

Abstract
SIRT1 is a nicotinamide adenosine dinucleotide-dependent deacetylase that orchestrates key metabolic adaptations to nutrient deprivation in peripheral tissues. SIRT1 is induced also in the brain by reduced energy intake. However, very little is known about SIRT1 distribution and the biochemical phenotypes of SIRT1-expressing cells in the neuraxis. Unknown are also the brain sites in which SIRT1 is regulated by energy availability and whether these regulations are altered in a genetic model of obesity. To address these issues, we performed in situ hybridization histochemistry analyses and found that Sirt1 mRNA is highly expressed in metabolically relevant sites. These include, but are not limited to, the hypothalamic arcuate, ventromedial, dorsomedial, and paraventricular nuclei and the area postrema and the nucleus of the solitary tract in the hindbrain. Of note, our single-cell reverse transcription-PCR analyses revealed that Sirt1 mRNA is expressed in pro-opiomelanocortin neurons that are critical for normal body weight and glucose homeostasis. We also found that SIRT1 protein levels are restrictedly increased in the hypothalamus in the fasted brain. Of note, we found that this hypothalamic-specific, fasting-induced SIRT1 regulation is altered in leptin-deficient, obese mice. Collectively, our findings establish the distribution of Sirt1 mRNA throughout the neuraxis and suggest a previously unrecognized role of brain SIRT1 in regulating energy homeostasis.
Keywords: SIRT1, distribution, regulation, activity, brain, obesity
Introduction
Sirtuins belong to a family of proteins with pleiotropic effects that are conserved across evolution from bacteria to humans (Michan and Sinclair, 2007). The first sirtuin studied is the yeast Saccharomyces cerevisiae silent information regulator 2 (Sir2), a nicotinamide adenosine dinucleotide (NAD+)-dependent histone deacetylase (Imai et al., 2000) that positively regulates longevity (Kaeberlein et al., 1999; Lin et al., 2000). SIRT1 is the closest Sir2 homolog out of the seven mammalian sirtuins that have been identified thus far (Frye, 2000; Imai et al., 2000). In addition to histones, SIRT1 deacetylates numerous transcription factors and cofactors that govern a wide range of different biological programs (Michan and Sinclair, 2007).
Several studies have shown that SIRT1 is a key regulator of metabolism. SIRT1 contents increase in many tissues during conditions of nutrient deprivation (Cohen et al., 2004). In fat cells (adipocytes), reduced food intake triggers the release of free fatty acid into the bloodstream to provide fuel supply to other tissues. By negatively regulating peroxisome proliferator-activated receptor-γ (PPAR-γ), a key adipogenic transcription factor (Tontonoz et al., 1994), SIRT1 plays an important role in this adaptive response to nutrient scarcity (Picard et al., 2004). Also, by positively regulating PGC1-α (PPAR-γ coactivator 1-α), SIRT1 is thought to coordinate the increase in hepatic glucose production and the switch from glucose to lipid oxidation in skeletal muscle that occur during starvation (Rodgers et al., 2005; Gerhart-Hines et al., 2007). Moreover, SIRT1 improves pancreatic β-cell function in part by inhibiting UCP2 (uncoupling protein 2), which is a negative player of glucose-induced insulin secretion (Zhang et al., 2001; Moynihan et al., 2005). Collectively, these findings support the view that SIRT1 links the status of energy content with cellular function in many metabolically relevant tissues.
Rapidly emerging evidences suggest that brain SIRT1 has diverse roles (e.g., controlling neurogenesis and neuroprotection). For example, increased Sirt1 gene dosage seems to activate pathways aimed at reducing β-amyloid deposition (Qin et al., 2006). This deposition is typical in Alzheimer's disease (AD). Moreover, pharmacological and genetic SIRT1 manipulations prevent neurodegeneration in animal models of AD and amyotrophic lateral sclerosis (D. Kim et al., 2007). SIRT1 is also a critical player in the protective mechanisms against axonal degeneration (Araki et al., 2004). Furthermore, SIRT1 has been suggested to be part of the molecular pathways determining the fate of neuronal progenitor cells (Prozorovski et al., 2008).
Because of the established importance of peripheral SIRT1 in metabolism, we hypothesized that SIRT1 may be expressed and regulated by energy availability in brain sites governing energy balance. To date, very little is known regarding the anatomical distribution of this sirtuin throughout the neuraxis. To address these issues, we have generated sensitive antisense RNA probes for in situ hybridization histochemistry (ISHH) analyses. In addition, to unravel the biochemical phenotypes of SIRT1-expressing neurons, single-cell reverse transcription (RT)-PCR analyses were performed. Finally, we sought to determine the brain sites in which SIRT1 expression/activity is regulated by changes in energy availability and whether these regulations are altered in a genetic model of obesity.
Materials and Methods
Animals.
Fed ad libitum male mice were used for all experiments unless otherwise specified. Mice were housed with food and water available ad libitum in light- and temperature-controlled environments. Care of mice was within the Institutional Animal Care and Use Committee (IACUC) guidelines, and all the procedures were approved by the University of Texas Southwestern Medical Center IACUC.
Generation of Sirt1 cRNA probe.
Sirt1-specific cRNA probes were generated as follows. Mouse hypothalamic RNAs were extracted using Trizol Reagent (Invitrogen). RT-PCR (One-Step RT-PCR Kit from Invitrogen) was performed using these RNAs as templates, and the following deoxy-oligonucleotides as primers: T3-SIRT1, 5′-AATTAACCCTCACTAAAGGGAGActataccccatgaagtgcctc-3′; T7-SIRT1, 5′-TAATACGACTCACTATAGGGAGAgacaagacgtcatcttcagag-3′.
Primers were designed according to the mouse Sirt1 mRNA sequence NM_019812 from the National Center for Biotechnology Information database. Lowercase letters are nucleotides 1337–1357 (Sirt1 exon 8) in T3-SIRT1 and nucleotides 1950–1970 (Sirt1 exon 9) in T7-SIRT1. RT-PCR DNA products were loaded on a 1.5% agarose gel and resolved by electrophoresis. The predicted amplicon size was visualized. RT-PCR DNA products were purified using the QIAquick PCR purification kit (Qiagen), sequenced at the McDermott Center core DNA sequencing facility, University of Texas Southwestern Medical Center confirming that amplicons were mouse Sirt1 sequences. These RT-PCR DNA products were subsequently used as templates for generation of RNA probes. Sense or antisense 35S-labeled RNA probes were generated by in vitro transcription with either T3 or T7 RNA polymerase, respectively.
ISHH analysis.
Eight- to twelve-week-old C57BL/6 mice were deeply anesthetized with 7% chloral hydrate and perfused with 10% buffered formalin. Brains were removed, equilibrated with 20% sucrose solution, and cut at 25 μm into five equal series. ISHH for Sirt1 (35S) mRNA was performed as described previously (Elias et al., 1999). Brain sections were mounted onto SuperFrost Plus slides, air dried, and stored in desiccated boxes at −20°C until used for experiments. Before hybridization, sections were fixed in 4% formaldehyde in diethylpyrocarbonate (DEPC)-treated PBS, pH 7, for 20 min at 4°C. Then, sections were dehydrated in increasing concentrations of ethanol, cleared in xylenes for 15 min, rehydrated in decreasing concentrations of ethanol, and placed in prewarmed sodium citrate buffer (95–100°C, pH 6). While in sodium citrate buffer, slices were placed in a microwave oven for 10 min at 20–70% power. Afterward, slices were dehydrated as before and then air dried. One of the five series was hybridized with T7-transcribed, antisense 35S-labeled RNA probes (106 cpm/ml) in a hybridization solution containing 50% deionized formamide, 10 mm Tris-HCl, pH 7.5, 1% sheared salmon sperm DNA, 2.5% total yeast RNA, 5 mg of tRNA, 0.1% SDS, 0.1% sodium thiosulfate, 100 mm DTT, 10% dextran sulfate, 0.6 m NaCl, 0.5 mm EDTA, pH 8, and 1× Denhardt's solution. Coverslips were applied to sections, which were then incubated at 57°C for 12–16 h. Next, coverslips were removed and sections washed in 2× SSC buffer and incubated in 0.002% RNase A with 0.5 m NaCl, 40 mm Tris-HCl, pH 8, and 0.1 mm EDTA, pH 8 for 30 min followed by 30 min incubation in the same buffer minus the RNase. Stringency washes were applied as follows: 2× SSC, 1 mm DTT at 50°C for 1 h; 0.2× SSC, 1 mm DTT at 55°C for 1 h; 0.2× SSC, 1 mm DTT at 60°C for 1 h. Afterward, sections were dehydrated, air dried, and placed in x-ray film cassettes with BMR-2 film from Kodak for 1–3 d. Films were developed and slices dipped in Kodak NTB autoradiographic emulsion, dried, and stored in desiccated, foil-wrapped boxes at 4°C for 2–3 weeks. Finally, slices were developed with Kodak Dektol developer, counterstained with thionin, dehydrated, cleared in xylenes, and coverslipped with Permaslip. Another series was hybridized with T3-transcribed, sense 35S-labeled RNA probes as described above and used as negative control. ISHH patterns were visualized first on autoradiographic films and then by observing slides dipped in photographic emulsion. Reference brain series were stained with thionin for determining nuclei boundaries. Pictures were taken using either a Zeiss Axioskop or a Zeiss Stemi 2000-C dissecting microscope with a Zeiss digital camera.
Single-neuron RT-PCR.
Transgenic mice expressing humanized Renilla green fluorescent protein (hrGFP) only in pro-opiomelanocortin (POMC) neurons were used (Parton et al., 2007). Mice were deeply anesthetized and transcardially perfused with an artificial CSF (aCSF) solution (in mm: 126 sucrose, 21.4 NaHCO3, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, and 11 d-glucose). Mice were then decapitated and the entire brain removed. After removal, brains were immediately submerged in ice-cold, carbogen-saturated (95% O2/5% CO2) aCSF (in mm: 2.5 NaCl, 126 KCl, 1.2 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 21.4 NaHCO3, and 11.1 glucose). A brain block containing the hypothalamus was made. Coronal sections (200–250 μm) were cut with a Leica VT1000S Vibratome, and epifluorescence was used to target fluorescent cells using a Zeiss Axioskop FS2 Plus. Individual cells were patched with a glass pipette and harvested by applying a negative pressure. After reverse transcription, the cDNAs for SIRT1, POMC, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were amplified by PCR using the following primers (these pairs of primers are all designed across at least one intron, allowing discrimination between genomic- and cDNA-specific amplicons): SIRT1, 5′-GCAGATTAGTAAGCGGCTTGAGG-3′ and 5′-AGCACATTCGGGCCTCTCCGTA-3′; POMC, 5′-CAGACCTCCATAGATGTGTGGAGC-3′ and 5′-CTCAGCAACGTTGGGGTACAC-3′; GAPDH, 5′-CCCGTAGACAAAATGGTGAAGGTC-3′ and 5′-GCCAAAGTTGTCATGGATGACC-3′.
Colocalization of β-endorphin and GFP.
Pomc-GFP transgenic mice were perfused with 10% formalin, and the brains were sectioned at 25 μm (1:5 series) on a freezing sliding microtome. Immunofluorescence was performed using rabbit β-endorphin antisera followed by Cy3-conjugated donkey anti-rabbit-IgG antibodies as previously described (Parton et al., 2007).
Western blot analysis.
Eight-week-old wild-type FVB/N and C57BL/6 mice and ob/ob C57BL/6 mice were fed ad libitum or fasted for 24 h. Fasted mice were starved for 24 h by removing only food (water was provided ad libitum) 2 h before the onset of the dark cycle the day before they were killed. Fed and fasted mice were all killed 2 h before the onset of the dark cycle, and brains were quickly removed. After hypothalamus was dissected out, the remaining brain tissue was divided into two parts by a coronal cut at the border between the midbrain and hypothalamus. The rostral and caudal parts are referred to as forebrain (without hypothalamus) and hindbrain, respectively. All procedures were performed under a stereomicroscope. Samples were then snap frozen in liquid nitrogen and subsequently stored at −80°C. Proteins were extracted by homogenizing samples in lysis buffer [20 mm Tris, 5 mm EDTA, 1% (v/v) NP-40, and protease inhibitors (P2714-1BTL from Sigma)], then resolved by SDS-PAGE, and finally transferred to a nitrocellulose membrane by electroblotting. SIRT1 was detected using rabbit polyclonal antisera against the N terminus (amino acids 1–131) of mouse SIRT1 (Millipore). Acetylated p53 was detected using rabbit polyclonal antisera against the lysine 379 of mouse p53 (Cell Signaling). β-Actin, used as loading control, was detected with rabbit polyclonal antisera (Abcam).
Laser-capture microdissection.
Eight- to twelve-week-old C57BL/6 mice were anesthetized, and brains were quickly removed and frozen on dry ice on a bed of OCT on aluminum foil. Brains were stored −80°C until ready to cryosection. Brains were sectioned at 7–25 μm in a cryostat at approximately −15°C onto cold silanized slides. Afterward, sections were stored at −80°C. Then, sections were dehydrated and stained as follows: 75% ethanol for 1 min, DEPC water for 30 s, drip thionin onto slide for 30 s, DEPC water for 30 s, 75% ethanol for 1 min, 95% ethanol for 1 min, 100% ethanol for 1 min, xylenes for 10 min, and then air dry briefly until xylenes are evaporated. Single nuclei were captured as described in the Arcturus manual (Molecular Devices). RNA extraction buffer (50 μl; PicoPure RNA Isolation Kit; Molecular Devices) was added to the collected tissues, which were then incubated at 42°C for 30 min. Samples were stored at −80°C. RNA isolation was done according to the PicoPure protocol.
Assessment of mRNA content.
Eight- to twelve-week-old C57BL/6 mice were fed ad libitum or fasted for 24 h. Fasted mice were starved for 24 h by removing only food (water was provided ad libitum) 2 h before the onset of the dark cycle the day before they were killed. Fed and fasted mice were all killed 2 h before the onset of the dark cycle, and brains were quickly removed. Hypothalamus, forebrain (without hypothalamus), and hindbrain were dissected as described above. Samples were snap frozen in liquid nitrogen and subsequently stored at −80°C. RNAs were extracted using Trizol reagent (Invitrogen). Complementary DNA was generated by Superscript II (Invitrogen) and used with SYBR Green PCR master mix (Applied Biosystem) for quantitative real-time PCR (q-RTPCR) analysis. Sirt1 mRNA contents were normalized to β-actin mRNA contents. Sequences of deoxy-oligonucleotides primers used are outlined here: Sirt1, 5′-TGCTGGCTTAATAGACTTGCA-3′ and 5′-GCACCGTGGAATATGTAACGA-3′; β-actin, 5′-CATCGTGGGCCGCTCTA-3′ and 5′-CACCCACATAGGAGTCCTTCTG-3′. For nucleus-specific q-RTPCR analyses, Sirt1 sense and antisense primers, MGB probe [5′-FAM; Applied Biosystems; assay on demand (Mn00490758_m1)] were used. Sirt1 mRNA levels were normalized to the housekeeping 18S ribosomal RNA. All assays were performed using an Applied Biosystems Prism 7900HT sequence detection system.
Results
Distribution of Sirt1 mRNA in brain
ISHH analyses with antisense 35S-labeled RNA probes spanning Sirt1 exons 8 and 9 sequences were performed to determine the anatomical distribution of Sirt1 mRNA in the mouse brain. These analyses revealed that in the forebrain Sirt1 mRNA is expressed at moderate to high levels in the islands of Calleja (ICj) (Fig. 1A). Also, we found that within the cerebral cortex, Sirt1 mRNA is expressed in virtually all structures with the piriform cortex displaying the highest levels of hybridization (Fig. 1A–F). Striatal structures exhibited low to moderate hybridization with no remarkable differences in expression levels between the nucleus accumbens, caudate–putamen, globus pallidus, and substantia innominata (Fig. 1A–F and data not shown). Low to moderate Sirt1 mRNA expression was present throughout the septum, with moderate to high levels of expression in the bed nucleus of the stria terminalis (BST), bed nucleus of anterior commissure (BAC), triangular septal nucleus (TS), and lateral septal nucleus (LS) (Fig. 1B–D). In the hippocampal formation, all fields, CA1, CA2, CA3, and the dentate gyrus exhibited intense hybridization (Fig. 1F). In the epithalamus, high levels of expression were observed in the medial habenular nucleus (MHb) (Fig. 1F). Within the thalamus, moderate to high levels of hybridization were present in the anterodorsal thalamic nucleus (AD) and the anterior paraventricular thalamic nucleus (PVA), and low to moderate Sirt1 mRNA expression was present in virtually all other sites (Fig. 1D–F and data not shown).
Figure 1.

A series of low-power photomicrographs summarizing the distribution of Sirt1 mRNA in the mouse brain. A–I, ISHH with antisense probes. J, ISHH with sense probes (negative control). Pir, Piriform cortex; Hipp, hippocampus. Scale bar: (in J) A–J, 1 mm.
In the hypothalamus, Sirt1 mRNA was preferentially abundant in medial structures such as arcuate (ARH), ventromedial (VMH), dorsomedial (DMH), paraventricular (PVH), supraoptic (SO), and suprachiasmatic (Sch) nuclei (Figs. 1D–F, 2A,B,K). Although at lower levels, Sirt1 mRNA was also expressed in lateral hypothalamus (Fig. 1F). Throughout the rostral-to-caudal and dorsal-to-ventral extent of midbrain, pons, and medulla oblongata, Sirt1 mRNA was expressed broadly (Fig. 1G–I and data not shown). However, the dorsal raphe nucleus (DR) in the midbrain and the nucleus of the solitary tract (NTS) and the dorsal motor nucleus of the vagus (DMV) in the caudal hindbrain were found to display moderate levels of hybridization (Figs. 1G,I, 2C). Sirt1 mRNA was highly expressed in the cerebellum (Fig. 1H). We also found that Sirt1 mRNA is moderately to highly expressed in several circumventricular organs, such as the organum vasculosum of the lamina terminalis (OVLT) (Fig. 1B), the subfornical organ (SFO) (Fig. 1D), and the area postrema (AP) in the caudal hindbrain (Figs. 1J, 2C). We observed no specific hybridization when sense 35S-labeled RNA probes were used as a negative control (Fig. 1J).
Figure 2.
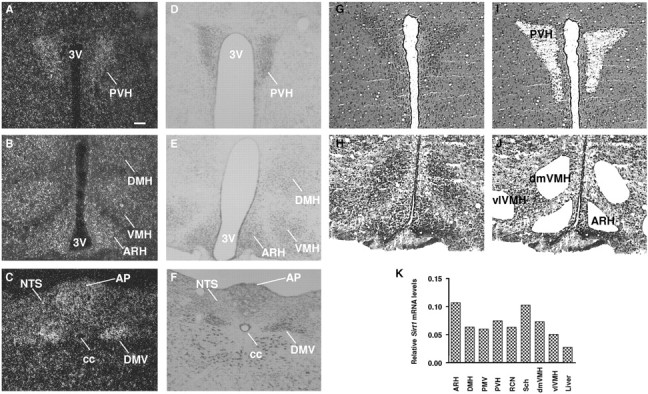
Sirt1 mRNA in metabolically relevant nuclei of the mouse brain. A–C, Dark-field photomicrographs demonstrating the expression of Sirt1 mRNA (white dots) in PVH, DMH, ARH, VMH, NTS, AP, and DMV. D–F, Nissl staining of reference sections appearing in A–C. G–J, Photomicrographs of thionin-stained brain slices before (G, H) and after (I, J) laser-capture microdissection. K, Results of nucleus-specific q-RTPCR analysis of Sirt1 mRNA contents in samples pooled from four to six different animals. Liver was used as a positive control. PMV, Ventral premammillary hypothalamic nucleus; RCN, retrochiasmatic hypothalamic nucleus; dmVMH, dorsomedial, ventromedial hypothalamic nucleus; vlVMH, ventrolateral, ventromedial hypothalamic nucleus; 3V, third ventricle; cc, central canal. Scale bar: (in A) A–J, 100 μm.
To independently test whether Sirt1 mRNA is expressed in the aforementioned nuclei, we used laser-capture microdissection techniques (Segal et al., 2005). This approach allowed us to obtain enriched RNA from specific nuclei as represented by photomicrographs of brain slices before (Fig. 2G,H) and after (Fig. 2I,J) hypothalamic nuclei were isolated. Nucleus-specific RNA samples were then used for q-RTPCR assays for Sirt1 mRNA contents. Liver was used as a positive control because Sirt1 mRNA has been previously reported to be expressed in this organ (Rodgers et al., 2005). As shown in Figure 2K, nuclei scored positive for Sirt1 mRNA expression by ISHH were also found to express this messenger RNA when independently assayed by q-RTPCR. Collectively, these findings reveal that Sirt1 mRNA is broadly expressed throughout the mouse neuraxis and at higher levels mainly in metabolically relevant sites such as nuclei in medial hypothalamus and caudal hindbrain.
SIRT1 is expressed in POMC neurons
Because POMC neurons are localized in the ARH and the NTS in the rodent brain (Bronstein et al., 1992; Elias et al., 1999), and they have been shown to be important regulators of energy metabolism (Yaswen et al., 1999; Balthasar et al., 2004; Parton et al., 2007), we asked whether SIRT1 is expressed in these cells. The generation of genetically engineered mice in which fluorescent proteins are expressed in a cell-specific manner has greatly facilitated the ability to assess whether a gene is expressed in a given cell of interest (Liu et al., 2003; Adamantidis et al., 2007). Thus, mice expressing GFP only in POMC cells (Pomc-GFP mice) were used to address this question. These mice were generated by cloning the coding sequence of humanized Renilla GFP into a Pomc bacterial artificial chromosome (Parton et al., 2007). Colocalization studies validated Pomc-GFP mice, because the vast majority of neurons containing β-endorphin (one of POMC products) also expressed GFP (Fig. 3A). By single-cell RT-PCR analysis, we found that 9 of 10 fluorescent cells express Pomc mRNA (Fig. 3B), a finding that further corroborates the colocalization studies. Importantly, Sirt1 mRNA was expressed in eight of nine ARH POMC neurons (Fig. 3B). Also, in agreement with the wide distribution of Sirt1 mRNA within the ARH (Fig. 2B), other POMC neighboring cells were found to express this sirtuin (Fig. 3B). These results suggest that ARH POMC neurons express SIRT1.
Figure 3.

POMC neurons express Sirt1 mRNA. A, Photomicrographs of brain slices from Pomc-GFP mice stained for β-endorphin. Green represents GFP fluorescence, and red represents β-endorphin immunofluorescence (IF). Arrows indicate neurons expressing both GFP and β-endorphin. B, Agarose-gel analysis of single-cell RT-PCR. Fluorescent (GFP) and POMC neighboring, nonfluorescent (no-GFP) cells were harvested from Pomc-GFP hypothalami. PCR products corresponding to SIRT1, POMC, and GAPDH were visualized. All pairs of primers were designed across at least one intron, allowing discrimination between genomic- and cDNA-specific amplicons. Hypothalamic cDNA was used as positive control (+), whereas the same PCR mix without template was used as negative control (−).
Fasting increases SIRT1 protein levels only in hypothalamus within the brain
Reduced nutrient availability leads to increased SIRT1 protein contents in the brain (Cohen et al., 2004). However, unknown are the sites at which this regulation occurs. To gather more insights in regards to the brain regions in which SIRT1 expression is regulated by energy availability, we compared Sirt1 mRNA levels and SIRT1 protein contents and activity in the hypothalamus, the forebrain (without the hypothalamus), and the hindbrain between the fed and fasted states in adult C57BL/6 mice. q-RTPCR analysis revealed no differences at the mRNA level in all these regions (data not shown). It is known that the expression of specific hypothalamic genes is oppositely regulated by fasting. For example, ARH Pomc and VMH Bdnf mRNA contents diminish, whereas ARH Agrp and Npy mRNA contents increase after fasting (Sanacora et al., 1990; Chua et al., 1991; Kim et al., 1996; Mizuno et al., 1998; Mizuno and Mobbs, 1999; Xu et al., 2003). Thus, to test whether the lack of differences in Sirt1 mRNA levels may have been the result of opposite fluctuations in the heterogeneous hypothalamic populations, nucleus-specific q-RTPCR assays were performed. Our results showed that Sirt1 mRNA levels in ARH and VMH are similar between the fed and fasted states (data not shown). Together, these data suggest that brain SIRT1 is not induced by fasting at the mRNA level.
In contrast, Western blot analysis revealed an increase in SIRT1 protein contents specifically in the hypothalamus, but not in the rest of the forebrain or in the hindbrain after 24 h of food deprivation (Fig. 4) (SIRT1/β-actin in hypothalamus, mean ± SEM: fed = 1.628 ± 0.197; fasted = 2.227 ± 0.169; n = 5; p < 0.05). Of note, this hypothalamic-specific, fasting-induced SIRT1 regulation was also observed in mice that are on a different genetic background (i.e., FVB/N mice) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Because SIRT1 biological effects are mainly determined by its deacetylation activity, we next asked whether fasting would lead to an increase in SIRT1 activity. The acetylation status of SIRT1's target p53 is considered a marker of SIRT1 activity in vivo (E. J. Kim et al., 2007b; Kim et al., 2008; Zhao et al., 2008). Therefore, we investigated SIRT1 activity by using the levels of acetylated lysine 379 (K379) in p53 as a readout. In accordance with an increase in SIRT1 protein contents, SIRT1 activity seemed also to be elevated in the fasted hypothalamus. Indeed, the levels of acetylated K379 in p53 were reduced in this brain site (Fig. 4) [p53(Ac-K379)/β-actin in hypothalamus, mean ± SEM: fed = 2.456 ± 0.255; fasted = 1.514 ± 0.180; n = 5; p < 0.05]. Surprisingly, this effect was even more pronounced in the fasted hindbrain (Fig. 4) [p53(Ac-K379)/β-actin in hindbrain, mean ± SEM: fed = 6.919 ± 1.340; fasted = 2.738 ± 0.440; n = 5; p < 0.05]. Of note, these fasting-induced changes in the levels of acetylated K379 in p53 were brain-area specific, because no differences were detected in the rest of the forebrain between the fed and fasted states (Fig. 4). Collectively, these data suggest that SIRT1 protein contents increase only in the hypothalamus in the fasted brain and that fasting-induced regulation of SIRT1 activity is brain-area specific.
Figure 4.

Nutritional regulation of SIRT1 protein and activity levels in the brain of C57BL/6 mice. SIRT1, acetylated lysine (K) 379 in p53 and β-actin (used as loading control) protein levels were assessed in the hypothalamus, the forebrain (without the hypothalamus), and the hindbrain between the fed and fasted states by Western blot analysis. Western blot data represent the results of 5–10 samples.
SIRT1 regulation by fasting is aberrant in leptin-deficient, obese mice
We found that SIRT1 protein contents are specifically increased in the hypothalamus after fasting (Fig. 4; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). It has been shown that fasting-dependent regulations of several genes known to govern body weight homeostasis are altered in obesity (Sanacora et al., 1990; Korner et al., 2000). Thus, to gather insights into whether SIRT1 may play a role in body energy homeostasis, we tested whether fasting-induced regulation of SIRT1 is intact in the brains of obese mice. Mice lacking the adipostatic hormone leptin (ob/ob mice) are hyperphagic and hypometabolic and, as a consequence of both, develop obesity (Zhang et al., 1994). Therefore, we assayed SIRT1 protein contents in the hypothalamus, the forebrain (without the hypothalamus), and the hindbrain of fed and fasted ob/ob mice. As observed in wild types, fasting did not cause any change in the levels of SIRT1 protein in the forebrain (without the hypothalamus) of ob/ob mice (Fig. 5). In contrast to the nonobese mice, fasting failed to increase SIRT1 protein levels in the hypothalamus of ob/ob mice (Fig. 5). Furthermore, fasting caused an abnormal reduction in SIRT1 protein contents in the hindbrain of ob/ob mice (Fig. 5) (SIRT1/β-actin in hindbrain, mean ± SEM: fed = 2.600 ± 0.262; fasted = 1.382 ± 0.172; n = 5; p < 0.005). The levels of acetylated K379 in p53 were also abnormally regulated in the brain of fasted ob/ob mice. Indeed, the levels of acetylated K379 in p53 were elevated (not reduced as in wild types) in the fasted hindbrain (Fig. 5) [p53(Ac-K379)/β-actin in hindbrain, mean ± SEM: fed = 1.228 ± 0.116; fasted = 2.815 ± 0.266; n = 5; p < 0.005]. Also, these were unchanged (not reduced as in wild types) in the fasted hypothalamus of ob/ob mice (Fig. 5). Collectively, our results suggest that SIRT1 regulation by reduced energy availability is altered in the brain of leptin-deficient, obese mice.
Figure 5.

Nutritional regulation of SIRT1 protein and activity levels in the brain of obese, ob/ob C57BL/6 mice. SIRT1, acetylated lysine (K) 379 in p53 and β-actin (used as loading control) protein levels were assessed in the hypothalamus, the forebrain (without the hypothalamus), and the hindbrain of obese mice between the fed and fasted statuses by Western blot analysis.
Discussion
The yeast Sir2 is a NAD+-dependent histone deacetylase (Imai et al., 2000). The enzymatic activity of the mammalian Sir2 ortholog (SIRT1) is not limited to histones but includes also transcription factors and cofactors that govern different biological programs (Michan and Sinclair, 2007). By deacetylating these factors, SIRT1 orchestrates critical metabolic adaptations to reduced energy availability in peripheral tissues (Picard et al., 2004; Rodgers et al., 2005; Gerhart-Hines et al., 2007). To date, very little has been reported regarding SIRT1 distribution and regulation by energy availability within the CNS. Here, we performed ISHH analyses and found that Sirt1 mRNA is widely expressed throughout the mouse neuraxis. Importantly, Sirt1 mRNA was abundantly expressed in metabolically relevant areas, such as, for example, the ARH, VMH, DMH, and PVH and the AP and NTS in the hindbrain. In addition to these sites, Sirt1 mRNA was also abundant in the piriform cortex, hippocampus, medial habenular nucleus, and cerebellum. Moreover, we found that Sirt1 mRNA is expressed in neurons critical for normal energy and glucose balance such as the POMC neurons. Finally, our studies identified the hypothalamus as the CNS site in which SIRT1 is induced after fasting and that this response is aberrant in leptin-deficient, obese mice.
In our study, the anatomical distribution of SIRT1 in the brain was determined by ISHH analyses. In other studies, commercially available SIRT1 antisera were used for immunohistochemistry (IHC) analyses. These showed expression of SIRT1 protein in neurons (Qin et al., 2006; Michan and Sinclair, 2007). Thus, ICC analysis may have been an ideal approach to determine the distribution of this sirtuin in the CNS. However, when we tested the specificity of these antisera in ICC applications, we found indistinguishable staining between Sirt1 knock-out and control wild-type tissues (G. Ramadori and R. Coppari, unpublished observation). Thus, we focused our efforts on mRNA distribution studies and developed antisense RNA probes specific for Sirt1 mRNA. As shown in Figure 1J, the specificity of our ISHH protocol is suggested by the fact that no hybridization was observed in brain sections that were used for ISHH with sense Sirt1 RNA probes. Moreover, the reliability of our assay is further supported by the fact that nuclei in which Sirt1 mRNA was readily detected by ISHH analysis were also found positive for the expression of this messenger RNA when independently tested by q-RTPCR (Fig. 2K).
In addition, we used acetylated K379 in p53 to measure SIRT1 activity in vivo. There are inherent difficulties with assessing the activity of an enzyme in vivo, and especially in nonhomogeneous tissues as the brain. Thus, these issues must be taken into account while considering our results. Nevertheless, SIRT1 regulates p53 acetylation both, directly by deacetylating p53 and indirectly by inhibiting p300, which is one of the enzymes that acetylates p53 (Luo et al., 2001; Vaziri et al., 2001; Langley et al., 2002; Bouras et al., 2005). Also, p53 is hyperacetylated in the absence of SIRT1 in vivo (Cheng et al., 2003). Therefore, SIRT1 activity has been previously assessed by using the acetylation status of p53 as a readout (E. J. Kim et al., 2007; Kim et al., 2008; Zhao et al., 2008). However, without direct experiments on samples from mice lacking SIRT1 in specific CNS sites, we cannot conclude that the changes in p53 acetylation status we have observed are solely dependent on the activity of SIRT1.
Our study is the first to reveal the anatomical distribution of Sirt1 mRNA in the CNS. Given the wide range of biological programs in which SIRT1 is involved, these findings are relevant for multiple reasons. For example, SIRT1 has been suggested to play a role in neuroprotection (Qin et al., 2006; D. Kim et al., 2007). However, relatively little is known about whether SIRT1 is expressed in neurons. As reviewed above, previous studies have shown expression of this protein in neurons (Qin et al., 2006; Michan and Sinclair, 2007), although we found immunoreactivity in Sirt1 knock-out samples when similar antisera were used. Nonetheless, our single-cell RT-PCR analysis results demonstrate that SIRT1 is expressed in neurons (Fig. 3B). It is worth noting at this point that SIRT1 neuroprotective effects are thought to be mediated in part by deacetylation and hence inhibition of the tumor suppressor p53 (D. Kim et al., 2007). We found that reduced energy availability leads to diminished levels of acetylated K379 in p53 only in hypothalamus and hindbrain within the brain, an effect that is altered in leptin-deficient, obese mice (Figs. 4, 5). If fasting-induced changes in p53 acetylation status are part of pathways required to cope with reduced energy supply, then the hypothalamus and/or the hindbrain of obese mice should be more susceptible to stress-induced neuronal damages. Future research efforts will be necessary to investigate these possibilities.
The present data may also have implications regarding the possible role of SIRT1 in regulating longevity. It is known that calorie restriction (CR), that is, a dietary regimen of at least 20% fewer calories than an organism would normally consume, prolongs lifespan (Koubova and Guarente, 2003). In lower metazoans, SIRT1 homologues are required for this effect (Lin et al., 2000; Rogina and Helfand, 2004; Lamming et al., 2005). However, in mammals, the molecular players and cell types that mediate CR's effects are still unknown. One potential site is the brain, as suggested by the fact that only two neurons in the head of C. elegans mediate CR-induced longevity, possibly by raising body metabolism (Bishop and Guarente, 2007). SIRT1 is increased in the brain of CR rodents (Cohen et al., 2004), but whether these increases occur in all brain structures or are restricted to specific sites has not been established yet. Although we have not analyzed SIRT1 in brains of CR mice, our data in Figure 4 and supplemental Figure 1 (available at www.jneurosci.org as supplemental material) suggest that reduced energy availability triggers SIRT1 accumulation in hypothalamus. Because neurons in this site are known to control body metabolism, we anticipate that future work will be aimed at directly testing whether this metabolic-sensor protein in these metabolic-sensor neurons plays a key role in mediating CR-induced longevity in mammals.
Of note, Sirt1 mRNA is abundantly expressed not only in sites known to govern body metabolism. For example, the piriform cortex, also called “paleocortex” because a similar structure is found in fish, amphibians, and reptiles, is enriched in Sirt1 mRNA. Thus, from an evolutionary point of view, it may not be surprising that Sirt1, an ancient gene, is highly expressed in this ancient brain cortical structure. Recently, SIRT1 has been shown to be part of the molecular pathways determining the fate of neuronal progenitor cells (NPCs) (Prozorovski et al., 2008). In the adult brain, within the hippocampal formation, the dentate gyrus subgranular zone is rich in NPCs. Sirt1 mRNA is also abundantly expressed in this region, and further studies directed to test whether this gene in this region plays a role in neurogenesis and learning and memory processes are warranted. The fact that SIRT1 is expressed in brain neurons is also important in respect to its suggested protective roles against axonal degeneration (Araki et al., 2004) and neurodegenerative diseases (D. Kim et al., 2007). Because axonal degeneration precedes neuronal death in neurodegenerative states such as Parkinson's and Alzheimer's diseases, pharmacological activation of SIRT1 may represent a novel and potentially more effective approach in the fight against these pathological conditions.
As discussed above, SIRT1 is a metabolic-sensor protein that links the status of energy availability with cellular function in many tissues (Picard et al., 2004; Rodgers et al., 2005; Gerhart-Hines et al., 2007). Thus, it is of note that this protein is regulated by energy availability in hypothalamus that has been long recognized as critical for normal energy balance (Fig. 4; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Importantly, SIRT1 is also expressed in POMC neurons in the hypothalamus (Fig. 3B). Therefore, it is formally possible that fasting-induced SIRT1 accumulation in hypothalamus includes POMC neurons. If this is true, then SIRT1 in these neurons may be part of CNS mechanisms that coordinate energy balance. At the molecular level, SIRT1 could suppress Pomc gene expression through deacetylation (hence activation) of its target FoxO1 (Kitamura et al., 2005). It is indeed known that activated FoxO1 squelches Pomc transcription (Kitamura et al., 2006). The fact that fasting-induced regulation of hypothalamic SIRT1 is abolished in leptin-deficient, obese mice (Fig. 5) further suggests that SIRT1 may play a role in brain-mediated control of energy homeostasis. Future studies on neuron-specific, Sirt1 gene knock-out mice will be necessary to assess the physiological relevance of SIRT1 in these neurons on body energy homeostasis.
In conclusion, our study provides the anatomical landscape of SIRT1 distribution and regulations by reduced energy availability within the CNS. Also, our data establish that these regulations are lost in a genetic model of obesity, suggesting a novel potential role for brain SIRT1 in regulating energy balance.
Footnotes
This work was supported by the Department of Internal Medicine, Division of Hypothalamic Research, University of Texas Southwestern Medical Center (start-up to R.C.), by the Italian Ministry of University and Research (PRIN 2005 to R.C.), and by National Institutes of Health Grants DK53301, MH61583, and DK081185 (J.K.E.) and DA07290 and GM007062 (A.L.B.). We thank Drs. Carol Elias and Claudia R. Vianna (University of Texas Southwestern Medical Center, Dallas, TX) for helpful discussions and Drs. Frederick Alt, Hwei-Ling Chen, and Raul Mostoslavsky (Harvard Medical School, Boston, MA) for providing Sirt1 knock-out tissues.
References
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr, Elmquist JK, Lowell BB. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem. 2005;280:10264–10276. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- Bronstein DM, Schafer MK, Watson SJ, Akil H. Evidence that beta-endorphin is synthesized in cells in the nucleus tractus solitarius: detection of POMC mRNA. Brain Res. 1992;587:269–275. doi: 10.1016/0006-8993(92)91007-2. [DOI] [PubMed] [Google Scholar]
- Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua SC, Jr, Leibel RL, Hirsch J. Food deprivation and age modulate neuropeptide gene expression in the murine hypothalamus and adrenal gland. Brain Res Mol Brain Res. 1991;9:95–101. doi: 10.1016/0169-328x(91)90134-j. [DOI] [PubMed] [Google Scholar]
- Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA, Sui G, Armour SM, Puigserver P, Sinclair DA, Tsai LH. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer's disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- Kim EM, Welch CC, Grace MK, Billington CJ, Levine AS. Chronic food restriction and acute food deprivation decrease mRNA levels of opioid peptides in arcuate nucleus. Am J Physiol. 1996;270:R1019–R1024. doi: 10.1152/ajpregu.1996.270.5.R1019. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Korner J, Wardlaw SL, Liu SM, Conwell IM, Leibel RL, Chua SC., Jr Effects of leptin receptor mutation on Agrp gene expression in fed and fasted lean and obese (LA/N-faf) rats. Endocrinology. 2000;141:2465–2471. doi: 10.1210/endo.141.7.7580. [DOI] [PubMed] [Google Scholar]
- Koubova J, Guarente L. How does calorie restriction work? Genes Dev. 2003;17:313–321. doi: 10.1101/gad.1052903. [DOI] [PubMed] [Google Scholar]
- Lamming DW, Latorre-Esteves M, Medvedik O, Wong SN, Tsang FA, Wang C, Lin SJ, Sinclair DA. HST2 mediates SIR2-independent life-span extension by calorie restriction. Science. 2005;309:1861–1864. doi: 10.1126/science.1113611. [DOI] [PubMed] [Google Scholar]
- Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, Friedman JM, Elmquist JK. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. J Neurosci. 2003;23:7143–7154. doi: 10.1523/JNEUROSCI.23-18-07143.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno TM, Mobbs CV. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology. 1999;140:814–817. doi: 10.1210/endo.140.2.6491. [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–297. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Méneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prozorovski T, Schulze-Topphoff U, Glumm R, Baumgart J, Schröter F, Ninnemann O, Siegert E, Bendix I, Brüstle O, Nitsch R, Zipp F, Aktas O. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat Cell Biol. 2008;10:385–394. doi: 10.1038/ncb1700. [DOI] [PubMed] [Google Scholar]
- Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem. 2006;281:21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Kershaw M, Finkelstein JA, White JD. Increased hypothalamic content of preproneuropeptide Y messenger ribonucleic acid in genetically obese Zucker rats and its regulation by food deprivation. Endocrinology. 1990;127:730–737. doi: 10.1210/endo-127-2-730. [DOI] [PubMed] [Google Scholar]
- Segal JP, Stallings NR, Lee CE, Zhao L, Socci N, Viale A, Harris TM, Soares MB, Childs G, Elmquist JK, Parker KL, Friedman JM. Use of laser-capture microdissection for the identification of marker genes for the ventromedial hypothalamic nucleus. J Neurosci. 2005;25:4181–4188. doi: 10.1523/JNEUROSCI.0158-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- Zhang CY, Baffy G, Perret P, Krauss S, Peroni O, Grujic D, Hagen T, Vidal-Puig AJ, Boss O, Kim YB, Zheng XX, Wheeler MB, Shulman GI, Chan CB, Lowell BB. Uncoupling protein-2 negatively regulates insulin secretion and is a major link between obesity, beta cell dysfunction, and type 2 diabetes. Cell. 2001;105:745–755. doi: 10.1016/s0092-8674(01)00378-6. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]