

Abstract
To better understand the roles of TGF-β in bone metabolism, we investigated osteoclast survival in response TGF-β and found that TGF-β inhibited apoptosis. We examined the receptors involved in promotion of osteoclast survival and found that the canonical TGF-β receptor complex is involved in the survival response. The upstream MEK kinase TAK1 was rapidly activated following TGF-β treatment. Since osteoclast survival involves MEK, AKT, and NFκB activation, we examined TGF-β effects on activation of these pathways and observed rapid phosphorylation of MEK, AKT, IKK, IκB, and NFκB. The timing of activation coincided with SMAD activation and dominant negative SMAD expression did not inhibit NFκB activation, indicating that kinase pathway activation is independent of SMAD signaling. Inhibition of TAK1, MEK, AKT, NIK, IKK, or NFκB repressed TGF-β-mediated osteoclast survival. Adenoviral-mediated TAK1 or MEK inhibition eliminated TGF-β-mediated kinase pathway activation and constitutively active AKT expression overcame apoptosis induction following MEK inhibition. TAK1/MEK activation induces pro-survival BclXL expression and TAK1/MEK and SMAD pathway activation induces pro-survival Mcl-1 expression. These data show that TGF-β-induced NFκB activation is through TAK1/MEK-mediated AKT activation, which is essential for TGF-β to support of osteoclast survival.
Keywords: osteoclast, apoptosis, TGF-β, signal transduction, TAK1MEK, AKT, NFκB, SMAD, BclXL, Mcl-1
Introduction
TGF-β has been shown to have a myriad of influences on bone metabolism. Among its complex influences, TGF-β differentially affects osteoblast differentiation by both promoting early stages of differentiation while represses final differentiation (reviewed in [1]). TGF-β has also been shown to either stimulate or repress osteoclast differentiation, depending on the TGF-β concentration and model system used [2-7]. One of the key mechanisms by which early tumor progression is suppressed is due to pro-apoptotic effects of TGF-β [8-15]. In most cell types examined to date, TGF-β has proven to be a potent inducer of apoptosis [12, 16, 17]. An exception to this response is in neuronal cells [18]. TGF-β protects these cells from apoptosis by stimulating expression of the non-canonical Activin-Like receptor Kinase 1 (ALK1) receptor within one hour of TGF-β treatment. Subsequent activation of Nuclear Factor kappa B (NFκB) is also mediated through this receptor. The authors proposed that TGF-β promoted cell survival through increasing expression of ALK1 whereas pro-apoptotic responses are mediated by canonical TGF-β signaling. Canonical TGF-β receptor complex involves TGF-β Receptor type I, ALK5 (ALK5/TβRI) and TGF-β Receptor type II (TβRII). TβRII binds TGF-β, stimulating association with ALK5/ TβRI and subsequent SMAD dependent and SMAD independent signaling (reviewed in [19-25]). SMAD dependent signaling has been the focus of TGF-β signaling studies since their discovery whereas SMAD independent signaling involving kinase cascades has more recently come to the fore as important in TGF-β influences [26-30].
Murine osteoclasts have been reported to respond to TGF-β with increased apoptosis [31]. In this study, osteoclasts derived from un-fractionated marrow cultured with vitamin D in the absence of exogenous stromal cells responded to TGF-β treatment with increased apoptosis. Since TGF-β influences on osteoclast differentiation differ in the presence verses absence of stromal cells [5], we have investigated TGF-β effects on osteoclast survival in osteoclast cultures that are stromal cell-free and co-cultures of osteoclasts with stromal cells. Using osteoclasts derived from RANKL and M-CSF treatment or co-culture of precursors with stromal cells, we examined the influences of TGF-β on osteoclast survival. Here we report a pro-survival response to TGF-β. The pro-survival response requires signaling from the canonical TGF-β receptor signaling complex independent of ALK1. We and others have documented the critical role of intracellular kinase cascades involving MEK1/2, AKT, and NFκB in regulation of osteoclast survival [32-35]. Therefore we have investigated the downstream kinase signaling events involved in TGF-β-mediated osteoclast survival and revealed a novel MEK-dependent pathway activating the AKT/NFκB survival pathway. Activation of this pathway is downstream of TGF-β Activated Kinase 1 (TAK1), a MAPKKK activated by both TGF-β and BMPs [36]. SMAD 2 and SMAD3 activation and nuclear localization are also increased by TGF-β treatment. These pathways act independently to increase transcription of pro-survival Bcl2 family members Bcl-XL and Mcl-1.
Materials and Methods
Unless otherwise indicated, all chemicals are from Sigma Chemical Co., St Louis, MO. TGF-β1 was purchased from R&D (Minneapolis, MN) and suspended in PBS/0.01% BSA (vehicle).
Osteoclast Culture without stromal cell addition
Marrow cells were obtained from Balb/c mice as outlined previously [35]. Briefly, marrow was flushed from the long bones with PBS, and red blood cells lysed. Cells were incubated at 37° C with 5% CO2 in a humidified incubator overnight in alpha modified Minimal Essential Medium (α-MEM) supplemented with 10% (v/v) fetal bovine serum (FBS, Hyclone, Logan UT), antibiotic/antimycotic, and 25 ng/ml M-CSF (R&D, Minneapolis, MN). The following day, non-adherent cells were plated at 2 × 105 cells/cm2 in 24 well plates (Fischer, Pittsburgh, PA) in the above medium with 100 ng/ ml RANKL (Calbiochem, La Jolla, CA or recombinant GST-RANKL generated and purified by affinity chromatography using Glutathione Sepharose 4B pre-packed disposable columns according to the provider (GE Healthcare Life Sciences)). Cells were fed fresh medium on day 3 and treated as described in the figure legends for specific experiments.
Stromal cell co-culture generation of osteoclasts
ST2 cells (Riken Cell Bank, Tsukuba, Japan) were cultured in α-MEM supplemented with 10% (v/v) FBS and antibiotic/antimycotic. ST2 cells (passage 5-25) were plated at 8 × 104 cells/cm2 in 24 well plates. Twenty-four hours later (day 0 of differentiation) Balb/c marrow cells were added at 2.1 × 104 cells/cm2 to the ST2 cell cultures in MEM (Invitrogen, Carlsbad, CA), 10% (v/v) FBS, 1% (v/v) non-essential amino acids, antibiotic/antimycotic, 7 × 10-3 M ascorbic acid (Invitrogen), 1 × 10-7 M dexamethasone, 1 × 10-5 M, vitamin D3 (Biomol, Plymouth Meeting, PA) 100 ng/ml biotin, 1.36 ug/ml vitamin B12, and 299 ng/ml lipoic acid [37]. Medium was changed every three days through day 9. On day 13 the co-cultures were treated as outlined in Figure 2 legend.
Hoechst and TRAP stains and apoptosis quantitation
Cells cultured on coverslips were fixed with 1% (w/v) paraformaldehyde in phosphate buffered saline (PBS) for at least 10 minutes, rinsed with PBS containing 0.01% (v/v) Tween 20 (PBST) three times and stained for 1 hour with 5 μg/ml Hoechst 33258 in PBST, followed by rinsing 3 times with PBST. Cells were then stained for tartrate resistant acid phosphatase (TRAP) as we have outlined using a kit from Sigma Chemical Co. according to the provider's instructions [35]. Coverslips were mounted on slides and visualized using light and epifluorescence. Total numbers of osteoclast were obtained by counting TRAP positive cells containing at least 3 nuclei. Of this population, apoptotic osteoclasts were delineated on the basis of chromatin condensation as we have reported [35]. Data were then expressed as the percentage of apoptotic osteoclasts displaying condensed nuclei out of the total number of osteoclasts.
Western Blotting
Osteoclasts were treated as outlined in the figure legends. At the end of treatments, cells were placed on ice and rinsed 3 times with cold PBS. Laemmli sample buffer lacking bromphenol blue and β-mercaptoethanol was added and cell extracts collected following scraping. Protein concentrations were determined using BioRad's Protein Quantitation In Detergent Analysis Kit. Forty micrograms of protein per lane was used for analysis. Prior to boiling and western blotting, β-mercaptoethanol and bromophenol blue were added to the samples. Western blotting was carried out as previously outlined [35]. Anti phospho- and total antibodies directed toward Tak1 (Thr184/187), MEK 1/2 (Ser217/221), AKT (Ser473), Inhibitor of IκB Kinase (IKK)α/β (Ser176/180), Inhibitor of NFκB (IκB) (Ser32), and NFκB (Ser536) (Cell Signaling, Beverly, MA) were used at a 1:1,000 dilution for Western blotting according to the product directions as we have reported [35]. Anti-tubulin hybridoma supernatant (E7) from The Developmental Studies Hybridoma Bank at the University of Iowa was also used at a 1:1,000 dilution. Peroxidase conjugated secondary antibodies (Cell Signaling) were used at a 1:5,000 dilution with chemiluminescent detection using ECL Plus according to the product directions (GE Health Care). Following detection of phospho-proteins, the membrane was stripped using Pierce's Restore PLUS Western Blot Stripping Buffer according to the product literature and the blots were re-probed for total proteins and tubulin.
Quantitative Real Time Polymerase Chain Reaction
Cells were either vehicle or TGF-β treated or infected with adenovirus expressing empty vector (Strategene Ad Easy, La Jolla, CA), Cre recombinase (Vector BioLabs, Philadelphia, PA), or harvested without infection as detailed in Figure 4 legend. Cells were rinsed with PBS and RNA harvested using TriZol Reagent according to the product literature (Invitrogen). Following quantitation, cDNA was synthesized and Real Time PCR analysis carried out as we have reported [5]. Primers were:
| GENE | LEFT PRIMER | RIGHT PRIMER |
| ALK1 | 5′-CGGGAGACGGAGAYCTACAA-3′ | 5′-GGAGCCGTGTTCATGGTAGT-3′ |
| ALK5/TβRI | 5′-TTGCTCCGTTGTATTTGTGC-3′ | 5′-CAGGACTTAACACCGCCTTT-3′ |
| TβRII exon 2 | 5′-GCAAGTTTTGTGATGCGAGA-3′ | 5′-GACTTCATGCGGCTTCTCA-3′ |
| BclXL | 5′-GATGGAGGAACCAGGTTGTG-3′ | 5′-CCCCGGAAGGTCTTTTGTAT-3′ |
| Mcl-1 | 5′-GCAGAGCCTGTTGTGTGTGT-3′ | 5′-AGTGAAGAGCACAGGGAGGA-3′ |
| TUBULIN | 5′-CTGCTCATCAGCAAGATCAGAG-3′ | 5′-GCATTATAGGGCTCCACCACAG-3′ |
NFκB Reporter Assay
An adenovirus system that has three NFκB consensus sites driving luciferase gene expression (gift from P.B. McCray Jr., The University of Iowa, Iowa City) was used to infect cells at a MOI of 100 for 24 hours. Cell lysates were harvested and processed as described [38]. Protein concentrations were determined as above and 75 μg in 20 μl was used to measure luciferase activity as described [38].
Adenoviral Preparation and Treatments
Dominant negative AKT and mutant IκB were purchased from Vector Biolabs; Dominant negative TAK1 was a gift from Dr. Brenner, University of North Carolina Gene Therapy Center; Dominant negative IKK was a gift from Dr. Rainer de Martin; Dominant negative MEK was a gift from Dr. Tanaka; Dominant negative NIK generation has been previously reported [39]. Virus was expanded and titered using Ad 293 cells using standard protocols. A Multiplicity of Infection (MOI) of 100 was used in all studies unless indicated.
Chemical Inhibition
ALK5/TβRI inhibition: Cells were pre-treated with 2 mM (final concentration of 2 μm) SB-431542 or vehicle (DMSO) for 1 hour prior to TGF-β treatment as detailed in Figure 4 legend. MEK inhibition: The MEK1/2 inhibitor UO126 was used at a concentration of 10 μM where indicated during one-hour serum starvation and subsequent treatment.
Nuclear localization
Duplicate sets of wells were treated as outlined in the figure legends. Cells were washed, scraped and pelleted in cold PBS. The pellet was resuspended in 10 mM Tris-HCL pH 8.0, 10 mM KCl, 0.1 mM EGTA, 0.1 mM EDTA containing 1× Inhibitor cocktail (Roche). After incubation on ice for 15 minutes, 10% (v/v) NP-40 was added for a final concentration of 0.7% (v/v) and the cell suspension vortexed. Cells were spun at 14000 rpm for 15 minutes at 4° C. The pellet was washed twice with the above buffer and resuspended in 20 mM Tris-HCL pH 8.0, 0.4 M NaCl, 0.1 mM EGTA, 0.1 mM EDTA, and 1× Inhibitor cocktail (Roche). Nuclei were incubated on ice for 15 minutes and spun at 14000 rpm for 15 minutes at 4° C. Protein was quantitated as above and 15 μg/lane analyzed by western blotting using an antibody against total SMAD2 and SMAD3 (R&D), total NFκB (Cell Signaling) and actin (Sigma) to verify equal loading of the samples.
Statistics
Details of replicate experiments are in the figure legends. Data were analyzed using a one way analysis of variance (ANOVA) as compared to controls. Significance was determined at p<0.05 using KaleidaGraph software.
Results
TGF-β suppresses osteoclast apoptosis
We have examined the influences of TGF-β on osteoclast survival using mature osteoclasts that were differentiated in vitro from BalB/c marrow precursors cultured with RANKL and M-CSF (Figure 1). Cultures mature after 3.5 days following RANKL and M-CSF treatment. On day 5, the mature osteoclasts were treated with vehicle or TGF-β. Within four hours of treatment, the percentage apoptotic osteoclasts increased in vehicle treated cultures compared to either time zero or TGF-β treatment. The difference in the percentage of apoptotic osteoclasts comparing vehicle to TGF-β treatment persisted through 24 hours of culture. The total number of osteoclasts did not significantly differ between vehicle and TGF-β treated osteoclasts at any time point, but there were significantly fewer osteoclasts than time zero (data not shown). These data suggested that TGF-β could either be supporting osteoclast survival, increasing new osteoclast differentiation to replace apoptosed osteoclasts that had lifted off of the culture surface, or a combination of these events. To investigate these alternatives, we used several complementary approaches (Figure 2). Since RANKL is required for late osteoclast precursor fusion to form multinucleated cells [40], we examined osteoclasts differentiated with RANKL and M-CSF for TGF-β influences on mature osteoclasts with either RANKL or M-CSF removed (Figure 2A). Mature osteoclasts were either fixed or cultured for an additional 18 hours with fresh medium with RANKL and M-CSF, M-CSF alone, or RANKL alone added. TGF-β treatment repressed apoptosis in all culture conditions. Next we examined TGF-β influences using culture conditions where TGF-β repressed osteoclast differentiation. We and others have shown that, unlike RANKL/M-CSF cultures, co-cultures of osteoclast precursors and stromal cells respond to TGF-β treatment by a total suppression of differentiation through influences on stromal cell RANKL and OPG expression [3, 5]. We therefore examined the influences of TGF-β on mature osteoclast survival in co-culture (Figure 2B). Once cultures were mature (day 13), vehicle or TGF-β was added for an additional 24 (day 14) or 48 (day 15) hours of culture. Apoptosis significantly increased between days 13, 14 and 15 in vehicle treated cultures, whereas TGF-β treatment significantly suppressed this increase. We have demonstrated that osteoclast apoptosis involves proteolytic cleavage and activation of caspase 9 [41]. To determine whether TGF-β suppressed osteoclast apoptosis by blocking caspase 9 activation, we initially investigated whether blocking caspase 9 mimicked the influences of TGF-β (Figure 2C). Caspase 9 inhibition reduced osteoclast apoptosis to a level similar to TGF-β treatment and was not additive to TGF-β treatment when the caspase 9 inhibitor and TGF-β were both present. To further explore the influences of TGF-β on caspase 9 activation, we examined caspase 9 proteolytic activation after 6 hours of vehicle or TGF-β treatment and found that TGF-β reduced the proteolytic cleavage of caspase 9 to its active 34 kDa size (Figure 2D).
Fig. 1.
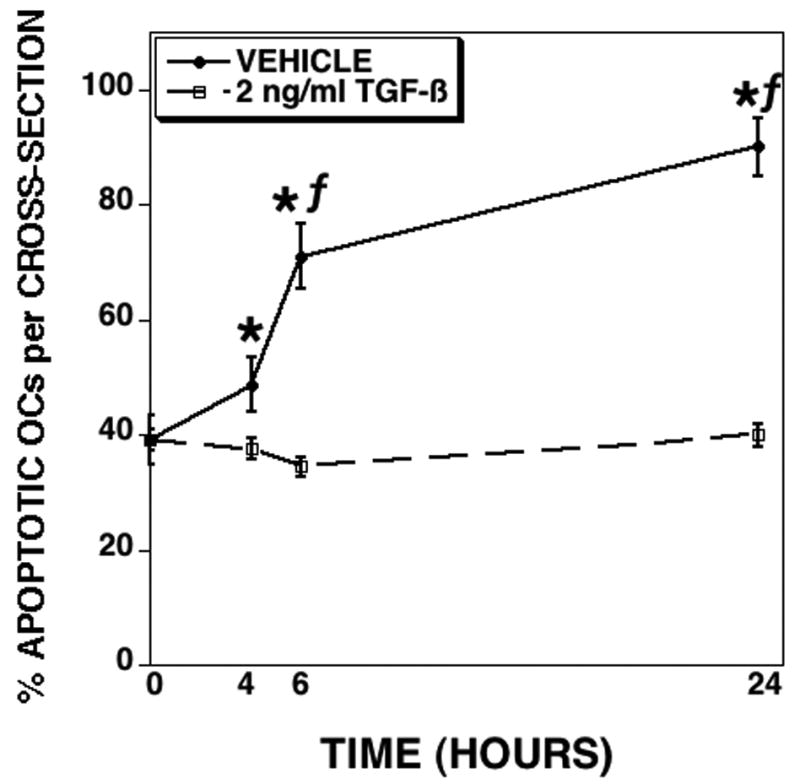
TGF-β promotes osteoclast survival. Mature osteoclasts were differentiated in vitro on coverslips in the presence of RANKL and M-CSF and assessed for responses to TGF-β. Osteoclasts were treated for the indicated time with vehicle or 2 ng/ml TGF-β. Cells were fixed and stained with Hoechst and TRAP. Each data point is the mean ± SEM of 6 replicate coverslips. This experiment was repeated 5 times and these data are from one experiment representative of the results. * p<0.05 comparing vehicle to TGF-β treatment. f p<0.05 comparing treatment to time zero.
Fig. 2.

TGF-β suppresses osteoclast apoptosis. (A) Mature osteoclasts differentiated in the presence of RANKL and M-CSF were fixed on day 5 (zero) or rinsed and fresh base medium. Medium was supplemented with M-CSF and RANKL (RL), M-CSF, or RANKL. Either vehicle or 2 ng/ml TGF-β were then added for an additional 18 hours of culture. The percentage of apoptotic osteoclasts was determined as in Figure 1. (B) Osteoclasts that were differentiated by co-culture with stromal cells for 13 days were either fixed (Day 13) or treated with vehicle or 2 ng/ml TGF-β for 24 (Day 14) or 48 (Day 15) hours. The percentage of apoptotic osteoclasts was determined as in Figure 1. (C) Mature osteoclasts differentiated in the presence of RANKL and M-CSF were pre-incubated with vehicle (DMSO) or a caspase 9 inhibitor for 1 hour prior to the addition of vehicle or 2 ng/ml TGF-β for 24 hours. The percentage of apoptotic osteoclasts was determined as in Figure 1. Each data point is the mean ± SEM of 6 (A&B) or 4 (C) replicate coverslips. Each experiment was repeated 2 times. These data are from one experiment representative of the results. * p<0.05 comparing vehicle to TGF-β treatment. f p<0.05 comparing time zero to individual treatment (A), or treatment to Day 13 (B), or treatment to DMSO (C). (D) Mature osteoclasts differentiated in the presence of RANKL and M-CSF were treated with vehicle (V) or 2 ng/ml TGF-β (T) for 6 hours prior to harvest. Forty μg of protein was loaded into each lane and Western blots were probed for the presence of latent (46 kDa) and cleaved active (34 kDa) caspase 9. This analysis was repeated twice and these data are representative of the results.
Apoptosis suppression requires ALK5/TβRI activation
We sought to resolve the receptors involved in TGF-β-mediated promotion of osteoclast survival. Initially, we examined the influences of TGF-β on expression of ALK1, the non-canonical receptor, and ALK5/TβRI in mature osteoclasts and found that TGF-β significantly increased ALK5/TβRI expression (Figure 3A). ALK1 expression was not significantly increased. As TGF-β is known to regulate tubulin expression polymerization and expression, we also determined if changes in tubulin were evident with TGF-β treatment [42, 43]. No change in tubulin expression was observed with TGF-β treatment (data not shown). This observation is not surprising given that TGF-β-mediated changes in tubulin expression occur primarily after long treatments with TGF-β (6-24 hours) whereas the data presented here are in response to short TGF-β treatments. To determine whether ALK5/TβRI was involved in TGF-β mediated osteoclast survival, we employed a chemical inhibitor that selectively blocks ALK5/TβRI without blocking ALK1 signaling [44] (Figure 3B). Blocking ALK5/TβRI increased the percentage of apoptotic osteoclasts by 24 hours of treatment in both vehicle and TGF-β treated cultures, thus blocking the influences of TGF-β on osteoclast survival. To explore the role of TβRII in TGF-β-mediated osteoclast survival, we examined osteoclasts in which the ligand binding domain of TβRII was excised. Mature osteoclasts from mice with the ligand binding domain of TβRII (exon 2) floxed were infected with adenoviral Cre to excise the binding domain and inactivate TβRII prior to TGF-β treatment. Figure 3C documents that we had effective reduction in full length TβRII mRNA with expression of the Cre recombinase. TGF-β treatment of Cre infected osteoclasts failed to suppress osteoclast apoptosis (Figure 3D).
Fig. 3.

Survival response involves ALK5/TβRI, not ALK1. (A) On day 5 of culture, mature osteoclasts differentiated with RANKL and M-CSF were treated with vehicle or 2 ng/ml TGF-β for 1 or 2 hours as indicated. RNA was harvested and assessed for ALK1 and ALK5/TβRI gene expression by Real Time PCR as outlined in the Materials and Methods section. (B) Mature osteoclasts were treated with either vehicle (DMSO) or specific ALK5/TβRI inhibitor SB-431542 for 1 hour prior to vehicle or TGF-β treatment for 8 or 24 hours. The percentage of apoptotic osteoclasts was determined as in Figure 1. (C and D) Mature osteoclasts differentiated with RANKL and M-CSF were cultured uninfected (NONE) or infected with empty vector (VECTOR) or Cre-expressing adenovirus (CRE) for 24 hours. (C) RNA was harvested and assessed for Cre-mediated excision of the TβRII ligand binding domain by Real Time PCR. (D) Cells were fixed and the percentage of apoptotic osteoclasts was determined as in Figure 1. Each of these experiments was repeated 2 times. Data are from one experiment representative of the results. A and C: Each data point is the mean ± SEM of 4 separate cultures. B and D: Each data point is the mean ± SEM of 6 replicate coverslips. * p<0.05 comparing vehicle to TGF-β treatment. f p<0.05 comparing treatment to DMSO (B) or no infection (NONE; E). These analyses were repeated 2 times and these data are representative of the results.
TGF-β activation of MEK and AKT pathways
We have shown that MEK, AKT, and NFκB are critical signals involved in osteoclast survival [35]. TAK1 is a MEK kinase that is activated by TGF-β, we examined its activation following TGF-β treatment of mature osteoclasts and observed rapid phosphorylation in response to treatment (Figure 4A). We and others have shown that osteoclast survival is dependent on activation of MEK, AKT, and NFκB [32-35]. Since TGF-β had been shown to activate these signaling pathways in a variety of cell types, we also examined TGF-β influences on osteoclast MEK and AKT pathway activation (Figure 4A). Within 5 minutes of TGF-β treatment, rapid phosphorylation of MEK1/2, AKT, IKKα/β, IκB, and NFκB and a decrease in total IκB protein were seen. To verify that TGF-β was activating NFκB, we infected mature osteoclasts with an NFκB reporter construct and found that 30 minutes of TGF-β treatment resulted in a 2-fold increase in reporter activity (Figure 4B). We also examined NFκB and SMAD activation as measured by nuclear localization (Figure 4C). Within five minutes of TGF-β treatment, NFκB and SMADs 2 and 3 levels were elevated in the nucleus and nuclear localization was sustained for up to 60 minutes. We were unable to detect nuclear localization of SMADs 1, 5, or 8 (data not shown). To control for contamination with non-nuclear proteins, blots were re-probed with tubulin and these were consistently negative (data not shown). We examined the influences of ALK5/TβRI blocking on nuclear localization of NFκB, SMAD2, and SMAD3 and found that blocking the receptor suppressed TGF-β-induced nuclear localization of these proteins (Figure 4D). To determine SMAD activation influences on kinase pathway activation of NFκB, we infected mature osteoclasts with dominant negative SMAD2 and SMAD3 (Figure 4E). Following TGF-β treatment, there was detectable nuclear localization of NFκB in the presence of dominant negative SMAD expression.
Fig. 4.

TGF-β rapidly activates signaling pathway components. On day 5 of culture, mature osteoclasts were treated with 2 ng/ml TGF-β for the indicated times. (A) Cells were rinsed on ice with cold PBS, extracts were harvested, and forty μg of protein was analyzed by Western blotting for the indicated phosphor-(p), total (t) protein, or tubulin. This experiment was repeated 6 times and these data are representative results. (B) Mature osteoclasts were infected with NFκB promoter reporter construct for 24 hours as outlined and then treated with 2 ng/ml TGF-β for the indicated time. Seventy-five micrograms of cell lysates were analyzed for luciferase activity as described above. This experiment was repeated twice and these data are from one experiment representative of the results. *p<0.05 compared to time zero. (C) Mature osteoclasts were treated for the indicated times with 2 ng/ml TGF-β prior to harvest, nuclear extraction and and five μg were analyzed by western blot analysis for total NFκB, SMAD2 or SMAD3. (D) Mature osteoclasts were harvested (0) or treated as above with vehicle (V), SB-431542 (SB), or 2 ng/ml TGF-β (T) alone or in combination as indicated for 5 minutes. Nuclear extracts were obtained as outlined in the Materials and Methods section. Five μg of protein were analyzed for total NFκB, SMAD2, or SMAD3 by western blotting. (E) Mature osteoclasts were infected with vector (MOI=100) or dominant negative SMADs 2 and 3 (MOI=50 each) for 18 hours prior to treatment with 2 ng/ml TGF-β for 5 minutes. Nuclear extracts were obtained as outlined in the Materials and Methods section. Five μg of protein were analyzed for total NFκB by western blotting. (C, D, and E) Each experiment was repeated twice and data are representative of the results.
Activation of TAK1, MEK, and AKT pathways are involved in TGF-β-mediated osteoclast survival signaling
Since MEK and AKT/NFκB are known osteoclast survival pathways [35] and TAK1, MEK and AKT/NFκB were rapidly activated with TGF-β treatment, we next examined the influences of blocking these pathways on TGF-β-mediated promotion of osteoclast survival (Figure 5). We used adenoviral delivery of dominant negative TAK1, MEK1, AKT, NIK, and IKK and mutant IκB (which blocks NFκB activation) to block these signaling components (Figure 5A). The dominant negative constructs increased osteoclast apoptosis in the vehicle treated cultures and TGF-β treatment could not overcome the induction of apoptosis. Examination of total TAK1, MEK, AKT, NIK, IKK, and IκB verify that dominant negative expression was successful (Figure 5B). Chemical blocking of MEK, AKT, and NFκB prior to TGF-β treatment had a similar impact on survival (data not shown).
Fig. 5.

Blocking TAK1, MEK, AKT, NIK, IKK, NFκB, SMAD32, or SMAD3 blocks TGF-β-mediated promotion of osteoclast survival. (A&B) On day 4 of culture, mature osteoclasts were infected for 18 hours with the indicated adenovirus prior to the addition of vehicle or 2 ng/ml TGF-β for 8 hours (vec = empty vector). (A) Cells were fixed and the percentage of apoptotic osteoclasts was determined as in Figure 1. Each data point is the mean ± SEM of 6 replicate coverslips. * p<0.05 comparing vehicle to TGF-β treatment. f p<0.05 comparing treatment to vector infected cells. (B) Parallel cultures were harvested for western blotting of 40 μg of cell protein to verify over-expression of recombinant proteins. Subsequent re-probing of the blots for tubulin levels confirmed equal lane loading (data not shown). These experiments were repeated 2 times and these data are from one experiment representative of the results.
TAK1 and MEK activation are required for TGF-β-mediated activation of the AKT/NFκB pathway
Because blocking TAK1, MEK, and AKT/NFκB blocked TGF-β-mediated promotion of osteoclast survival, we investigated the influences of adenoviral-mediated blocking of TAK1 or MEK1 on the AKT/NFκB signaling pathway (Figure 6A and B). In vector-infected osteoclasts, we again observed rapid phosphorylation of MEK1/2, AKT, IKK, IκB, and NFκB as well as auto-phosphorylation of TAK1 at Thr184 and Thr187 within the activation loop of TAK1. Importantly, infection with dominant negative TAK1 (Figure 6A) or MEK1 (Figure 6B) reduced or blocked phosphorylation of pathway components without affecting TAK1 activation. These data indicate that TGF-β activation of TAK1 is upstream of MEK and AKT activation and MEK is upstream of AKT activation. A similar pattern was observed with 1 hour of chemical inhibition of MEK1/2 activation prior to TGF-β treatment (data not shown). To verify influences of blocking TAK1 or MEK on pathway activation, we examined the impact of dnTAK1 or dnMEK1 expression on nuclear localization of NFκB (Figure 6C). Within 5 minutes of TGF-β treatment, nuclear NFκB levels are increased in vehicle treated cells. Expression of dnTAK1 or dnMEK1 abrogated this response. We examined the influences of blocking the AKT/NFκB pathway on constitutively active MEK activity (Figure 6D). Constitutively active MEK reduced osteoclast apoptosis and TGF-β treatment did not further reduce apoptosis with this treatment. Expression of dominant negative forms of AKT, NIK, IKK, or mutant IκB in osteoclasts expressing constitutively active MEK eliminated the influences of MEK activation on osteoclast survival while also blocking the influences of TGF-β on osteoclast survival. To further clarify whether the AKT/NFκB pathway is downstream of MEK in TGF-β-mediated suppression of osteoclast apoptosis, osteoclasts were infected with constitutively active AKT prior to treatment with a pharmacological inhibitor of MEK1/2, followed by treatment with either vehicle or TGF-β (Figure 6E). MEK inhibition increased osteoclast apoptosis while constitutively active AKT suppressed apoptosis. Importantly, constitutively active AKT abrogated the influence of MEK1/2 inhibition on osteoclast apoptosis.
Fig. 6.

TGF-β-mediated activation of TAK1 and MEK are required for downstream activation of AKT, IKK, IκB and NFκB. (A and B) On day 4 of culture, mature osteoclasts differentiated with RANKL and M-CSF were infected with vector or (A) dnTAK1 or (B) dnMEK1 (multiplicity of infection =100) for 18 hours prior to treatment with vehicle (-) or 2 ng/ml TGF-β (+) for 5 minutes. Cells were rinsed, extracts were harvested, and 40 μg of protein was analyzed by Western blotting for phospho-TAK1 Thr184 and Thr187 (pTAK 184 and pTAK 187), total TAK1 (tTAK), phospho-MEK (pMEK), total MEK (tMEK), phospho-AKT (pAKT), total AKT (tAKT), phospho-IKKα/β (pIKKα/β), total IKKα (tIKKα), phospho-IκB (pIκB), total IκB (tIκB), phospho-NFκB, (pNFκB), total NFκB (tNFκB), or tubulin as indicated. This experiment was repeated 4 times and these data are representative of the results. (C) Osteoclasts were infected with vector, dnTAK1, or dnMEK1 as above and treated for the indicted time with 2 ng/ml TGF-β prior to isolation and extraction of nuclei. Western blot analysis was performed as described for total NFκB. (D) On day 4 of culture, mature osteoclasts were infected with vector, constitutively active MEK, or dominant negative AKT, NIK, IKK or mutant IκB as indicated (MOI = 50 each for all double infections and MOI = 100 for single infections). Eighteen hours later, osteoclasts were treated with vehicle or 2 ng/ml TGF-β treatment for 8 hours. Cells were fixed and analyzed as in figure 1. This experiment was repeated 2 times and these data are representative of the results. *p<0.05 comparing vehicle to TGF-β treatment. f p<0.05 comparing treatment to vector infected cells. (E) On day 4 of culture, mature osteoclasts were infected with constitutively active AKT (MOI = 100). Eighteen hours later, osteoclasts were treated for 1 hour with DMSO or the MEK1/2 inhibitor U0126 prior to vehicle or 2 ng/ml TGF-β treatment for 8 hours. Cells were fixed and analyzed as in figure 1. This experiment was repeated 2 times and these data are representative of the results. *p<0.05 comparing vehicle to TGF-β treatment. f p<0.05 comparing treatment to vector infected cells. #p<0.05 compared to CA AKT/DMSO/vehicle treatment.
We have shown that expression of Bcl-2 protects osteoclasts from apoptosis induced by suppression of protein synthesis [41]. Because of this, investigated the mechanisms by which TGF-β promotes osteoclast survival by examining influences on expression of pro-survival members of the Bcl2 family. Within one hour of TGF-β treatment, there was increased BclXL (Figure 7A) and Mcl-1 (Figure 7B) expression. We were unable to detect rapid increases in expression of Bcl2 or the Inhibitor of Apoptosis family member Survivin (data not shown). Moreover, we did not see any decrease in expression of pro-apoptosis Bcl2 family members Bax, Bid, Bik, Bim, Box, Bcl2-like 11,12,13,or 14, or Bnip1,2,or 3 (data not shown). To determine the roles of TAK1, MEK, and SMAD2/3 signaling in these responses, mature osteoclast were infected with dnTAK1, dnMEK1, or combined dnSMAD2 and dnSMAD3. Repression of TAK1 or MEK1 reduced TGF-β induction of BclXL (Figure 7C). In contrast, repression of TAK1, MEK1 or SMAD2/3 reduced TGF-β induction of Mcl-1 (Figure 7D).
Fig 7.

(A&C) Mature osteoclasts were treated with vehicle or 2 ng/ml TGF-β for the indicated times prior to RNA harvest and Real Time PCR analysis. (B&D). Mature osteoclasts were infected with vector or the indicated dominant negative adenovirus (M)I = 100) for 18 hours. Cultures were treated for 1 hour with vehicle or TGF-β prior to RNA harvest and Real Time PCR analysis. Threshold cycles were normalized to tubulin and expression was determined relative to vehicle treatment. These experiments were repeated 2 times. These data are representative of the results. *p<0.05 comparing vehicle to TGF-β treatment. f p<0.05 comparing treatment to vector infected cells.
Discussion
In these studies we have documented that physiological TGF-β concentrations suppress osteoclast apoptosis through rapid activation of MEK, which activates the AKT/NFκB pathway. Biologically, this is likely to be crucial in the sustained resorption that takes place during conditions of excessive bone loss such as tumor-driven osteolysis or post-menopausal osteoporosis. TGF-β is released from the bone by osteoclastic activity, and we have shown that osteoclasts synthesize and activate TGF-β [45]. Thus, during periods of high bone turnover, active TGF-β levels are relatively high. If elevated levels of TGF-β promoted osteoclast apoptosis, the expectation would be that osteoclast apoptosis would occur during periods of high bone turnover, which has never been observed. Indeed, apoptosis of resorbing osteoclasts would provide negative feedback to repress resorption, which has also never been documented.
It is likely that the differences between our results and those of Hughes et al. [31] is due to altered sensitivity to TGF-β between the two model systems and/or the presence of stromal cells in the un-fractionated cells used by Hughes et al.. There are other differences between our protocols as well, notably the differentiation stimulus (we used RANKL and M-CSF and Hughes et al. used vitamin D). We attempted to examine this by replicating the protocol in Hughes et al, but were unable to obtain osteoclasts by this method (data not shown). In addition, the Hughes et al. osteoclasts were derived from C57BL/6-derived marrow. We therefore examined this strain of mice using RANKL and M-CSF-derived osteoclasts, which behaved in a manner similar to the Balb/c-derived cells reported here (data not shown). Different mouse vendors could also be contributing to differences, a possibility that is strengthened by the observation of variability in mouse colonies between vendors [46]. The approach to assess apoptosis also differed in that Hughes et al. observed that apoptotic osteoclasts lifted off of the culture surface and chromatin condensation of floating cells was used as the apoptosis marker. All remaining adherent cells were scored as viable. We have observed that intact osteoclasts obtained with RANKL and M-CSF remain adhered to the culture surface whether alive or apoptotic [35]. We have also published that no intact cells can be recovered from the medium, underscoring a basic difference in these two model systems [35].
We examined whether we were measuring a replacement of apoptotic osteoclasts with newly differentiated osteoclasts since TGF-β stimulates osteoclast differentiation in the presence of RANKL and M-CSF (3,5). We used several approaches to address this question. Our initial experiment was to examine responses when RANKL was removed from the cultures, as RANKL signaling is required for the final events in osteoclast differentiation. The observation that TGF-β suppressed osteoclast apoptosis in the absence of RANKL supports a pro-survival response. We also examined osteoclasts co-cultured with stromal cells since the dose of TGF-β used suppresses differentiation of these cultures [5]. In these studies, TGF-β also suppressed apoptosis. Since TGF-β completely suppresses osteoclast differentiation in this model system, these results clearly support the hypothesis that TGF-β promotes osteoclast survival. We also found that inhibition of caspase 9 mimicked TGF-β effects, but was not additive with TGF-β, and that TGF-β treated cultures had less activated caspase 9. These data confirm that TGF-β represses caspase 9-mediated osteoclast apoptosis. Published studies of human osteoclast lineage cell responses to TGF-β have been restricted to differentiation responses. With respect to these responses, TGF-β has been reported to induce differentiation of precursors when cultured with RANKL and M-CSF, similar to murine osteoclast precursor responses [6, 47].
Interestingly, human osteoclasts have been shown to require NFκB for survival, supporting that targeting this survival factor is a common mechanism to support survival of human and murine osteoclasts [48]. Recently, this same group published that human osteoclasts differentiated from cord blood responded differently to apoptosis induction than human osteoclasts differentiated from peripheral blood, suggesting that the source of human cells must be carefully evaluated in human osteoclast survival studies [49]. The studies reported here were carried out with murine osteoclasts cultured on plastic and glass surfaces. Since osteoclasts release and activate bone-bound TGF-β, it will be important to resolve the role of TGF-β effects on osteoclasts that are in their natural environment on bone in a mixed cell milieu, which is a future direction of this project.
As noted above, TGF-β induces apoptosis of many cell types. The best studied exception to this observation occurs in tumor cells in which various alterations in the TGF-β signaling pathway allows for escape from the pro-apoptotic effects of TGF-β. TGF-β has been shown to promote neuronal cell survival by stimulation and activation of ALK1, an alternate TGF-β receptor, resulting in activation and phosphorylation of SMADs 1 and 5 (data not shown) [50-52]. In studies of Konig et al [18], TGF-β stimulated neuronal cell ALK1, but not ALK5/TβRI, expression within one hour of TGF-β treatment. In contrast, TGF-β stimulated expression of ALK5/TβRI in osteoclasts between 1 and 2 hours of TGF-β treatment. There was no significant influence on ALK1 expression in osteoclasts. We also were unable to detect TGF-β-mediated increase in nuclear localization of SMADs 1 or 5, which are the downstream effectors of ALK1. These data further support that osteoclast survival signaling in response to TGF-β is not through activation of the ALK1/SMADs 1 and 5 pathways. To determine the role of ALK5/TβRI in TGF-β-mediated promotion of osteoclast survival, we examined responses in osteoclasts in the presence of an inhibitor of ALK5/TβRI that does not inhibit ALK1. We found that ALK5/TβRI inhibition blocked TGF-β-mediated osteoclast survival and inhibition of ALK5/TβRI blocked nuclear localization of NFκB, SMAD2, and SMAD3. These data support that TGF-β activates ALK5/TβRI to repress apoptosis, unlike reports in neuronal cells. To further investigate canonical signaling, we examined osteoclasts in which the TβRII ligand binding domain was excised. In cells unable to respond to TGF-β by activation of ALK5/TβRI - TβRII complex, TGF-β did not suppress apoptosis. Taken together, these data confirm the requirement of the canonical receptor complex in TGF-β support of osteoclast survival.
TGF-β-mediated phosphorylation of osteoclast TAK1, MEK1/2, AKT, IKK, and NFκB occurred rapidly, within 5 minutes of treatment. Transient phosphorylation of IκB was observed and total IκB decreased with time, which is expected since phosphorylation leads to proteasome-mediated degradation of IκB [53]. Thus, loss of total IκB is the most likely explanation for the reduced phospho-IκB. Phosphorylation of NFκB was also slightly reduced by 30 minutes of treatment, although direct measurement of NFκB activity with a reporter construct after 30 minutes of TGF-β treatment clearly showed significant retained activation. We observed nuclear localization of SMADs 2 and 3 within the same time period as activation of the MEK/AKT pathway, supporting that these pathways are activated in parallel and not sequentially. This is further supported by the observation that dnSMAD2/3 were unable to block TGF-β-mediated induction of NFκB nuclear localization. In some cell types the effects of Smad2 and Smad3 activation in response to TGF-β are different. For instance, Smad3 preferentially regulates expression of c-Myc, Id1 and p21Cip1 over Smad2 in response to TGF-β treatment. This preferential response occurs through a higher affinity of Smad3 to co-factors such as E2F4/5, ATF3 and FoxO required for Smad targeting to these gene promoters [54-56]. In addition, Smad3 induces CTGF expression and downregulates E-cadherin in response to TGF-β whereas induction of MMP-2 is Smad2-dependent in proximal-tubule epithelial cells [57]. However, a number of studies document a role for both Smad2 and Smad3 in mediating the effects of TGF-β, including the data described here. Studies have shown that MEK/ERK activation can block SMAD nuclear localization in some cell types [28, 58]. Since we observed parallel SMAD and MEK pathway activation and sustained SMAD nuclear localization, these interactions are not likely to be important in TGF-β influences on osteoclasts.
Since we observed rapid phosphorylation of MEK and AKT/NFκB and these pathways are known to promote osteoclast survival, we pursued the roles of these pathways in TGF-β-mediated osteoclast survival. In the absence of TGF-β treatment, dominant negative blocking of SMAD2, SMAD3, TAK1, MEK, AKT, or IKK as well as blocking NFκB activation using a mutant IκB led to significant increase in osteoclast apoptosis. Since osteoclasts secrete and activate TGF-β, this could be, at least in part, due to blocking autocrine TGF-β effects. Dominant negative NIK did not appear to significantly increase basal apoptosis, perhaps due to large variability in these cultures. Importantly, each of these inhibitory constructs blocked TGF-β-mediated repression of osteoclast apoptosis, indicating that these signaling components are downstream effectors of TGF-β promoted osteoclast survival.
TAK1 has been shown to activate MEK3, MEK6, MEK4, and MEK7, leading to JNK activation [59-61]. We have been unable to document TGF-β–mediated JNK activation in osteoclasts (data not shown). Previous studies have addressed potential TAK1-mediated activation of the MEK/ERK pathway, but have not demonstrated activation of MEK downstream of TGF-β-induced TAK1. In studies by Hirota et al., the effects of TGF-β in an articular chrondrocyte cell line and a hepatoma cell line were contrasted [62]. The authors were able to illustrate a MEK-dependent transcriptional response mediated by TGF-β in chondroctyes. This effect was not altered by modulation of TAK1 function. In contrast, while the effects in hepatoma cells were MEK-independent, TGF-β induced transcription could be blocked through expression of dnTAK1. Thus, these studies suggested that TGF-β-mediated MEK activation was not accomplished via TAK1. Additional studies have also shown that although TAK1 activation is required to induce Akt activation in response to iron accumulation, blocking the function of TAK1 does not block ERK activation mediated by iron accumulation [63], again showing that MEK-mediated responses are not TAK1-dependent To our knowledge, this is the first report that TAK1 also targets MEK1/2 signaling. Therefore, TAK1 may be a general MEK kinase capable of activating multiple MEK signaling pathways. Our investigation of the interactions between MEK and AKT/NFκB pathways following TGF-β treatment lead to the unexpected observation that MEK activation causes phosphorylation and activation of the AKT/NFκB pathway. Our initial observations were from chemical inhibition studies, which resulted in a similar blocking. Given the possibility of nonspecific influences of pharmacological inhibitors, we elected to employ the more selective adenoviral blocking strategy, which revealed the identical response. Blocking MEK increased osteoclast apoptosis, which was reversed by expression of constitutively active AKT. Likewise, blocking the AKT/NFκB pathway reduced the ability of constitutively active MEK to promote osteoclast survival. Our results, summarized in Figure 8, reveal that TGF-β-mediated promotion of osteoclast survival is mediated by a novel TGF-β RI/RII and downstream TAK1/MEK/AKT/NFκB and SMAD2/3 pathways. Induction of these pathways induces expression of pro-survival Bcl2 family members BclXL and Mcl-1 to block caspase-mediated osteoclast apoptosis. This scenario is in concordance with our observation that blocking protein synthesis rapidly induces osteoclast apoptosis and we conjecture that synthesis of BclXL and Mcl-1 are pivotal in this response [41]. These results also suggest that studies published of the roles of MEK and AKT on osteoclast survival and, perhaps, differentiation may need to be re-evaluated to determine whether MEK is likewise regulating AKT following stimulation with other ligands in addition to TGF-β.
Fig. 8.

Model of proposed signaling pathway. In mature osteoclasts, TGF-β rapidly activates both SMAD and TAK1. TAK1 activation leads to sequential activation of MEK1/2, AKT, NIK, and IKKα/β, leading to phosphorylation of IκB. This causes targeted degradation of IκB, allowing subsequent NFκB nuclear localization to promote osteoclast survival. TGF-β-dependent SMAD2/3 activation and nuclear localization also occurs in parallel. NFκB activates transcription of BCLXL and Mcl-1 whereas SMAD2/3 activate transcription of Mcl-1.
Acknowledgments
Support for this work was provided by the NIH grant R01 DE14680 and The Mayo Foundation. We thank Dr. Beth Lee for the gift of the GST RANKL expression construct. We also thank Drs. Patricia Collin-Osdoby and Philip Osdoby for their advice on the GST RANKL purification as well as the Mayo Clinic Bone Histomorphometry Core. We are grateful to Dr. Hal Moses for his gift of the TβRII exon 2 floxed mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-beta1 to the bone. Endocr Rev. 2005;26:743–774. doi: 10.1210/er.2004-0001. [DOI] [PubMed] [Google Scholar]
- 2.Hattersley G, Chambers TJ. Effects of transforming growth factor beta 1 on the regulation of osteoclastic development and function. J Bone Miner Res. 1991;6:165–172. doi: 10.1002/jbmr.5650060210. [DOI] [PubMed] [Google Scholar]
- 3.Quinn JM, Itoh K, Udagawa N, Hausler K, Yasuda H, Shima N, Mizuno A, Higashio K, Takahashi N, Suda T, Martin TJ, Gillespie MT. Transforming growth factor beta affects osteoclast differentiation via direct and indirect actions. J Bone Miner Res. 2001;16:1787–1794. doi: 10.1359/jbmr.2001.16.10.1787. [DOI] [PubMed] [Google Scholar]
- 4.Sells Galvin RJ, Gatlin CL, Horn JW, Fuson TR. TGF-beta enhances osteoclast differentiation in hematopoietic cell cultures stimulated with RANKL and M-CSF. Biochem Biophys Res Commun. 1999;265:233–239. doi: 10.1006/bbrc.1999.1632. [DOI] [PubMed] [Google Scholar]
- 5.Karst M, Gorny G, Galvin RJ, Oursler MJ. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J Cell Physiol. 2004;200:99–106. doi: 10.1002/jcp.20036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karsdal MA, Hjorth P, Henriksen K, Kirkegaard T, Nielsen KL, Lou H, Delaisse JM, Foged NT. Transforming growth factor-beta controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. J Biol Chem. 2003;278:44975–44987. doi: 10.1074/jbc.M303905200. [DOI] [PubMed] [Google Scholar]
- 7.Fiorelli G, Ballock RT, Wakefield LM, Sporn MB, Gori F, Masi L, Frediani U, Tanini A, Bernabei PA, Brandi ML. Role for autocrine TGF-beta 1 in regulating differentiation of a human leukemic cell line toward osteoclast-like cells. J Cell Physiol. 1994;160:482–490. doi: 10.1002/jcp.1041600311. [DOI] [PubMed] [Google Scholar]
- 8.Rotello RJ, Lieberman RC, Purchio AF, Gerschenson LE. Coordinated regulation of apoptosis and cell proliferation by transforming growth factor beta 1 in cultured uterine epithelial cells. Proc Natl Acad Sci U S A. 1991;88:3412–3415. doi: 10.1073/pnas.88.8.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel P, Varghese E, Ding G, Fan S, Kapasi A, Reddy K, Franki N, Nahar N, Singhal P. Transforming growth factor beta induces mesangial cell apoptosis through NO- and p53-dependent and -independent pathways. J Investig Med. 2000;48:403–410. [In Process Citation] [PubMed] [Google Scholar]
- 10.Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci U S A. 1992;89:5408–5412. doi: 10.1073/pnas.89.12.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haufel T, Dormann S, Hanusch J, Schweiger A, Baur G. Three distinct roles for TGF-beta during intercellular induction of apoptosis: a review. Anticancer Research. 1999;19:105–111. [PubMed] [Google Scholar]
- 12.Dormann S, Schwieger A, Hanusch J, Haufel T, Engelmann I, Bauer G. Intercellular induction of apoptosis through modulation of endogenous survival factor concentration: a review. Anticancer Res. 1999;19:87–103. [PubMed] [Google Scholar]
- 13.Brown TL, Patil S, Howe PH. Analysis of TGF-beta-inducible apoptosis. Methods Mol Biol. 2000;142:149–167. doi: 10.1385/1-59259-053-5:149. [DOI] [PubMed] [Google Scholar]
- 14.Arsura M, FitzGerald MJ, Fausto N, Sonenshein GE. Nuclear factor-kappaB/Rel blocks transforming growth factor beta1- induced apoptosis of murine hepatocyte cell lines. Cell Growth Differ. 1997;8:1049–1059. [PubMed] [Google Scholar]
- 15.Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. TGF-beta-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol. 2001;3:708–714. doi: 10.1038/35087019. [DOI] [PubMed] [Google Scholar]
- 16.Fink SP, Swinler SE, Lutterbaugh JD, Massague J, Thiagalingam S, Kinzler KW, Vogelstein B, Willson JK, Markowitz S. Transforming growth factor-beta-induced growth inhibition in a Smad4 mutant colon adenoma cell line. Cancer Res. 2001;61:256–260. [PubMed] [Google Scholar]
- 17.Lotem J, Sachs L. Hematopoietic cytokines inhibit apoptosis induced by transforming growth factor beta 1 and cancer chemotherapy compounds in myeloid leukemic cells. Blood. 1992;80:1750–1757. [PubMed] [Google Scholar]
- 18.Konig HG, Kogel D, Rami A, Prehn JH. TGF-{beta}1 activates two distinct type I receptors in neurons: implications for neuronal NF-{kappa}B signaling. J Cell Biol. 2005;168:1077–1086. doi: 10.1083/jcb.200407027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serra R, Crowley MR. Mouse models of transforming growth factor beta impact in breast development and cancer. Endocr Relat Cancer. 2005;12:749–760. doi: 10.1677/erc.1.00936. [DOI] [PubMed] [Google Scholar]
- 20.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 21.Liu F. Receptor-regulated Smads in TGF-beta signaling. Front Biosci. 2003;8:s1280–1303. doi: 10.2741/1149. [DOI] [PubMed] [Google Scholar]
- 22.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 23.Schiffer M, von Gersdorff G, Bitzer M, Susztak K, Bottinger EP. Smad proteins and transforming growth factor-beta signaling. Kidney Int Suppl. 2000;77:S45–52. doi: 10.1046/j.1523-1755.2000.07708.x. [DOI] [PubMed] [Google Scholar]
- 24.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 25.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. Embo J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reimann T, Hempel U, Krautwald S, Axmann A, Scheibe R, Seidel D, Wenzel KW. Transforming growth factor-beta1 induces activation of Ras, Raf-1, MEK and MAPK in rat hepatic stellate cells. FEBS Lett. 1997;403:57–60. doi: 10.1016/s0014-5793(97)00024-0. [DOI] [PubMed] [Google Scholar]
- 27.Axmann A, Seidel D, Reimann T, Hempel U, Wenzel KW. Transforming growth factor-beta1-induced activation of the Raf-MEK-MAPK signaling pathway in rat lung fibroblasts via a PKC-dependent mechanism. Biochem Biophys Res Commun. 1998;249:456–460. doi: 10.1006/bbrc.1998.9188. [DOI] [PubMed] [Google Scholar]
- 28.Hayashida T, Decaestecker M, Schnaper HW. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-beta-dependent responses in human mesangial cells. Faseb J. 2003;17:1576–1578. doi: 10.1096/fj.03-0037fje. [DOI] [PubMed] [Google Scholar]
- 29.Inoki K, Haneda M, Ishida T, Mori H, Maeda S, Koya D, Sugimoto T, Kikkawa R. Role of mitogen-activated protein kinases as downstream effectors of transforming growth factor-beta in mesangial cells. Kidney Int. 2000;58 77:S76–80. doi: 10.1046/j.1523-1755.2000.07712.x. [DOI] [PubMed] [Google Scholar]
- 30.Wilkes MC, Leof EB. Transforming growth factor beta activation of c-Abl is independent of receptor internalization and regulated by phosphatidylinositol 3-kinase and PAK2 in mesenchymal cultures. J Biol Chem. 2006;281:27846–27854. doi: 10.1074/jbc.M603721200. [DOI] [PubMed] [Google Scholar]
- 31.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2:1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- 32.Miyazaki T, Katagiri H, Kanegae Y, Takayanagi H, Sawada Y, Yamamoto A, Pando MP, Asano T, Verma IM, Oda H, Nakamura K, Tanaka S. Reciprocal role of ERK and NF-kappaB pathways in survival and activation of osteoclasts. J Cell Biol. 2000;148:333–342. doi: 10.1083/jcb.148.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee ZH, Lee SE, Kim CW, Lee SH, Kim SW, Kwack K, Walsh K, Kim HH. IL-1alpha Stimulation of Osteoclast Survival through the PI 3- Kinase/Akt and ERK Pathways. J Biochem (Tokyo) 2002;131:161–166. doi: 10.1093/oxfordjournals.jbchem.a003071. [DOI] [PubMed] [Google Scholar]
- 34.Lee SE, Chung WJ, Kwak HB, Chung CH, Kwack KB, Lee ZH, Kim HH. Tumor necrosis factor-alpha supports the survival of osteoclasts through the activation of Akt and ERK. J Biol Chem. 2001;276:49343–49349. doi: 10.1074/jbc.M103642200. [DOI] [PubMed] [Google Scholar]
- 35.Gingery A, Bradley E, Shaw A, Oursler MJ. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89:165–179. doi: 10.1002/jcb.10503. [DOI] [PubMed] [Google Scholar]
- 36.Jadrich JL, O'Connor MB, Coucouvanis E. Expression of TAK1, a mediator of TGF-beta and BMP signaling, during mouse embryonic development. Gene Expr Patterns. 2003;3:131–134. doi: 10.1016/s1567-133x(03)00012-7. [DOI] [PubMed] [Google Scholar]
- 37.Ragab AA, Lavish SA, Banks MA, Goldberg VM, Greenfield EM. Osteoclast differentiation requires ascorbic acid. J Bone Miner Res. 1998;13:970–977. doi: 10.1359/jbmr.1998.13.6.970. [DOI] [PubMed] [Google Scholar]
- 38.Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC. TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene. 2002;21:5783–5790. doi: 10.1038/sj.onc.1205681. [DOI] [PubMed] [Google Scholar]
- 39.Smith C, Andreakos E, Crawley JB, Brennan FM, Feldmann M, Foxwell BM. NF-kappaB-inducing kinase is dispensable for activation of NF-kappaB in inflammatory settings but essential for lymphotoxin beta receptor activation of NF-kappaB in primary human fibroblasts. J Immunol. 2001;167:5895–5903. doi: 10.4049/jimmunol.167.10.5895. [DOI] [PubMed] [Google Scholar]
- 40.Ishii M, Iwai K, Koike M, Ohshima S, Kudo-Tanaka E, Ishii T, Mima T, Katada Y, Miyatake K, Uchiyama Y, Saeki Y. RANKL-induced expression of tetraspanin CD9 in lipid raft membrane microdomain is essential for cell fusion during osteoclastogenesis. J Bone Miner Res. 2006;21:965–976. doi: 10.1359/jbmr.060308. [DOI] [PubMed] [Google Scholar]
- 41.Oursler MJ, Bradley EW, Elfering SL, Giulivi C. Native, not nitrated, cytochrome c and mitochondria-derived hydrogen peroxide drive osteoclast apoptosis. Am J Physiol Cell Physiol. 2005;288:C156–168. doi: 10.1152/ajpcell.00092.2004. [DOI] [PubMed] [Google Scholar]
- 42.Gundersen GG, Kim I, Chapin CJ. Induction of stable microtubules in 3T3 fibroblasts by TGF-beta and serum. J Cell Sci. 1994;107(Pt 3):645–659. doi: 10.1242/jcs.107.3.645. [DOI] [PubMed] [Google Scholar]
- 43.Lomri A, Marie PJ. Effects of transforming growth factor type beta on expression of cytoskeletal proteins in endosteal mouse osteoblastic cells. Bone. 1990;11:445–451. doi: 10.1016/8756-3282(90)90141-k. [DOI] [PubMed] [Google Scholar]
- 44.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 45.Oursler MJ. Osteoclast synthesis and secretion and activation of latent transforming growth factor beta. J Bone Miner Res. 1994;9:443–452. doi: 10.1002/jbmr.5650090402. [DOI] [PubMed] [Google Scholar]
- 46.Terszowski G, Muller SM, Bleul CC, Blum C, Schirmbeck R, Reimann J, Pasquier LD, Amagai T, Boehm T, Rodewald HR. Evidence for a functional second thymus in mice. Science. 2006;312:284–287. doi: 10.1126/science.1123497. [DOI] [PubMed] [Google Scholar]
- 47.Massey HM, Scopes J, Horton MA, Flanagan AM. Transforming growth factor-beta1 (TGF-beta) stimulates the osteoclast-forming potential of peripheral blood hematopoietic precursors in a lymphocyte-rich microenvironment. Bone. 2001;28:577–582. doi: 10.1016/s8756-3282(01)00432-x. [DOI] [PubMed] [Google Scholar]
- 48.Penolazzi L, Lambertini E, Borgatti M, Piva R, Cozzani M, Giovannini I, Naccari R, Siciliani G, Gambari R. Decoy oligodeoxynucleotides targeting NF-kappaB transcription factors: induction of apoptosis in human primary osteoclasts. Biochem Pharmacol. 2003;66:1189–1198. doi: 10.1016/s0006-2952(03)00470-2. [DOI] [PubMed] [Google Scholar]
- 49.Penolazzi L, Pocaterra B, Tavanti E, Lambertini E, Vesce F, Gambari R, Piva R. Human osteoclasts differentiated from umbilical cord blood precursors are less prone to apoptotic stimuli than osteoclasts from peripheral blood. Apoptosis. 2008;13:553–561. doi: 10.1007/s10495-008-0188-7. [DOI] [PubMed] [Google Scholar]
- 50.Ottaviani E, Barbieri D, Malagoli D, Kletsas D. Involvement of PI 3-kinase, PKA and PKC in PDGF- and TGF-beta-mediated prevention of 2-deoxy-D-ribose-induced apoptosis in the insect cell line, IPLB-LdFB. Cell Biol Int. 2001;25:171–177. doi: 10.1006/cbir.2000.0582. [DOI] [PubMed] [Google Scholar]
- 51.Saile B, Matthes N, El Armouche H, Neubauer K, Ramadori G. The bcl, NFkappaB and p53/p21WAF1 systems are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-beta or TNF-alpha on activated hepatic stellate cells. Eur J Cell Biol. 2001;80:554–561. doi: 10.1078/0171-9335-00182. [DOI] [PubMed] [Google Scholar]
- 52.Zhu Y, Culmsee C, Klumpp S, Krieglstein J. Neuroprotection by transforming growth factor-beta1 involves activation of nuclear factor-kappaB through phosphatidylinositol-3-OH kinase/Akt and mitogen-activated protein kinase-extracellular-signal regulated kinase1,2 signaling pathways. Neuroscience. 2004;123:897–906. doi: 10.1016/j.neuroscience.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 53.Magnani M, Crinelli R, Bianchi M, Antonelli A. The ubiquitin-dependent proteolytic system and other potential targets for the modulation of nuclear factor-kB (NF-kB) Curr Drug Targets. 2000;1:387–399. doi: 10.2174/1389450003349056. [DOI] [PubMed] [Google Scholar]
- 54.Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 55.Kang Y, Chen CR, Massague J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003;11:915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- 56.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 57.Phanish MK, Wahab NA, Colville-Nash P, Hendry BM, Dockrell ME. The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGFbeta1 responses in human proximal-tubule epithelial cells. Biochem J. 2006;393:601–607. doi: 10.1042/BJ20051106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blanchette F, Rivard N, Rudd P, Grondin F, Attisano L, Dubois CM. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor beta 1-induced furin gene transactivation. J Biol Chem. 2001;276:33986–33994. doi: 10.1074/jbc.M100093200. [DOI] [PubMed] [Google Scholar]
- 59.Moriguchi T, Kuroyanagi N, Yamaguchi K, Gotoh Y, Irie K, Kano T, Shirakabe K, Muro Y, Shibuya H, Matsumoto K, Nishida E, Hagiwara M. A novel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J Biol Chem. 1996;271:13675–13679. doi: 10.1074/jbc.271.23.13675. [DOI] [PubMed] [Google Scholar]
- 60.Shirakabe K, Yamaguchi K, Shibuya H, Irie K, Matsuda S, Moriguchi T, Gotoh Y, Matsumoto K, Nishida E. TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J Biol Chem. 1997;272:8141–8144. doi: 10.1074/jbc.272.13.8141. [DOI] [PubMed] [Google Scholar]
- 61.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 62.Hirota Y, Tsukazaki T, Yonekura A, Miyazaki Y, Osaki M, Shindo H, Yamashita S. Activation of specific MEK-ERK cascade is necessary for TGFbeta signaling and crosstalk with PKA and PKC pathways in cultured rat articular chondrocytes. Osteoarthritis Cartilage. 2000;8:241–247. doi: 10.1053/joca.1999.0297. [DOI] [PubMed] [Google Scholar]
- 63.Chen L, Xiong S, She H, Lin SW, Wang J, Tsukamoto H. Iron causes interactions of TAK1, p21ras, and phosphatidylinositol 3-kinase in caveolae to activate IkappaB kinase in hepatic macrophages. J Biol Chem. 2007;282:5582–5588. doi: 10.1074/jbc.M609273200. [DOI] [PubMed] [Google Scholar]