

Abstract
Objective
To describe the clinical, neuropsychological and radiological features of a family with a C31LfsX35 mutation in the progranulin gene (PGRN).
Design
Case series
Patients
A large British kindred (DRC255) with a PGRN mutation was assessed. Affected individuals presented with a mean age of 57.8 (54 to 67) and mean duration of disease of 6.1 years (2 to 11).
Results
All cases exhibited a clinical and radiological phenotype compatible with FTLD based on current consensus criteria. However, unlike sporadic FTLD, parietal deficits consisting of limb apraxia, dyscalculia, visuoperceptual and/or visuospatial impairment were a common feature, and brain imaging showed posterior extension of frontotemporal atrophy to involve the parietal lobes. Other common clinical features included language output impairment with either dynamic aphasia or non-fluent aphasia, and a behavioural syndrome dominated by apathy.
Conclusions
We propose that parietal features may be a prominent feature of PGRN mutations and that this may be due to disruption of fronto-parietal functional pathways.
INTRODUCTION
Frontotemporal lobar degeneration (FTLD) is a group of disorders characterized by focal atrophy of the frontal and temporal lobes. Three clinical syndromes are described by consensus criteria (1): frontotemporal dementia (FTD), progressive non-fluent aphasia (PNFA) and semantic dementia (SD). FTD is characterized by early personality change and progressive behavioural symptoms. PNFA presents with speech production difficulties. SD presents characteristically with anomia, poor single-word comprehension and fluent aphasia. These syndromes overlap clinically and radiologically with one another and with other neurodegenerative disorders including corticobasal degeneration (CBD), progressive supranuclear palsy (PSP) and motor neuron disease (MND) (2,3).
Approximately 30-50% of patients with FTLD have a family history of dementia with autosomal dominant inheritance (4). A proportion of these families have a mutation in the tau gene (MAPT) on chromosome 17q21.1 (www.molgen.ua.ac.be/FTDMutations). However, tau mutations collectively account for only around 4% of all patients with FTLD and 25% of cases with a positive family history (5,6). Recently, mutations in the progranulin gene (PGRN), located 1.7Mb away from MAPT at 17q21.32, have been identified in families with tau-negative, ubiquitin-positive inclusions on neuropathological examination (FTLD-U). Over 30 mutations have been detected to date (6,7,8,9), and together may account for around 5-10% of FTLD cases (6). Initial descriptions suggest that PGRN mutations are particularly associated with PNFA (7,8), however the phenotype continues to be defined.
METHODS
Here we report a large British family (DRC255) comprising ten affected individuals in three successive generations (fig 1) with a clinical FTLD phenotype and a genetically confirmed PGRN mutation in two affected individuals. Five of the family members were seen either in the Specialist Cognitive Disorders Clinic of the National Hospital for Neurology and Neurosurgery, London, UK or as part of a longitudinal study of patients ‘at-risk’ for developing FTLD. We describe clinical, neuropsychological, radiological, neuropathological and genetic findings in this family.
Figure 1.

Pedigree of the DRC255 family. Circles represent females and squares males. Diamonds are anonymized individuals. Filled symbols represent affected family members.
RESULTS
Clinical features
The average age at onset of disease in the family is 57.8 years (range 54 to 67) and mean length of clinical history from onset of symptoms to death is 6.1 years (range 2 to 11). All affected family members developed a clinical syndrome in keeping with FTLD.
Historical cases
Little information is available about family members from earlier generations. Case I.1 died in his 50’s with a diagnosis of presenile dementia. Case II.1 died aged 56 and was said to have both Pick’s disease and Parkinson’s disease on clinical grounds at the time of death. Case II.4 was noted to have word-finding difficulties aged 56. He deteriorated cognitively over the next two years with apraxia a prominent feature, dying aged 58. Case II.7 had a diagnosis of presenile dementia from the age of 54 and died aged 58. Case II.8 developed a marked personality change in her mid 50’s becoming initially socially withdrawn and apathetic. Behavioural problems progressed and she became mute prior to her death aged 66.
Detailed case descriptions
Detailed clinical, neuropsychological and brain imaging data are available for five affected individuals from the kindred and their case descriptions are presented below.
Case III.2
This right-handed man developed behavioural symptoms and personality change at the age of 65. He initially became socially withdrawn and apathetic. Around the same time his wife also noticed that he had lost empathy: he remained uncharacteristically indifferent after she had fractured her arm. He developed a sweet tooth, hypersomnia and his conversational speech diminished. Around a year into the illness he developed route-finding problems and became disinhibited, making sexual advances to strangers. Eighteen months after symptom onset he had difficulty putting clothes on correctly and was unable to perform simple calculations. He was first assessed two years into the illness and at this time Mini-Mental State Examination (MMSE) score was 23/30 (10). Spontaneous speech was reduced without evidence of aphasia. Neurological examination was normal apart from brisk facial reflexes and mild bilateral ideomotor limb apraxia. A clinical diagnosis of FTD was made. The patient continued to deteriorate and died aged 72.
Case III.3
This man (handedness uncertain) developed behavioural change aged 56. He became apathetic and hypersomnolent, stopped caring for himself and displayed aggression towards family members. Around the same time his speech decreased in quantity with increasing use of ‘stock phrases’. He was first assessed two years into the illness, at which time there was widespread cognitive impairment. Speech was reduced in quantity but without evidence of aphasia. Apart from a pout reflex, neurological examination was unremarkable. A clinical diagnosis of FTD was made. Behavioural and speech problems progressed and were accompanied by difficulties with episodic memory. Four years into the illness he was noted to be mute with generalized rigidity. He died aged 64.
Case III.4
This left-handed man developed behavioural disturbance and language impairment aged 56. He became increasingly apathetic and aggressive, and the quantity of propositional speech diminished. Over the next year, he developed inappropriate social behavior, sweet tooth and hyperphagia. Two years into the illness there were difficulties with episodic and topographical memory. The legibility of his writing deteriorated and he had increasing difficulty dressing and with calculation. He was first assessed two and half years into the illness when MMSE score was 10/30. He had little spontaneous speech and was echolalic with verbal perseverations, but there were no phonemic, grammatical or semantic errors. Neurological examination revealed severe bilateral ideomotor and ideational limb apraxia. Parkinsonian features were present with bradykinesia, rigidity and postural tremor of both upper limbs, slightly more marked on the left. There was a supranuclear gaze palsy affecting upgaze. A clinical diagnosis of FTD was made. The patient died aged 61.
Case III.9
This right-handed woman presented at the age of 58 with increasing difficulty using her hands over six months such that she became unable to dress herself, switch on the television or put on spectacles. She lost the ability to write, had problems with calculation and her speech output became effortful with a stammer and increasing difficulty finding words. Over the same period she became mildly apathetic and developed a sweet tooth. On examination MMSE score was 15/30 with evidence of anomia. Neurological examination revealed marked bilateral ideomotor and ideational limb apraxia and a predominantly left-sided asymmetrical extrapyramidal syndrome with tremor, rigidity and myoclonus. The tendon reflexes were symmetrically brisk with flexor plantar responses. Eye movements were abnormal with hypometric saccades and jerky pursuit. The clinical diagnosis was FTD with features of both CBD syndrome and non-fluent aphasia. The patient’s condition deteriorated over the next two years such that she became immobile, mute, unable to swallow and doubly incontinent. She died aged 63.
Patient III.11
This right-handed woman first developed problems with word-finding difficulty at the age of 67. On examination, six months after symptom onset, MMSE score was 28/30 and there was anomia. The cognitive and neurological examination was otherwise normal. When assessed a year later, anomia had progressed and she had developed phonemic paraphasias, agrammatism and difficulty repeating polysyllabic words. Her family reported that she had become apathetic and socially withdrawn. MMSE score had fallen to 22/30. There was mild bilateral ideomotor and ideational limb apraxia but the neurological examination was otherwise normal. The clinical diagnosis was PNFA.
Summary of clinical features
The clinical features in this series fall within the wide phenotypic spectrum described previously for sporadic and familial FTLD (1,11). Considering the affected individuals as a group (Table 1), the most consistent features are impaired language output, a behavioural syndrome with early and prominent apathy and limb apraxia. Language impairment consisted of non-fluent aphasia in some cases (III.9 and III.11) and reduced quantity of spontaneous speech in others (III.2, III.3 and III.4). Parkinsonian and other extrapyramidal features emerged in three of the cases. None of the patients had bulbar or limb features suggestive of motor neuron disease. Hypersomnia, an infrequent feature in most FTLD series (12,13) was prominent in two cases.
Table 1. Clinical features in affected individuals.
| Case | III.2 | III.3 | III.4 | III.9 | III.11 |
|---|---|---|---|---|---|
| Gender, handedness | M, R | M,NK | M, L | F, R | F, R |
| Age of onset | 65 | 56 | 56 | 58 | 67 |
| Length of history (onset to death) | 7 | 8 | 5 | 5 | N/A |
| First symptoms | Apathy Decreased speech Loss of empathy Sweet tooth Hypersomnia | Apathy Decreased speech Aggression Hypersomnia | Apathy Decreased speech Aggression Sweet tooth | Limb apraxia Dyscalculia Stuttering Anomia Apathy | Anomia Apathy |
| Behavioural features | |||||
| Apathy | + | + | + | + | + |
| Loss of empathy | + | - | + | - | - |
| Disinhibition | + | - | + | - | - |
| Poor judgment | + | + | + | - | - |
| Aggression | + | + | + | - | - |
| Impaired insight | + | + | + | + | + |
| Sweet tooth | + | - | + | + | - |
| Hypersomnia | + | + | - | - | - |
| Language features | |||||
| Decreased quantity of speech | + | + | + | + | + |
| Echolalia | - | - | + | - | - |
| Hesitant or stuttering speech | - | - | - | + | - |
| Anomia | - | - | - | + | + |
| Agrammatism | - | - | - | - | + |
| Phonemic paraphasias | - | - | - | - | + |
| Mutism (years to mutism) | NK | + (4) | + (4) | + (2) | - |
| Other clinical findings | |||||
| Limb apraxia | mild | NK | mild | severe | mild |
| Ideational | - | NK | + | + | + |
| Ideomotor | + | NK | + | + | + |
| Parkinsonism | - | + (late) | + (late) | + (early) | - |
| Symmetry | - | R=L | L>R | L>R | - |
| Rigidity | - | + | + | + | - |
| Bradykinesia | - | - | + | - | - |
| Tremor | - | - | + | + | - |
| Pyramidal signs | - | - | - | + | - |
| Abnormal eye movements | - | - | + | + | - |
| Clinical diagnosis | FTD | FTD | FTD | CBD/PNFA | PNFA |
NK =Not known
Neuropsychological findings
Four patients were assessed at their initial presentation (Table 2). Both episodic memory and executive function were impaired in cases III.2, III.4 and III.9. Case III.11 scored within the normal range for memory but showed deterioration in recognition memory for words from above the 50th percentile at diagnosis to between the 10th and 25th percentile one year later, and also subsequently developed reduced verbal fluency. Naming was relatively preserved in cases III.2 and III.4 but impaired in cases III.9 and III.11 (14). These latter two cases also had impaired word repetition consistent with non-fluent aphasia. Simple calculation was impaired in three cases (III.2, III.4 and III.9) and perceptual and spatial skills were impaired in two cases (III.2 and III.9).
Table 2. Summary of neuropsychological findings at presentation.
| Case | III.2 | III.4 | III.9 | III.11 |
|---|---|---|---|---|
| Duration since symptom onset (years) | 2 | 2 | 1 | 1 |
| General intellectual function | ||||
| Verbal IQ | 84 | 61*** | 72** | 85 |
| Performance IQ | 72** | <54*** | 60*** | 108 |
| Memory | ||||
| Verbal memorya | 32/50** | fail | 21/25 | 38/50 |
| Visual memoryb | 29/50*** | fail | 14/25*** | 44/50 |
| Language | ||||
| Namingc | 16/30 | 13/30* | 8/30*** | 3/30*** |
| Word comprehensiond | 36/50 | NT | NT | 33/50 |
| Word repetition | normal | normal | impaired | impaired |
| Other cognitive domains | ||||
| Spellinge | 14/30 | NT | 2/30*** | 17/30 |
| Calculationf | 1/24*** | 0/24*** | 0/24*** | 14/24 |
| Visuospatial/perceptual skillsg | <5th percentile | 25-50th percentile | <5th percentile | 75-90th percentile |
| Limb praxis | mild impairment | severe impairment | severe impairment | mild impairment |
| Executive functionh, i | fail | fail | fail | normal |
Recognition Memory Test for Words (out of 50) (35) [short version - out of 25]
Recognition Memory Test for Faces (out of 50) (35) [short version - out of 25]
Graded Naming Test (14)
Warrington Synonyms Test (out of 50) (36)
Graded Difficulty Spelling Test (37)
Graded Difficulty Arithmetic Test (out of 24) (38)
Visual Object and Spatial Perception (VOSP) battery (39)
Weigl test (40)
Modified card sorting test (41)
<1st percentile
=1st to < 5th percentile
=5th to <10th percentile; no stars ≥10th percentile
NT = not tested
The neuropsychometric profile of these cases, taken together, is noteworthy for the relatively early involvement of parietal lobe (calculation, visuoperceptual and visuospatial) functions. In contrast to features such as executive dysfunction, anomia and memory impairment, which are observed in a broad spectrum of FTLD cases, including familial cases with mutations in the tau gene (15), parietal lobe features are unusual in FTLD. Coupled with the high frequency of limb apraxia, this pattern argues for a more posterior extension of disease associated with this PGRN mutation than would be regarded as typical in FTLD.
Brain imaging findings
Four patients (cases III.2, III.4, III.9 and III.11) underwent brain imaging. Findings are summarized in Table 3 and representative magnetic resonance (MR) images are presented in Figure 2. Brain imaging findings show considerable individual variation in this series but atrophy involving the frontal, temporal and parietal lobes was present in all cases. Cerebral atrophy was strikingly asymmetrical in cases III.2, III.4 (both predominantly right-sided) and III.11 (predominantly left-sided).
Table 3. Summary of brain imaging findings.
| Case | Duration since symptom onset (years) | Modality | Key findings |
|---|---|---|---|
| III.2 | 2 | MRI | Marked asymmetrical frontal, temporal and parietal lobe atrophy predominantly affecting the right side. In the frontal lobes there is greater atrophy affecting the inferior frontal regions than the frontal convexities, although the convexity sulci are wider bilaterally. There is asymmetrical temporal lobe atrophy, with the whole right temporal lobe affected more than the left. There is also widespread right-sided parietal atrophy. |
| III.4 | 1 | CT | Asymmetrical generalized atrophy, affecting the right hemisphere more than the left. |
| III.9 | 1 | MRI | Mild diffuse atrophy with increased prominence of the parietal sulci. |
| III.11 | 1 | MRI | Asymmetrical frontal, temporal and parietal lobe atrophy predominantly affecting the left side. In the frontal lobes there is atrophy of the medial superior frontal and frontopolar regions, and involvement of the anterior cingulate gyrus. There is marked atrophy of the left temporal pole. The left superior temporal, middle and inferior temporal and fusiform gyri are significantly affected, with some atrophy of left amygdala and hippocampus. In the parietal lobes there is relatively selective atrophy of the left angular gyrus posteriorly. |
Figure 2.


Representative brain images in affected individuals (III.2 and III.11)
Figs 2a,2b: Coronal T1 MR images showing asymmetrical right-sided fronto-temporo-parietal atrophy (III.2)
Figs 2c, 2d: Coronal T1 MR images showing asymmetrical left-sided fronto-temporo-parietal atrophy (III.11)
Neuropathological findings
Three members of the kindred (cases III.2, III.3 and III.4) have come to post-mortem examination with similar appearances seen in all cases. Macroscopically, all showed severe bilateral atrophy of the frontal and temporal lobes with moderate atrophy of the parietal lobes and relative sparing of the occipital lobes. Histological investigation showed superficial spongiosis, nerve cell loss and gliosis of the frontal, temporal and parietal cortices. There were numerous ubiquitin-positive neurites and neuronal cytoplasmic inclusions (NCI) in the superficial cortical laminae and striatum. Scattered ubiquitin-positive neuronal intranuclear inclusions (NII) were found, which were lentiform or round in shape. The dentate fascia granule cells also contained scattered NCI, which often had a granular appearance, as well as occasional NII. Figure 4 shows the findings in case III.2. All three cases showed features of type 1 FTLD-U according to the classification described by Mackenzie et al. (16, 17) which corresponds to previously reported cases with PGRN mutations (18). Of note, the neurites, NCI and NII also stained for TDP-43 which has recently been described to be a component of the ubiquitin-positive neurites and inclusions in FTLD-U (17).
Figure 4.

Histological features in case III.2. A = dentate fascia ubiquitin-positive inclusions; B = ubiquitin-positive neuronal cytoplasmic inclusions (NCI) and neurites in the frontal cortex; C & D = ubiquitin-positive neuronal intranuclear inclusions (NII) in the frontal cortex. The NII on C is of the lentiform type.
Genetic analyses
All 13 exons of the PGRN gene were sequenced in cases III.3 and III.11 in at least one direction. Analysis of the electropherogram traces revealed a c.90_91insCTGC mutation in the first coding exon on the reverse complement strand in both cases (figure 3 shows the electropherogram for III.3), which would be predicted to cause a frameshift and premature termination (C31LfsX35). This mutation was originally described in the UBC17 family (7,19). Analysis of four microsatellites near to PGRN in patient III.3 identified rare alleles linked to the UBC17 mutation (17), suggesting a common ancestry.
Figure 3.
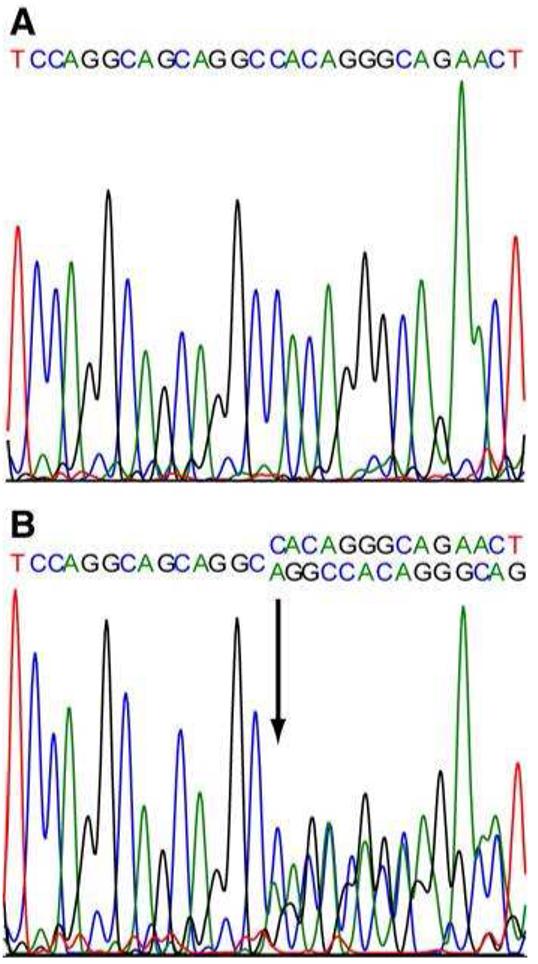
Electropherogram traces from the first coding exon of PGRN in the reverse complement. (A) healthy population control DNA (B) patient III.3. Arrow indicates the presence of a c.90_91insCTGC mutation.
COMMENT
Here we describe the phenotype of a large family with autosomal dominant TDP-43-positive FTLD-U due to a PGRN mutation. Common clinical features include language output impairment leading to mutism, behavioural disturbance with early prominent apathy, and extrapyramidal features. In addition to these features, which are commonly associated with FTLD, we propose that parietal deficits (dyscalculia, visuospatial/perceptual dysfunction, and/or limb apraxia), which are infrequently described in FTLD, may also be a feature of the progranulin phenotype. Our case III.9 was diagnosed during life with a CBD syndrome in which parietal dysfunction is well recognized (20) and which is pathologically heterogeneous (21). Indeed, the CBD syndrome has been described previously in association with a PGRN mutation (9). However, it is notable that all of the cases here had evidence of parietal dysfunction. Neuropsychometry may expose parietal lobe deficits that might otherwise be overlooked.
We suggest that language and behavioural features may have greater value in differentiating these cases from other causes of FTLD when accompanied by parietal deficits. Furthermore, the specific features of the speech or behavioural syndrome may also have diagnostic potential. All five of the patients described in detail in family DRC255 had impairment of language output. Cases III.9 and III.11 had features consistent with PNFA (1). In contrast, cases III.2, III.3 and III.4 exhibited a decrease in quantity of spontaneous speech in the absence of semantic, grammatical or phonemic errors and with relatively intact naming and verbal comprehension. This would be in keeping with dynamic aphasia (22) rather than the PNFA or SD subtypes of FTLD (1). Our cases suggest that the common endpoint for both the PNFA and the dynamic aphasia presentations is mutism. Behavioral features were early and prominent in cases III.2, III.3 and III.4 and less prominent in the other two cases. However, all cases developed apathy as the earliest and most salient behavioral feature.
How does our family (DRC255) compare clinically with previously described families with PGRN mutations (1083: 23, DR2-DR8 Belgian founder family: 24, HDDD2: 25,26, HFTD3: 27,28, F53 and F337: 29, and UBC17 which appears to be the same family as DRC255: 17) and with other genetically mediated causes of FTLD, notably mutations in the tau gene? The most commonly described features in patients with PGRN mutations are language output impairment with features suggestive of either dynamic aphasia or non-fluent aphasia and a behavioural syndrome often characterized by apathy. Published data on the clinical phenomenology of patients with PGRN mutations are limited, however parietal lobe features have been described in many families (19,25, 27, 29). These clinical observations are further supported by histopathological evidence of significant parietal lobe involvement in association with PGRN mutations (19,25,26,29). Tau mutations are associated with wide phenotypic variation in published series (30,31). However, in detailed studies of patients with a tau 10+16 mutation (15), disinhibition rather than apathy dominated the behavioural syndrome and while ‘language deficits’ occurred in these patients, features of a non-fluent aphasia were not reported and language impairment was not a prominent feature. Of particular note is that parietal features are generally not described. Parkinsonism and other extrapyramidal features appear to occur commonly in association with both PGRN and tau mutations (19,29,30,31).
The cases described in the present series showed variability in the pattern of radiological atrophy, but it is notable that each of the cases with detailed MR imaging showed frontotemporal atrophy with associated involvement of the parietal lobes. Longitudinal structural imaging in one patient with a PGRN mutation revealed progressive asymmetrical fronto-temporo-parietal atrophy more marked on the right (32), while a recent study specifically comparing PGRN cases with ubiquitin-positive PGRN-negative cases showed that PGRN-positive cases had greater gray matter loss in the parietal lobes (as well as the frontal lobes) (33). These findings combined with the results of this study support the hypothesis that more posterior extension of atrophy to involve the parietal lobes may be a radiological marker for PGRN-associated disease, consistent with the clinical and neuropsychological profile. The involvement of the parietal lobe as well as more anterior areas suggests that PGRN mutations may disrupt functional pathways linking the frontal and parietal lobes, for which anatomical substrates exist in the human brain (34). Preferential disease spread within hemispheric pathways (rather than between hemispheres) would also be in keeping with the striking asymmetry seen early in the disease both in our own and other cases reported in the literature.
Conclusions
The mutation in the PGRN gene in this family produces a clinico-radiological phenotype that overlaps substantially with the spectrum of FTLD due to other sporadic and genetically mediated pathologies. However, certain clinical and radiological features common to a majority of affected individuals suggest that there may be a core phenotype of progranulin-associated disease. In particular, parietal lobe deficits are a salient feature of the progranulin phenotype. Future work will be directed toward testing these observations in other affected families, assessment of their specificity and predictive value, and elucidation of the mechanisms by which PGRN mutations produce their phenotypic effects.
Acknowledgments
Funding/Support
This work was supported by the Alzheimer Research Trust and the Medical Research Council UK and by an EC Contract awarded to the APOPIS Consortium. JDR is supported by a Wellcome Trust Research Training Fellowship. JDW is supported by a Wellcome Trust Intermediate Clinical Fellowship. SPB is supported by the Medical Research Council UK.
MNR had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Financial Disclosure
Nil
References
- 1.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998 Dec;51(6):1546–54. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 2.Kertesz A, Davidson W, Munoz DG. Clinical and pathological overlap between frontotemporal dementia, primary progressive aphasia and corticobasal degeneration: the Pick complex. Dement Geriatr Cogn Disord. 1999;10(Suppl 1):46–9. doi: 10.1159/000051212. [DOI] [PubMed] [Google Scholar]
- 3.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002 Oct 8;59(7):1077–9. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 4.Stevens M, van Duijn CM, Kamphorst W, et al. Familial aggregation in frontotemporal dementia. Neurology. 1998;50(6):1541–5. doi: 10.1212/wnl.50.6.1541. [DOI] [PubMed] [Google Scholar]
- 5.Stanford PM, Brooks WS, Teber ET, et al. Frequency of tau mutations in familial and sporadic frontotemporal dementia and other tauopathies. J Neurol. 2004 Sep;251(9):1098–104. doi: 10.1007/s00415-004-0489-x. [DOI] [PubMed] [Google Scholar]
- 6.Gass J, Cannon A, Mackenzie IR, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006 Oct 15;15(20):2988–3001. doi: 10.1093/hmg/ddl241. Epub 2006 Sep 1. [DOI] [PubMed] [Google Scholar]
- 7.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006 Aug 24;442(7105):916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 8.Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006 Aug 24;442(7105):920–4. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 9.Masellis M, Momeni P, Meschino W, et al. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006 Oct 9; doi: 10.1093/brain/awl276. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 10.Folstein M, Folstein S, McHugh P. The “Mini Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 11.Neary D, Snowden J, Mann D. Frontotemporal dementia. Lancet Neurol. 2005 Nov;4(11):771–80. doi: 10.1016/S1474-4422(05)70223-4. [DOI] [PubMed] [Google Scholar]
- 12.Liu W, Miller BL, Kramer JH, et al. Behavioral disorders in the frontal and temporal variants of frontotemporal dementia. Neurology. 2004 Mar 9;62(5):742–8. doi: 10.1212/01.wnl.0000113729.77161.c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boxer AL, Miller BL. Clinical features of frontotemporal dementia. Alzheimer Dis Assoc Disord. 2005 Oct-Dec;19(Suppl 1):S3–6. doi: 10.1097/01.wad.0000183086.99691.91. [DOI] [PubMed] [Google Scholar]
- 14.McKenna P, Warrington EK. Testing for nominal dysphasia. J Neurol Neurosurg Psychiatry. 1980;43(9):781–8. doi: 10.1136/jnnp.43.9.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janssen JC, Warrington EK, Morris HR, et al. Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. 2002 Apr 23;58(8):1161–8. doi: 10.1212/wnl.58.8.1161. [DOI] [PubMed] [Google Scholar]
- 16.Mackenzie IR, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DM. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol (Berl) 2006 Nov;112(5):539–49. doi: 10.1007/s00401-006-0138-9. Epub 2006 Sep 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol (Berl) 2007 Jan 12; doi: 10.1007/s00401-006-0189-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 18.Mackenzie IR, Baker M, Pickering-Brown S, et al. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006 Nov;129(Pt 11):3081–90. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- 19.Mackenzie IR, Baker M, West G, et al. A family with tau-negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain. 2006 Apr;129(Pt 4):853–67. doi: 10.1093/brain/awh724. [DOI] [PubMed] [Google Scholar]
- 20.Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S15–9. doi: 10.1002/ana.10570. [DOI] [PubMed] [Google Scholar]
- 21.Boeve BF, Maraganore DM, Parisi JE, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology. 1999 Sep 11;53(4):795–800. doi: 10.1212/wnl.53.4.795. [DOI] [PubMed] [Google Scholar]
- 22.Warren JD, Warren JE, Fox NC, Warrington EK. Nothing to say, something to sing: primary progressive dynamic aphasia. Neurocase. 2003 Apr 9;(2):140–55. doi: 10.1076/neur.9.2.140.15068. [DOI] [PubMed] [Google Scholar]
- 23.Rademakers R, Cruts M, Dermaut B, et al. Tau negative frontal lobe dementia at 17q21: significant finemapping of the candidate region to a 4.8 cM interval. Mol Psychiatry. 2002;7(10):1064–74. doi: 10.1038/sj.mp.4001198. [DOI] [PubMed] [Google Scholar]
- 24.van der Zee J, Rademakers R, Engelborghs S, et al. A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain. 2006 Apr;129(Pt 4):841–52. doi: 10.1093/brain/awl029. [DOI] [PubMed] [Google Scholar]
- 25.Lendon CL, Lynch T, Norton J, et al. Hereditary dysphasic disinhibition dementia: a frontotemporal dementia linked to 17q21-22. Neurology. 1998 Jun;50(6):1546–55. doi: 10.1212/wnl.50.6.1546. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee O, Pastor P, Cairns NJ, et al. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006 Sep;60(3):314–22. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosso SM, Kamphorst W, de Graaf B, et al. Familial frontotemporal dementia with ubiquitin-positive inclusions is linked to chromosome 17q21-22. Brain. 2001 Oct;124(Pt 10):1948–57. doi: 10.1093/brain/124.10.1948. [DOI] [PubMed] [Google Scholar]
- 28.Bronner IF, Rizzu P, Seelaar H, et al. Progranulin mutations in Dutch familial frontotemporal lobar degeneration. Eur J Hum Genet. 2007 Jan 17; doi: 10.1038/sj.ejhg.5201772. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 29.Snowden JS, Pickering-Brown SM, Mackenzie IR, et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain. 2006 Nov;129(Pt 11):3091–102. doi: 10.1093/brain/awl267. Epub 2006 Sep 26. [DOI] [PubMed] [Google Scholar]
- 30.van Swieten JC, Rosso SM, van Herpen E, Kamphorst W, Ravid R, Heutink P. Phenotypic variation in frontotemporal dementia and parkinsonism linked to chromosome 17. Dement Geriatr Cogn Disord. 2004;17(4):261–4. doi: 10.1159/000077150. [DOI] [PubMed] [Google Scholar]
- 31.Baba Y, Tsuboi Y, Baker MC, et al. The effect of tau genotype on clinical features in FTDP-17. Parkinsonism Relat Disord. 2005 Jun;11(4):205–8. doi: 10.1016/j.parkreldis.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Boeve BF, Baker M, Dickson DW, et al. Frontotemporal dementia and parkinsonism associated with the IVS1+1G->A mutation in progranulin: a clinicopathologic study. Brain. 2006 Oct 9; doi: 10.1093/brain/awl268. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Whitwell JL, Jack CR, Baker M, et al. Voxel-Based Morphometry in Frontotemporal Lobar Degeneration With Ubiquitin-Positive Inclusions With and Without Progranulin Mutations. Arch Neurol. 2007;64:371–376. doi: 10.1001/archneur.64.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Catani M, Jones DK, ffytche DH. Perisylvian language networks of the human brain. Ann Neurol. 2005 Jan;57(1):8–16. doi: 10.1002/ana.20319. [DOI] [PubMed] [Google Scholar]
- 35.Warrington EK. Manual for the Recognition Memory Test for words and faces. NFER-Nelson; Windsor, UK: 1984. [Google Scholar]
- 36.Warrington EK, McKenna P, Orpwood L. Single word comprehension: a concrete and abstract word synonym test. Neuropsychol Rehabil. 1998;8:143–54. [Google Scholar]
- 37.Baxter DM, Warrington EK. Measuring dysgraphia: a graded-difficulty spelling test. Behav Neurol. 1994;7:107–16. doi: 10.3233/BEN-1994-73-401. (3-4) [DOI] [PubMed] [Google Scholar]
- 38.Jackson M, Warrington EK. Arithmetic skills in patients with unilateral cerebral lesions. Cortex. 1986;22(4):611–20. doi: 10.1016/s0010-9452(86)80020-x. [DOI] [PubMed] [Google Scholar]
- 39.Warrington EK, James M. The visual object and space perception battery. Thames Valley Test Company; Bury St Edmunds, UK: 1991. [Google Scholar]
- 40.Weigl E. On the psychology of the so called process of abstraction. J Abnorm Soc Psychol. 1948;36:3–33. [Google Scholar]
- 41.Nelson HE. A modified card sorting test sensitive to frontal lobe defects. Cortex. 1976;12:313–24. doi: 10.1016/s0010-9452(76)80035-4. [DOI] [PubMed] [Google Scholar]