Google Scholar]
- 71.Cape JL, Bowman MK, Kramer DM. A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: Importance for the Q-cycle and superoxide production. Proc. Natl. Acad. Sci. (U.S.A.) 2007;104:7887–7892. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang H, Osyczka A, Dutton PL, Moser CC. Exposing the Complex III Qo semiquinone radical. Biochim. Biophys. Acta. 2007;1767:883–887. doi: 10.1016/j.bbabio.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu J, Egawa T, Yeh S-R, Yu L, Yu C-A. Simultaneous reduction of iron-sulfur protein and cytochrome bL during ubiquinol oxidation in cytochrome bc1 complex. Proc. Natl. Acad. Sci. (U.S.A.) 2007;104:4864–4869. doi: 10.1073/pnas.0607812104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Junemann S, Heathcote P, Rich PR. On the mechanism of quinol oxidation in the bc1 complex. J. Biol. Chem. 1998;273:21603–21607. doi: 10.1074/jbc.273.34.21603. [DOI] [PubMed] [Google Scholar]
- 75.De Vries S, Albracht SPJ, Berden JA, Marres CAM, Slater EC. The effect of pH, ubiquinone depletion and myxothiazol on the reduction kinetics of the prosthetic groups of QH2:cytochrome c oxidoreductase. Biochim Biophys Acta. 1983;723:91–103. doi: 10.1016/0005-2728(83)90013-0. [DOI] [PubMed] [Google Scholar]
- 76.Kolling DRJ, Samoilova RI, Holland JT, Berry EA, Dikanov SA, Crofts AR. Exploration of ligands to the Qi-site semiquinone in the bc1 complex using high resolution EPR. J. Biol. Chem. 2003;278:39747–39754. doi: 10.1074/jbc.M305913200. [DOI] [PubMed] [Google Scholar]
- 77.De Vries S, Berden JA, Slater EC. Oxidation-reduction properties of an antimycin-sensitive semiquinone anion bound to QH2:cytochrome c oxidoreductase. In: Trumpower BL, editor. Function of Quinones in Energy Conserving Systems. New York: Academic Press; 1982. pp. 235–246. [Google Scholar]
- 78.Robertson DE, Prince RC, Bowyer JR, Matsuura K, Dutton PL, Ohnishi T. Thermodynamic properties of the semiquinone and its binding site in the ubiquinol:cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems. J. Biol. Chem. 1984;259:1758–1763. [PubMed] [Google Scholar]
- 79.Crofts AR, Barquera B, Bechmann G, Guergova M, Salcedo-Hernandez R, Hacker B, Hong S, Gennis RB. Structure and function in the bc1 complex of Rb. sphaeroides. In: Mathis P, editor. Photosynthesis: from light to biosphere. vol. II. Dordrecht: Kluwer Academic Publ; 1995. pp. 493–500. [Google Scholar]
- 80.Glaser EG, Meinhardt SW, Crofts AR. Reduction of cytochrome b561 through the antimycin-sensitive site of the ubiquinol:cytochrome c2 oxidoreductase complex of Rps. sphaeroides. FEBS Lett. 1984;178:336–342. doi: 10.1016/0014-5793(84)80629-8. [DOI] [PubMed] [Google Scholar]
- 81.Meinhardt SW, Crofts AR. A New Effect of Antimycin on the b-cytochromes of Rps. sphaeroides. In: Sybesma C, editor. Advances in Photosynthesis Research. vol. 1. The Hague: Martinus Nijhoff/Dr. W. Junk Publishers; 1984. pp. 649–652. [Google Scholar]
- 82.Salerno JC, Xu Y, Osgood P, C.H K, King TE. Thermodynamic and spectroscopic characteristics of the cytochrome bc1 complex. Role of quinone in the behavior of cytochrome b562. J. Biol. Chem. 1989;264:15398–15403. [PubMed] [Google Scholar]
- 83.Salerno JC, Osgood M, Liu Y, Taylor H, Scholes CP. Electron nuclear double resonance (ENDOR) of the Qc-ubiSQ radical in the mitochondrial electron transport chain. Biochemistry. 1990;29:6987–6993. doi: 10.1021/bi00482a006. [DOI] [PubMed] [Google Scholar]
- 84.Dikanov SA, Samoilova RI, Kolling DRJ, Holland JT, Crofts AR. Hydrogen bonds involved in binding the Qi-site semiquinone in the bc1 complex, identified through deuterium exchange using pulsed EPR. J. Biol. Chem. 2004;279:15814–15823. doi: 10.1074/jbc.M313417200. [DOI] [PubMed] [Google Scholar]
- 85.Rich PR, Jeal AE, Madgwick SA, Moody AJ. Inhibitor effects on redox-linked protonations of the b hemes of the mitochondrial bc1 complex. Biochim. Biophys. Acta. 1990;1018:29–40. doi: 10.1016/0005-2728(90)90106-e. [DOI] [PubMed] [Google Scholar]
- 86.Huang LS, Cobessi D, Tung EY, Berry EA. Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: A new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J. Mol. Biol. 2005;351:573–597. doi: 10.1016/j.jmb.2005.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esser L, Elberry M, Zhou F, Yu C-A, Yu L, Xia D. Inhibitor complexed structures of the cytochrome bc1 complex from the photosynthetic bacterium Rhodobacter sphaeroides. J. Biol. Chem. 2007 doi: 10.1074/jbc.M708608200. in press. [DOI] [PubMed] [Google Scholar]
- 88.Carugo O, Carugo KD. When X-rays modify the protein structure: radiation damage at work. Trends Biochem. Sci. 2005;30:213–219. doi: 10.1016/j.tibs.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 89.Weiss MS, et al. On the influence of the incident photon energy on the radiation damage in crystalline biological samples. Journal of Synchrotron Radiation. 2005;12:304–309. doi: 10.1107/S0909049505003328. [DOI] [PubMed] [Google Scholar]
- 90.Dikanov SA, Holland JT, Endeward B, Kolling DRJ, Samoilova RI, Prisner TF, Crofts AR. Hydrogen bonds between nitrogen donors and the semiquinone in the Qi-site of the bc1 complex. J. Biol. Chem. 2007 doi: 10.1074/jbc.M702333200. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dutton PL, Jackson JB. Thermodynamic and kinetics characterization of electron-transfer components in situ in Rps. spheroides and Rhodospirillum rubrum. Eur. J. Biochem. 1972;30:495–510. doi: 10.1111/j.1432-1033.1972.tb02121.x. [DOI] [PubMed] [Google Scholar]
- 92.Dutton PL, Wilson DM. Redox potentiometry in biological systems. Methods Enzymol. 1976;54:411–435. doi: 10.1016/s0076-6879(78)54026-3. [DOI] [PubMed] [Google Scholar]
- 93.Andrews KM. Ph.D. Thesis. University of Illinois at Urbana-Champaign; 1984. Purification and characterization of the cytochrome bc1 complex from Rhodobacter sphaeroides. [Google Scholar]
- 94.Crofts AR, Barquera B, Bechmann G, Guergova M, Salcedo-Hernandez R, Hacker B, Hong S, Gennis RB. Structure and function in the bc1-complex of Rb. sphaeroides. In: Mathis P, editor. Photosynthesis: from light to biosphere. vol. Vol. II. Dordrecht: Kluwer Academic Publ.; 1995. pp. 493–500. [Google Scholar]
- 95.Robertson D, Prince R, Bowyer J, Matsuura K, Dutton P, Ohnishi T. Thermodynamic properties of the semiquinone and its binding site in the ubiquinol-cytochrome c (c2) oxidoreductase of respiratory and photosynthetic systems. J. Biol. Chem. 1984;259:1758–1763. [PubMed] [Google Scholar]
- 96.Rich PR, Jeal AE, Madgwick SA, Moody AJ. Inhibitor effects on redox-linked protonations of the b haems of the mitochondrial bc1 complex. Biochim. Biophys. Acta. 1990;1018:29–40. doi: 10.1016/0005-2728(90)90106-e. [DOI] [PubMed] [Google Scholar]
- 97.Zweck A, Bechmann G, H W. The pathway of the quinol/quinone transhydrogenation reaction in ubiquinol: cytochrome-c reductase of Neurospora mitochondria. Eur. J. Biochem. 1989;183:199–203. doi: 10.1111/j.1432-1033.1989.tb14913.x. [DOI] [PubMed] [Google Scholar]
- 98.Rich PR, Madgwick SA, Moss DA. The interactions of duroquinol, DBMIB and NQNO with the chloroplast cytochrome bf complex. Biochim. Biophys. Acta. 1991;1058:312–328. [Google Scholar]
- 99.Shinkarev VP, Crofts AR, Wraight CA. The electric field generated by photosynthetic reaction center induces rapid reversed electron transfer in the bc(1) complex. Biochemistry. 2001;40:12584–12590. doi: 10.1021/bi011334j. [DOI] [PubMed] [Google Scholar]
- 100.Siedow JN, Power S, del la Rosa FF, Palmer G. The preparation and characterization of highly purified enzymically active Complex III from baker's yeast. J. Biol. Chem. 1978;253:2392–2399. [PubMed] [Google Scholar]
- 101.de la Rosa FF, Palmer G. Reductive titration of CoQ-depleted Complex III from baker’s yeast: Evidence for an exchange-coupled complex between QH· and low-spin ferricytochrome b. FEBS Lett. 1983;163:140–143. doi: 10.1016/0014-5793(83)81181-8. [DOI] [PubMed] [Google Scholar]
- 102.Cooley JW, Ohnishi T, Daldal F. Binding dynamics at the quinone reduction (Qi) site influence the equilibrium interactions of the iron sulfur protein and hydroquinone oxidation (Qo) site of the cytochrome bc1 complex. Biochemistry. 2005;44:10520–10532. doi: 10.1021/bi050571+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yu C-A, Wen X, Xiao K, Xia D, Yu L. Inter- and intra-molecular electron transfer in the cytochrome bc1 complex. Biochim. Biophys. Acta. 2002;1555:65–70. doi: 10.1016/s0005-2728(02)00256-6. [DOI] [PubMed] [Google Scholar]
- 104.Gong X, Yu L, Xia D, Yu C-A. Evidence for electron equilibrium between the two hemes bL in the dimeric cytochrome bc1 complex. J. Biol. Chem. 2005;280:9251–9257. doi: 10.1074/jbc.M409994200. [DOI] [PubMed] [Google Scholar]
- 105.Crofts AR, Berry EA. Structure and function of the cytochrome bc1 complex of mitochondria and photosynthetic bacteria. Curr. Opinions in Struc. Biol. 1998;vol. 8:501–509. doi: 10.1016/s0959-440x(98)80129-2. [DOI] [PubMed] [Google Scholar]
- 106.Berry E, Guergova-Kuras M, Huang L-S, Crofts AR. Structure and Function of Cytochrome bc complexes. Annu. Rev. Biochem. 2000;69:1007–1077. doi: 10.1146/annurev.biochem.69.1.1005. [DOI] [PubMed] [Google Scholar]
- 107.Bowyer JR, Crofts AR. On the Mechanism of Photosynthetic Electron Transfer in Rps. capsulata and Rps. sphaeroides. Biochim. Biophys. Acta. 1981;636:218–233. doi: 10.1016/0005-2728(81)90096-7. [DOI] [PubMed] [Google Scholar]
- 108.Meinhardt SW, Crofts AR. Kinetic and Thermodynamic Resolution of Cytochrome c1 and Cytochrome c2 from Rps. sphaeroides. FEBS Lett. 1982;149:223–227. [Google Scholar]
- 109.Meinhardt SW, Crofts AR. The Role of Cytochrome b566 in the Electron Transfer Chain of Rps. sphaeroides. Biochim. Biophys. Acta. 1983;723:219–230. doi: 10.1016/0005-2728(83)90120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Snozzi M, Crofts AR. Electron transport in chromatophores from Rhodopseudomonas sphaeroides GA fused with liposomes. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1984;766:451–463. doi: 10.1016/0005-2728(84)90261-5. [DOI] [PubMed] [Google Scholar]
- 111.Shinkarev VP, Crofts AR, Wraight CA. Spectral Analysis of the bc1 Complex Components in Situ: Beyond the Traditional Difference Approach. Biochim. Biophys. Acta. 2005;1757:67–77. doi: 10.1016/j.bbabio.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 112.Shinkarev VP, Crofts AR, Wraight CA. In situ kinetics of cytochromes c1 and c2. Biochemistry. 2006;45:7897–7903. doi: 10.1021/bi060172u. [DOI] [PubMed] [Google Scholar]
- 113.Shinkarev VP, Crofts AR, Wraight CA. Spectral and kinetic resolution of the bc1 complex components in situ: A simple and robust alternative to the traditional difference wavelength approach. Biochim. Biophys. Acta. 2006;1757:273–283. doi: 10.1016/j.bbabio.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 114.Fato R, Battino M, Degli Esposti M, Parenti Castelli G, Lenaz G. Determination of Partition and Lateral Diffusion Coefficients of Ubiquinones by Fluorescence Quenching of n-(9-Anthroyloxy)stearic Acids in Phospholipid Vesicles and Mitochondrial Membranes. Biochemistry. 1986;25:3378–3390. doi: 10.1021/bi00359a043. [DOI] [PubMed] [Google Scholar]
- 115.Cape JL, Strahan JR, Lenaeus MJ, Yuknis BA, Le TT, Shepherd JN, Bowman MK, Kramer DM. The respiratory substrate rhodoquinol induces Q-cycle bypass reactions in the yeast cytochrome bc1 complex - Mechanistic and physiological implications. J. Biol. Chem. 2005;280:34654–34660. doi: 10.1074/jbc.M507616200. [DOI] [PubMed] [Google Scholar]
- 116.Rieske JS, Baum H, Stoner CD, Lipton SH. On the antimycin-sensitive cleavage of complex III of the mitochondrial respiratory chain. J. Biol. Chem. 1967;242:4854–4866. [PubMed] [Google Scholar]
- 117.Crofts AR, Berry EA. Structure and function of the cytochrome bc1 complex of mitochondria and photosynthetic bacteria. Curr. Opinions in Struc. Biol. 1998;8:501–509. doi: 10.1016/s0959-440x(98)80129-2. [DOI] [PubMed] [Google Scholar]
- 118.Marcus RA, Sutin N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- 119.DeVault D. Quantum mechanical tunnelling in biological systems. Q. Rev. Biophys. 1980;13:387–564. doi: 10.1017/s003358350000175x. [DOI] [PubMed] [Google Scholar]
- 120.Moser CC, Page CC, Farid R, Dutton PL. Biological electron transfer. J. Bioenerg. Biomembranes. 1995;27:263–274. doi: 10.1007/BF02110096. [DOI] [PubMed] [Google Scholar]
- 121.Moser CC, Page CC, Chen XX, Dutton PL. Biological electron tunnelling through protein media. J. Biol. Inorg. Chem. 1997;2:393–398. [Google Scholar]
- 122.Moser CC, Farid TA, Chobot SE, Dutton PL. Electron tunnelling chains of mitochondria. Biochim. Biophys. Acta. 2006;1757:1096–1109. doi: 10.1016/j.bbabio.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 123.Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature of biological electron transfer. Nature. 1992;355:796. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 124.Moser CC, Page CC, Dutton PL. Darwin at the molecular scale: selection and variance in electron tunnelling proteins including cytochrome c oxidase. Phil. Trans. R. Soc. 2006;361:1295–1305. doi: 10.1098/rstb.2006.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shinkarev VP, Wraight CA. Intermonomer electron transfer in the bc1 complex dimer is controlled by the energized state and by impaired electron transfer between low and high potential hemes. FEBS Letters. 2007;581:1535–1541. doi: 10.1016/j.febslet.2007.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Covián R, Juan Pablo Pardo JP, Moreno-Sánchez R. Tight Binding of Inhibitors to bovine bc1 complex Is independent of the Rieske protein redox state - Consequences for semiquinone stabilization in the quinol oxidation site. J. Biol. Chem. 2002;277:48449–48455. doi: 10.1074/jbc.M208060200. [DOI] [PubMed] [Google Scholar]
- 127.Crofts AR. The Q-cycle, - a personal perspective. Photosynth. Res. 2003;80:223–243. doi: 10.1023/B:PRES.0000030444.52579.10. [DOI] [PubMed] [Google Scholar]
- 128.Kröger A, Klingenberg M. The kinetics of the redox reactions of ubiquinone related to the electron-transport activity in the respiratory chain. Eur. J. Biochem. 1973;34:358–368. doi: 10.1111/j.1432-1033.1973.tb02767.x. [DOI] [PubMed] [Google Scholar]
- 129.Kröger A, Klingenberg M. Further Evidence for the Pool Function of Ubiquinone as Derived from the Inhibition of the Electron Transport by Antimycin. Europ. J. Biochem. 1973;39:313–323. doi: 10.1111/j.1432-1033.1973.tb03129.x. [DOI] [PubMed] [Google Scholar]
- 130.Fernandez-Velasco J, Crofts AR. Complexes or super complexes: Inhibitor titrations show that electron transfer in chromatophores from Rb. Sphaeroides involves a dimeric ubiquinol: cytochrome c2 oxidoreductase, and is delocalized. Biochem. Soc. Trans. 1991;19:588–593. doi: 10.1042/bst0190588. [DOI] [PubMed] [Google Scholar]
- 131.Bechmann G, Weiss H, Rich P. Nonlinear inhibition curves for tight-binding inhibitors of dimeric ubiquinol-cytochrome c oxidoreductase - evidence for rapid inhibitor mobility. Eur. J. Biochem. 1992;208:315–325. doi: 10.1111/j.1432-1033.1992.tb17189.x. [DOI] [PubMed] [Google Scholar]
- 132.Prince RC, Dutton PL. Single and multiple turnover reactions in the ubiquinone-cytochrome b-c2 oxidoreductase of Rhodopseudomonas sphearoides: The physical chemistry of the major electron donor to cytochrome c2, and its coupled reactions. Biochim. Biophys. Acta. 1977;462:731–747. doi: 10.1016/0005-2728(77)90114-1. [DOI] [PubMed] [Google Scholar]
- 133.Venturoli G, Virgili M, Melandri BA, Crofts AR. Kinetic measurements of electron transfer in coupled chromatophores from photosynthetic bacteria: A method of correction for the electrochromic effects. FEBS Lett. 1987;219:477–484. doi: 10.1016/0014-5793(87)80276-4. [DOI] [PubMed] [Google Scholar]
- 134.Venturoli G, Fernandez-Velasco JG, Crofts AR, Melandri BA. The effect of the size of the quinone pool on the electrogenic reactions in the UQH2:cyt c2 oxidoreductase of Rhodobacter capsulatus. Pool behavior at the quinine reductase site. Biochim. Biophys. Acta. 1988;935:258–272. [Google Scholar]
- 135.Venturoli G, Fernandez-Velasco JG, Crofts AR, Melandri BA. Demonstration of a collisional interaction of ubiquinol with the ubiquinol-cytochrome c2 oxidoreductase complex in chromatophores from Rhodobacter sphaeroides. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1986;851:340–352. doi: 10.1016/0005-2728(86)90070-8. [DOI] [PubMed] [Google Scholar]
- 136.Kuras R, Guergova-Kuras M, Crofts AR. Steps Toward Constructing a Cytochrome b6f Complex in the Purple Bacteria Rhodobacter sphaeroides: An Example of the Structural Plasticity of a Membrane Cytochrome. Biochemistry. 1998;37:16280–16288. doi: 10.1021/bi9813476. [DOI] [PubMed] [Google Scholar]
- 137.Crofts AR, Rose S. Marcus treatment of endergonic reactions: A commentary. Biochim. Biophys. Acata. 2007;1767:1228–1232. doi: 10.1016/j.bbabio.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kuki A, Wolynes PG. Electron tunneling paths in proteins. Science. 1987;236:1647–1652. doi: 10.1126/science.3603005. [DOI] [PubMed] [Google Scholar]
- 139.Balabin IA, Onuchic JN. Dynamically Controlled Protein Tunneling Paths in Photosynthetic Reaction Centers. Science. 2000;290:114–117. doi: 10.1126/science.290.5489.114. [DOI] [PubMed] [Google Scholar]
- 140.Kawatsu T, Beratan DN, Kakitani T. Conformationally Averaged Score Functions for Electronic Propagation in Proteins. J. Phys. Chem. B. 2006;110:5747–5757. doi: 10.1021/jp052194g. [DOI] [PubMed] [Google Scholar]
- 141.Prytkova TR, Kurnikov IV, Beratan DN. Coupling Coherence Distinguishes Structure Sensitivity in Protein Electron Transfer. Science. 2007;315:622–625. doi: 10.1126/science.1134862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hoffman BM, Celis LM, Cull DA, Patel AD, Seifert JL, Wheeler KE, Wang J, Yao J, Kurnikov IV, Nocek JM. Differential influence of dynamic processes on forward and reverse electron transfer across a protein–protein interface. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3564–3569. doi: 10.1073/pnas.0408767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nishioka H, Kimura A, Yamato T, Kawatsu T, Kakitani T. Interference, Fluctuation, and Alternation of Electron Tunneling in Protein Media. 1. Two Tunneling Routes in Photosynthetic Reaction Center Alternate Due to Thermal Fluctuation of Protein Conformation. J. Phys. Chem. B. 2005;109:1978–1987. doi: 10.1021/jp046282x. [DOI] [PubMed] [Google Scholar]
- 144.Walker FA. Magnetic spectroscopic (EPR, ESEEM, Mössbauer, MCD and NMR) studies of low-spin ferriheme centers and their corresponding heme proteins. Coord. Chem. Rev. 1999;185–186:471–534. [Google Scholar]
- 145.Walker FA. Models of the bis-histidine-ligated electron-transferring cytochromes. Comparative geometric and electronic structure of ow-spin ferro- and ferrihemes. Chem. Rev. 2004;104:589–615. doi: 10.1021/cr020634j. [DOI] [PubMed] [Google Scholar]
- 146.Yun C-H, Crofts AR, Gennis RB. Assignment of the histidine axial ligands to the cytochrome bH and cytochrome bL components of the bc1 complex from Rb. sphaeroides by site-directed mutagenesis. Biochemistry. 1991;30:6747–6754. doi: 10.1021/bi00241a017. [DOI] [PubMed] [Google Scholar]
- 147.Holland JT. Center for Biophysics and Computational Biology, vol. Ph. D. Urbana-Champaign: University of Illinois; 2007. Investigation of the interaction between cytochrome bH and the Qi-site in Rhodobacter sphaeroides; p. 163. [Google Scholar]
- 148.Brandt U, Okun JG. Role of deprotonation events in ubihydroquinone:cytochrome c oxidoreductase from bovine heart and yeast mitochondria. Biochemistry. 1997;36:11234–11240. doi: 10.1021/bi970968g. [DOI] [PubMed] [Google Scholar]