

Abstract
Sporadic inclusion-body myositis (s-IBM) is the most common muscle disease of older persons. The muscle-fiber molecular phenotype exhibits similarities to both Alzheimer-disease (AD) and Parkinson-disease (PD) brains, including accumulations of amyloid-β, phosphorylated tau, α-synuclein and parkin, as well as evidence of oxidative stress and mitochondrial abnormalities. Early-onset autosomal-recessive PD can be caused by mutations in the DJ-1 gene, leading to its inactivation. DJ-1 has anti-oxidative and mitochondrial-protective properties. In AD and PD brains, DJ-1 is increased and oxidized.
We studied DJ-1 in 17 s-IBM and 18 disease-control and normal muscle biopsies by: 1) immunoblots of muscle homogenates and mitochondrial fractions; 2) real-time PCR; 3) oxyblots evaluating DJ-1 oxidation; 4) light- and electron-microscopic immunocytochemistry. Compared to controls, in s-IBM muscle fibers DJ-1 was: a) increased in the soluble fraction, monomer 2-fold (p=0.01), and dimer 2.8-fold (p=0.004); b) increased in the mitochondrial fraction; c) highly oxidized; and d) aggregated in about 15% of the abnormal muscle fibers. DJ-1 mRNA was increased 3.5-fold (p=0.034).
Accordingly, DJ-1 might play a role in human muscle disease, and thus not be limited to human CNS degenerations. In s-IBM muscle fibers, DJ-1 could be protecting these fibers against oxidative stress, including protection of mitochondria.
Keywords: DJ-1, inclusion-body myositis, Parkinson disease, oxidative stress, mitochondria
Introduction
Sporadic-inclusion body myositis (s-IBM), the most common, progressive muscle disease associated with aging, is manifested by pronounced muscle weakness and wasting, leading to severe disability [reviewed in 1]. There is no successful treatment. The pathology of s-IBM muscle biopsies is characterized by: a) vacuolar degeneration and atrophy of muscle fibers, accompanied by intra-muscle-fiber accumulations of misfolded, ubiquitinated, congophilic, multi-protein aggregates; and b) lymphocytic inflammation [reviewed in 2–4]. An intriguing feature of the s-IBM muscle-fiber phenotype is its similarity to both the Alzheimer-disease (AD) brain and Parkinson-disease (PD) Lewy bodies, including accumulation of aggregated amyloid-β (Aβ), phosphorylated tau, and several other Alzheimer-characteristic proteins, as well as α-synuclein (α-syn) and parkin [reviewed in 2,4]. Also similarly to AD and PD, oxidative stress and mitochondrial abnormalities are evident in s-IBM [4,5].
DJ-1 is a ubiquitously-expressed protein of the ThiJ/PfpI/DJ1 superfamily, which is highly conserved across species [reviewed in 6,7]. Mutations in the DJ-1 gene, preventing expression of DJ-1- protein, are a cause of early-onset autosomal-recessive PD [6–9]. In sporadic AD and PD brains, DJ-1 was reported to be increased and highly oxidized [10]. Although its precise functions are not yet known, DJ-1 has been proposed to act as an antioxidant [11–15] and be an important mitochondrial protective agent [14,15]. The role of DJ-1 in muscle diseases has not been studied, to our knowledge.
Because of the molecular and pathological similarities of s-IBM muscle fibers to AD and PD brains, we asked whether alterations of DJ-1 occur in s-IBM muscle fibers.
Material and Methods
Muscle Biopsies
Studies were performed on portions of diagnostic muscle biopsies obtained (with informed consent) from 17 s-IBM and 18 age-matched-control biopsies, including 2 dermatomyositis , 2 polymyositis , 1 morphologically-nonspecific myopathy, 2 amyotrophic lateral sclerosis, 2 peripheral neuropathy, and 9 normal muscles (considered normal after all diagnostic tests were performed). Not all studies were performed on all biopsies (details below). s-IBM patients were ages 61–87 years, median age 73; normal control patients were ages 61–86, median age 72. Diagnoses were based on clinical and laboratory investigations, including our routinely-performed 16-reaction diagnostic histochemistry of the biopsies. All s-IBM biopsies met s-IBM diagnostic criteria, as we described [16].
Immunoblots
These were performed to: a) evaluate the specificity of DJ-1 antibodies rabbit polyclonal [Abcam, Boston, MA] and mouse monoclonal [Stressgene, Victoria, BC, Canada] in human muscle, both having been previously characterized in human and mouse brain and in various cultured cells [9,15,17]; and b) determine the content of DJ-1 in s-IBM and normal-control muscle biopsies. Because previous studies reported a difference of DJ-1 content between the soluble and insoluble fractions of brain tissue and in cells overexpressing DJ-1 [17,18], in the present study we performed immunoblots of DJ-1 on soluble and insoluble fractions of total homogenates of 5 s-IBM and 5 age-matched normal-control muscle biopsies. The detergent-soluble/insoluble fractionation was modified from a method described previously [19]. In brief, 20 10µm-thick frozen-muscle sections were collected at −25°C, rapidly suspended in lysis buffer (20 mM HEPES [pH 7.4] containing 120 mM NaCl, 5 mM EDTA, 10% glycerol, 1% Triton X-100 and supplemented with complete protease inhibitors [Roche Diagnostics, Mannheim, Germany]). Muscle fibers and their intracellular organelles were disrupted using an ultrasonic homogenizer. After incubation for 30 min at 4°C, the suspensions were separated by centrifugation at 16,000g for 1 h at 4°C. Supernatants were collected as the soluble fraction, and were considered to contain cytoplasmic proteins, as well as the content of the disrupted organelles, including mitochondria.
The insoluble pellet was washed four times with ice-cold lysis buffer, solubilized in SDS (final concentration 1%) for 1 hr at 60°C, and centrifuged at 16,000g for 30 min at 4°C. The supernatants were collected as the insoluble fraction.
20 µg of protein (measured by the Bradford method) was loaded on 10% NuPAGE gels (Invitrogen, Carlsbad, CA), electrophoresed, transferred to nitrocellulose or PVDF membranes, and immunoprobed with an antibody against DJ-1 diluted 1:1500, as we described [20–22]. The blots were developed using the enhanced chemiluminescence system (Amersham Bioscience, Piscataway, NJ). Protein loading was evaluated by actin bands visualized with a rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), diluted 1:2000. Quantification of immunoreactivity was performed by densitometric analysis using NIH Image 1.310 software. Intensity of the band of interest was calculated in relation to intensity of the actin band. Omission of a primary antibody was the control for reaction specificity.
RNA isolation and qRT-PCR
Total RNA from 5 s-IBM and 6 age-matched control muscle biopsies was isolated using an RNA isolation kit (BD Pharmingen, San Diego, CA), as recently described [22]. 1µg of RNA was subjected to genomic-DNA removal, and then cDNA synthesis using the QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA), according to manufacturer’s instructions. Real-time PCR (qRT-PCR) was performed in duplicate at total volume of 25 µl, using 1 µl of cDNA, QuantiTect Primers (Qiagen) for PARK7/DJ1 or GAPDH, and QuantiFastSYBR Green PCR Master mix (Qiagen). PCR runs were performed on an Eppendorf Mastercycler® realplex2. Cycling conditions were 95°C for 5 minutes, followed by 40 cycles of 95°C for 10 seconds and 60°C for 30 seconds. Relative gene expression was calculated by using the 2−ΔΔCT method, in which the amount of DJ-1 mRNA was normalized to an endogenous reference (GAPDH). The results are expressed as fold induction relative to controls. Correct PCR products were confirmed by agarose gel electropheresis and melting curve analysis.
Light-microscopic immunocytochemistry
To determine the localization of DJ-1 in human muscle fibers, we performed immunofluorescence on 10-µm-thick unfixed sections of fresh-frozen diagnostic muscle biopsies, as described [20–23], using either the rabbit polyclonal or mouse monoclonal antibodies, diluted 1:100. In addition to the DJ-1 immunofluorescence, some sections were counterstained with the nuclear marker Hoechst 33342 (Invitrogen/Molecular Probes). Immunostainings were performed on 4 s-IBM, 4 age-matched normal-controls and all disease-controls listed above. To block nonspecific-binding of an antibody to Fc receptors, sections were preincubated with normal goat serum diluted 1:10 [20–23]. Controls for staining specificity were omission of the primary antibody, or its replacement with non-immune sera or irrelevant antibody; these were always negative.
Gold-immuno-electronmicroscopy
This was performed on 10-µm unfixed frozen sections adhered to the bottom of 35-mm Petri dishes, as detailed previously [20–23]. For double immunostaining, sections were incubated concurrently in rabbit polyclonal antibody against DJ-1, combined with mouse monoclonal antibody against the mitochondrial marker porin/VDAC (Invitrogen/Molecular Probes), followed by incubation in two different specie-specific secondary antibodies, one conjugated to 12-nm gold particles and the other conjugated to 6-nm gold particles. Subsequently, the sections were fixed in a 2% paraformaldehyde-1.25% glutaraldehyde mixture, postfixed in 1% osmium tetroxide, and Epon-embedded in situ in the Petri dish. The embedded sections in the dish were viewed under phase-contrast microscopy, and the muscle fibers of interest were marked, drilled-out, and processed for electron microscopy, as described [20–23].
Isolation of mitochondrial fractions
This was performed from four s-IBM and four age-matched control muscle biopsy samples using a four steps procedure, essentially as previously described by others [24]. Briefly, 30 10µm sections of fresh-frozen human muscle biopsies were placed in a pre-chilled microcentrifuge tube containing 200 µl of isolation medium (320 mM sucrose, 1 mM EDTA, 10 mM Trizma-base, pH 7.4). Step 1: after thorough but gentle manual homogenization on ice, the homogenate was centrifuged at 3000 g for 5 min at 4°C, and the pellet discarded. Step 2: the supernatant was centrifuged at 12,000 g for 10 min at 4°C. Step 3: the resulting pellet (crude mitochondrial fraction) was re-suspended in 1 ml isolation medium, and centrifuged at 3000 g to remove nuclear contamination. Step 4: the resulting pellet was discarded and the supernatant was centrifuged at 12, 000 g for a further 10 min. Steps 3–4 were repeated to further purify the mitochondrial fraction. The resulting pellet from the last 12, 000 g spin constituted the mitochondria-enriched fraction [24].
Immunoprecipitation, 2,4- Dinitrophenyl (DNP) derivatization, and Oxyblot
To evaluate the presence of carbonyl groups indicative of DJ-1 oxidation, we performed an immunoprecipitation/protein-derivatization/ immunoblot procedure in four s-IBM and four age-matched control muscle biopsies, and in one dermatomyositis muscle biopsy. Because human muscle biopsies are rather sparse, we were not able to perform this procedure on more samples. 100 µg of muscle homogenate prepared as for immunoblots (above), were immunoprecipitated for 1 hour at 4°C with the DJ-1 rabbit polyclonal antibody that had been cross-linked to protein-A Dynabeads (Invitrogen), according to the manufacturer’s protocol. After extensive washing, the DJ-1- immunoprecipitated complex was eluted from the beads by boiling for 10 minutes, followed by 5 minutes centrifugation at 16,000g. The supernatant was saved, and the pellet discarded. To determine the carbonyl content of DJ-1, we used the OxyBlot Protein Oxidation Detection Kit (Chemicon International, Temecula, CA), as directed by the manufacturer. The assay is based on an established method utilizing derivatization of carbonyls with 2,4-dinitrophenylhydrazine (DNPH) and subsequent immuno-detection of hydrazones with anti-DNP antibody [10]. To derivatize the sample, 10µl of DJ-1-immunoprecipitated complex was added to 10 µl of 10mM 2,4-dinitrophenylhydrazine (DNPH) solution for 15 minutes. As a control, 10µl of a duplicate sample was added to10 µl of a control solution not containing DNPH. Both samples were subsequently incubated with 7.5 µl of a neutralization solution to stop the derivatization reaction. After neutralization, the samples were loaded onto 10% NuPAGE gel (as above for immunoblots) and immunoprobed with an antibody against DNP (according to the manufacturer’s protocol). The immunoblots were then developed and processed as above.
Statistical Analysis
In all experiments these analyses were performed using Student t-test. Significance level was set at p<0.05. For all experimetns, data are reported as mean ± SEM.
Results
DJ-1 is increased in the soluble fraction of s-IBM muscle fibers
Both anti-DJ-1 antibodies recognized a 23 kDa DJ-1 monomer and 46 kDa DJ-1 dimer in both control and s-IBM muscle fibers. To determine whether, DJ-1 is increased in s-IBM muscle fibers, as was recently reported for AD and PD brains [10,18], we performed immunoblot analysis of soluble and insoluble fractions of five s-IBM and five control muscle biopsies. As compared to controls, DJ-1 monomer in the s-IBM soluble fraction was increased 2-fold (p=0.01), and DJ-1 dimer was increased 2.8-fold (p=0.004) (Figure 1). Those bands were specific since they were not present after the primary antibody was omitted from the immunoblotting (Figure 1). There was no difference in DJ-1 expression between the insoluble fractions of control and s-IBM muscle fibers (not shown).
Figure 1.
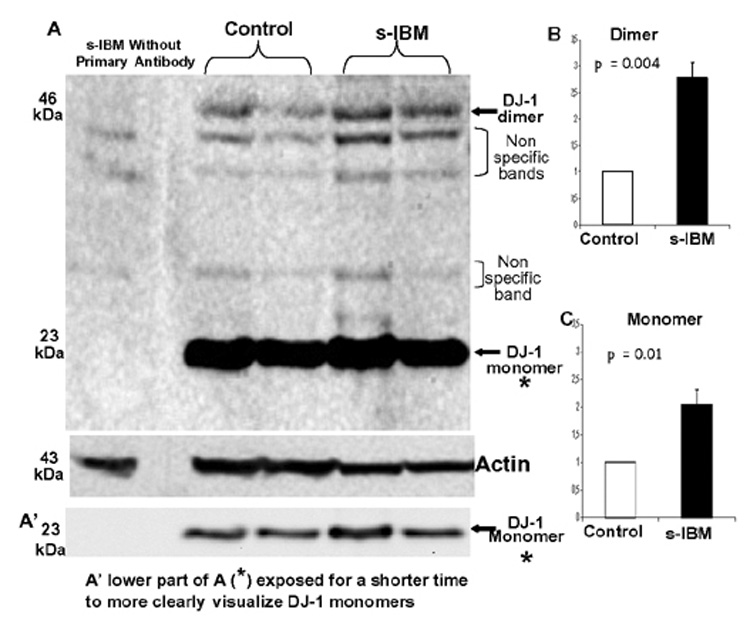
Immunoblots of DJ-1 in muscle homogenates of Control and s-IBM muscle biopsies. A - demonstrates that a 23 kDa band corresponding to DJ-1 monomer, and 46 kDa band corresponding to DJ-1 dimer are present in the soluble fractions of both control and s-IBM muscle biopsies. There are 3 non-specific bands, which are also present after a primary antibody has been omitted from the immunoblotting, of the soluble fractions of s-IBM and Control muscle. The actin bands (below) serve as the protein-loading control. A’ indicates the same monomer bands as in A but exposed for a shorter time. B, and C - densitometric analysis of DJ-1 dimer and monomer, both of which are significantly increased in s-IBM muscle fibers.
DJ-1 mRNA is increased in s-IBM muscle fibers
In addition to the increased DJ-1 protein, by quantitative RT-PCR, mRNA of DJ-1 in s-IBM muscle biopsies was increased 3.5-fold (p=0.034) as compared to the control human muscle biopsies (Figure 2).
Figure 2.

Real-time PCR of DJ-1 mRNA in Control and s-IBM muscle fibers. Data are presented as the mean ± SEM, normalized to the GAPDH expression.
DJ-1 is aggregated in only a minority of s-IBM muscle fibers
In all s-IBM muscle biopsies, only about 10 to 15% of the abnormal muscle fibers contained various-sized DJ-immunoreactive cytoplasmic aggregates (Figure 3 A, B,C). Those fibers also often had a slightly and diffusely increased cytoplasmic DJ-1 immunoreactivity (Figure 3 A,C). Aggregated DJ-1 was not associated with the nuclei (Figure 3 C). Mononuclear cell infiltrates were not immunoreactive for DJ-1 in either s-IBM, polymyositis or dermatomyositis. Under our staining conditions, in normal muscle biopsies DJ-1 was very weakly and diffusely distributed in the cytoplasm, and nuclei were not highlighted by the DJ-1 immunoreaction (Fig.3D). In none of the disease-control or normal-control biopsies were there muscle fibers containing DJ-1 immunoreactive-aggregates.
Figure 3.

Immunofluorescence of DJ-1 in s-IBM and Control normal muscle fibers. A and B - single-label immunofluorescence illustrates DJ-1-immunoreactive aggregates in two vacuolated muscle fibers. C – immunofluorescence for DJ-1 (red) in another muscle fiber co-stained with the nuclear marker Hoechst (blue), indicates that DJ-1 immunoreactivity is not related to nuclei. Some of the DJ-1 aggregates are indicated by arrows in A–C. D, Control normal muscle fibers, in which DJ-1 immunoreactivity is very weak and uniform. A–C ×1100; D × 850.
In both control and s-IBM muscle biopsies DJ-1 associates with mitochondria by immuno-electronmicroscopy, and its amount is increased in s-IBM mitochondrial fractions
To determine whether DJ-1 is present within mitochondria, we performed double-label gold immuno-electronmicroscopy of DJ-1 with the mitochondrial marker porin/VDAC. Co-labeling with porin was used to identify mitochondria, since our gold immunostaining performed on frozen sections often does not permit proper morphological preservation of mitochondria. We found that DJ-1 was present in porin-labeled mitochondria, in both control and s-IBM muscle fibers (Figure 4 A,B,C) - - however, the precise localization of DJ-1 within the mitochondria could not be determined. The non-mitochondrial part of the muscle fibers did not contain gold particles (Figure 4C). (Since cytoplasmic DJ-1 aggregates are present only in the minority of s-IBM muscle fibers, we were not able to localize them by immuno-electronmicroscopy.) Even though it appeared that some of the mitochondria in s-IBM biopsies had more gold particles representing DJ-1 that control mitochondria, immuno-electronmicroscopic preparations are not suitable for quantification. To verify our morphologic observations, and to determine whether mitochondrial DJ-1 might have contributed to the increased DJ-1 we observed on the immunoblots of the soluble fractions of muscle fibers, we isolated mitochondria from four s-IBM and four age matched control muscle biopsies (because human muscle biopsies are rather sparse we were not able to isolate mitochondria from more samples). Immunoblots of mitochondrial fractions revealed that DJ-1 monomer was substantially increased in s-IBM muscle mitochondria as compared to the mitochondria isolated from the age-matched normal muscle biopsies (Figure 4 D). DJ-1 dimer was not detectable in the mitochondrial fraction. Considering that the total amount of DJ-1 dimer is usually much lower than that of the DJ-1 monomer, we postulate that our inability to detect DJ-1 dimer in the mitochondrial fraction might be due to the small amount of mitochondrial proteins that we were able to obtain from our mitochondrial fractions; much more loading of of the mitochondrial protein might be required to visualize DJ-1 dimer in those preparations. Another possibility is that DJ-1 dimer is present in the cytoplasm but not in the mitochondria.
Figure 4.

A–C. Double-label gold-immuno-electronmicroscopy of DJ-1 and porin in mitochondria of control and s-IBM muscle fibers. This illustrates that DJ-1 (12 nm gold particles) and a mitochondrial marker porin (6 nm gold particles) are both associated with mitochondria of control (A) and s-IBM (B, C) muscle fibers. All × 85,000. D - Immunoblots of isolated mitochondrial fractions of four age-matched control and four s-IBM muscle fibers. DJ-1 monomer is substantially increased in s-IBM mitochondria as compared to controls, while dimer is not detectable. Immunoblots of porin were used as the mitochondria protein loading control.
DJ-1 is oxidized in s-IBM muscle fibers
Because in both AD and PD brains DJ-1 was reported to be oxidized [10], and was proposed to act as a redox-dependent chaperone [25], we performed immunoprecipitation/protein-oxidation detection-blot experiments to evaluate carbonyl groups of DJ-1 in s-IBM muscle fibers. The presence of carbonyls groups is a commonly-used indicator of oxidative damage to proteins [26]. Four control and four s-IBM muscle-biopsy homogenates were immunoprecipitated with anti-DJ-1 antibody (each sample in duplicate). Successful immunoprecipitation of DJ-1 is indicated in Figure 5A, which indicates a strong band corresponding to the DJ-1 monomer, and a very faint band corresponding to the DJ-1 dimer. For oxyblot procedure, one duplicate-sample underwent derivatization with 2,4-dinitrophenylhydrazine (DNPH+); the other one was not derivatized and served as a negative control (DNPH−) (Figure 5B). All samples, each from four s-IBM and four control biopsies, were electrophoresed and immunoblotted with an anti-DNP antibody. The resulting oxyblot, demonstrating protein carbonyl content, as an indicator of oxidation of the protein, showed that DJ-1 is highly oxidized in s-IBM muscle fibers, but not in the four control muscles (Figure 5B). Similarly to the immunoblots of the mitochondrial fractions, DJ-1 dimer was not detected on oxyblot preparations. This might be due to the fact that either a) only a part of the total DJ-1 present in s-IBM muscle is oxidized, or b) much more protein loading would be required to detect oxidized dimer.
Figure 5.

Immunopreciptitation (A) and oxidation (B) of DJ-1 in s-IBM and Control muscle biopsies. A- immunoprecipitation (IP) was performed using an anti-DJ-1 rabbit polyclonal antibody, followed by immunoblotting (IB) using an anti-DJ-1 mouse monoclonal antibody. #, the primary antibody was omitted from the immunoprecipitation reaction in order to ascertain the specificity of the reaction. B-The oxyblot, demonstrating protein carbonyl content, as an indicator of oxidation of the protein, shows that DJ-1 is highly oxidized in s-IBM muscle fibers, but not in age-matched Control muscles (details in text).
An additional oxyblot was also performed on one muscle biopsy from a 38-old dermatomyositis patient and an age-matched normal biopsy - - this did not reveal oxidized DJ-1 in either sample (not shown).
Discussion
In this study we demonstrate for the first time that in s-IBM muscle fibers DJ-1 is: a) significantly increased on the protein and mRNA levels; b) highly oxidized; c) increased in the mitochondria.
Previously, DJ-1 was studied in Parkinson and Alzheimer disease brains, mouse models, and in various non-muscle cultured cells, most of which were overexpressing DJ-1 [9,10,11,14,15,18,25]. Even though the exact biological functions of DJ-1 are not yet firmly established, it is now generally thought that DJ-1 plays a role in oxidative-stress responses, either as a redox or antioxidant protein [11–15, 25]. DJ-1 seems to oxidize itself to protect cells from oxidative stress, but its over-oxidation leads to its inactivation [13]. Experimental downregulation of DJ-1 sensitizes cells to oxidative stress, and its overexpression protects cells from oxidative-stress-induced cell death [11,12]. Oxidative stress is considered to play a role in PD and AD brains [reviewed in 6, 7,27–29], and in them DJ-1 has been reported increased and oxidatively damaged [10].
The presence of DJ-1 monomer and dimer in normal human muscle fibers suggests that this protein plays a physiological role in this tissue. It has been proposed that even under physiological conditions DJ-1 might be a free radical scavenger [15].
However, both DJ-1 mRNA, and its monomers and dimers are substantially increased in s-IBM muscle fibers as compared to control biopsies. We attribute these increases to a self-defense of s-IBM muscle fibers against cellular stresses. Previously demonstrated in s-IBM fibers were: a) nitric-oxide induced stress [30], and b) oxidative stress as indicated by increased redox factor -1 [31], increased anti-oxidative enzymes SOD-1, GSHPx and catalase [32,33], abnormalities of NF-κB [34,35], and oxidative nucleic acid damage [36]. Our present studies demonstrating in s-IBM fibers increase of DJ-1 mRNA and the increase of both the DJ-1 protein monomers and dimers, and DJ-1 oxidation, together strongly suggest that DJ-1 might play a novel role attempting to protect s-IBM muscle fibers from oxidative stress. However, in s-IBM it is not known whether oxidation of DJ-1 leads to its inactivation, and/or is responsible for its increased dimerization.
In s-IBM fibers, the increase of DJ-1 is mainly occurring in the soluble fraction of the whole muscle-fiber homogenate, and this correlates with the relative paucity of the immuno-histochemically detectable DJ-1 aggregates. By contrast, in previous studies DJ-1 monomer was reported increased in the insoluble fraction of PD brain. Whether this difference depends on the difference in the tissues examined or other factors remains unknown.
Another important aspect of our study is the demonstration that DJ-1 is substantially increased in s-IBM mitochondria as compared to age-matched controls. Previously, DJ-1 has been found in a) mitochondria of mouse brain, and b) mitochondria of cells overexpressing DJ-1, in which experimentally-induced oxidative stress increased the amount of mitochondrial DJ-1 [9,14]. The latter observation led to the proposal that oxidative-stress stimulates translocation of DJ-1 from cytoplasm into mitochondria in order to mitigate mitochondrial damage [9,14]. Overexpressing cells can be subject to artefacts, but our morphological and fractionation studies of human muscle biopsies clearly demonstrated that DJ-1 was localized within mitochondria, and DJ-1 was substantially increased in the mitochondrial fraction of s-IBM fibers. DJ-1 is believed to migrate to loci where oxidative stress occurs [37] - - this could explain its mitochondrial localization in s-IBM muscle fibers. Similarly to AD and PD brain [7,38,39], mitochondrial abnormalities are a prominent feature of s-IBM muscle fibers [5,40]. Ragged-red fibers [41], cytochrome-c-oxidase - negative muscle fibers, and multiple mitochondrial DNA deletions in muscle fibers are more common in s-IBM than in the age-matched normal subjects [5,40]. Even though the exact mechanisms leading to mitochondrial abnormalities in s-IBM are still not known, under experimental conditions amyloid-β precursor protein, amyloid-β and α-synuclein -- all of which are accumulated in s-IBM muscle fibers -- are capable of inducing mitochondrial abnormalities [38, 42–44]. We propose that DJ-1 is attempting to mitigate mitochondrial damage in s-IBM.
Conclusion
Our studies suggest that DJ-1 might play a novel role in human muscle disease, and thus is not limited to human CNS degenerations. Therapeutic enhancement of DJ-1 to diminish oxidative stress might provide a novel therapeutic opportunity in s-IBM.
Our future experimental studies employing our IBM human muscle culture model, which exhibits several aspects of biopsied s-IBM muscle fibers, could possibly provide a better understanding of the role of DJ-1, not only in s-IBM, but also, by analogy, in AD and PD.
Acknowledgements
Supported by grants (to VA) from the National Institutes of Health (AG 16768 Merit Award), the Muscular Dystrophy Association, and The Myositis Association. CT is on leave from Department of Neuroscience, University of Tor Vergata and Fondazione Santa Lucia, Rome, Italy. AN is on leave from Department of Biochemistry, Medical University of Gdansk, Gdansk, Poland. SW was on leave from the Department Anatomy and Neurobiology, Medical University of Gdansk, Gdansk, Poland. Maggie Baburyan provided technical assistance in electronmicroscopy. We are grateful to Drs. Wendy Gilmore and Michael Jakowec of the USC Department of Neurology for allowing us to use their real-time PCR equipment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Engel WK, Askanas V. Inclusion-body myositis: clinical, diagnostic, and pathologic aspects. Neurology. 2006;66 Suppl. 1:20–29. doi: 10.1212/01.wnl.0000192260.33106.bb. [DOI] [PubMed] [Google Scholar]
- 2.Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol. 2007;6:620–631. doi: 10.1016/S1474-4422(07)70171-0. [DOI] [PubMed] [Google Scholar]
- 3.Dalakas MC. Advances in the immunobiology and treatment of inflammatory myopathies. Curr Rheumatol. 2007;4:291–297. doi: 10.1007/s11926-007-0047-5. [DOI] [PubMed] [Google Scholar]
- 4.Askanas V, Engel WK. Inclusion-body myositis, a multifactorial muscle disease associated with aging: current concepts of pathogenesis. Curr Opin Rheumatol. 2007;19:550–559. doi: 10.1097/BOR.0b013e3282efdc7c. [DOI] [PubMed] [Google Scholar]
- 5.Oldfors A, Moslemi AR, Jonasson L, Ohlsson M, Kollberg G, Lindberg C. Mitochondrial abnormalities in inclusion-body myositis. Neurology. 2006;66 Suppl. 1:49–55. doi: 10.1212/01.wnl.0000192127.63013.8d. [DOI] [PubMed] [Google Scholar]
- 6.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nat Rev Neurosc. 2006;7:207–219. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 7.Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16:183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 8.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 9.Blackinton J, Ahmad R, Miller DW, van der Brug MP, Canet-Avilés RM, Hague SM, Kaleem M, Cookson MR. Effects of DJ-1 mutations and polymorphisms on protein stability and subcellular localization. Brain Res Mol Brain Res. 2005;134:76–83. doi: 10.1016/j.molbrainres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Choi J, Sullards MC, Olzmann JA, Rees HD, Weintraub ST, Bostwick DE, Gearing M, Levey AI, Chin LS, Li L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem. 2006;281:10816–10824. doi: 10.1074/jbc.M509079200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinat C, Shendelman S, Jonason A, Leete T, Beal MF, Yang L, Floss T, Abeliovich A. Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: an ES-derived cell model of primary Parkinsonism. PLoS Biol. 2004;2:1754–1763. doi: 10.1371/journal.pbio.0020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meulener MC, Xu K, Thomson L, Ischiropoulos H, Bonini NM. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci U S A. 2006;103:12517–12522. doi: 10.1073/pnas.0601891103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Canet-Avilés RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, Torp R, Torgner IA, Ottersen OP, Dawson TM, Dawson VL. Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet. 2005;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]
- 16.Askanas V, Engel WK. Inclusion-body myositis: newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:1–14. doi: 10.1093/jnen/60.1.1. [DOI] [PubMed] [Google Scholar]
- 17.Neumann M, Müller V, Görner K, Kretzschmar HA, Haass C, Kahle PJ. Pathological properties of the Parkinson's disease-associated protein DJ-1 in alpha-synucleinopathies and tauopathies: relevance for multiple system atrophy and Pick's disease. Acta Neuropathol. 2004;107:489–496. doi: 10.1007/s00401-004-0834-2. [DOI] [PubMed] [Google Scholar]
- 18.Moore DJ, Zhang L, Troncoso J, Lee MK, Hattori N, Mizuno Y, Dawson TM, Dawson VL. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum Mol Genet. 2005;14:71–84. doi: 10.1093/hmg/ddi007. [DOI] [PubMed] [Google Scholar]
- 19.Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 20.Vattemi G, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol. 2004;164:1–7. doi: 10.1016/S0002-9440(10)63089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fratta P, Engel WK, McFerrin J, Davies KJ, Lin SW, Askanas V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am J Pathol. 2005;167:517–526. doi: 10.1016/s0002-9440(10)62994-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nogalska A, Engel WK, McFerrin J, Kokame K, Komano H, Askanas V. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J Neurochem. 2006;96:1491–1499. doi: 10.1111/j.1471-4159.2006.03668.x. [DOI] [PubMed] [Google Scholar]
- 23.Askanas V, Engel WK, Alvarez RB, McFerrin J, Broccolini A. Novel immunolocalization of alpha-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J Neuropathol Exp Neurol. 2000;59:592–598. doi: 10.1093/jnen/59.7.592. [DOI] [PubMed] [Google Scholar]
- 24.Gandhi S, Muqit MM, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, Latchman DS, Holton JL, Wood NW, Revesz T. PINK1 protein in normal human brain and Parkinson's disease. Brain. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- 25.Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A. DJ-1 is a redox dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004;2:1764–1773. doi: 10.1371/journal.pbio.0020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–218. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 27.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding Q, Dimayuga E, Keller JN. Oxidative damage, protein synthesis, and protein degradation in Alzheimer's disease. Curr Alzheimer Res. 2007;4:73–79. doi: 10.2174/156720507779939788. [DOI] [PubMed] [Google Scholar]
- 29.Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci. 2007;64:2202–2210. doi: 10.1007/s00018-007-7218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang CC, Alvarez RB, Engel WK, Askanas V. Increase of nitric oxide synthases and nitrotyrosine in inclusion-body myositis. Neuroreport. 1996;8:153–158. doi: 10.1097/00001756-199612200-00031. [DOI] [PubMed] [Google Scholar]
- 31.Broccolini A, Engel WK, Alvarez RB, Askanas V. Redox factor-1 in muscle biopsies of patients with inclusion-body myositis. Neurosc Lett. 2000;287:1–4. doi: 10.1016/s0304-3940(00)01156-3. [DOI] [PubMed] [Google Scholar]
- 32.Askanas V, Sarkozi E, Alvarez RB, McFerrin J, Engel WK. Superoxide Dismutase-1 gene and protein in vacuolated muscle fibers of Sporadic Inclusion Body Myositis, Hereditary Inclusion Body Myopathy, and Cultured Human Muscle after β-Amyloid Precursor Protein gene transfer. Neurology. 1996;46:A487. [Google Scholar]
- 33.Broccolini A, Mirault ME, Engel WK, Askanas V. Abnormal accumulation of Seleno-Glutathione Peroxidase-1 and Catalase and their m-RNAs in Sporadic Inclusion Body Myositis. Neurology. 1999;52:A333. [Google Scholar]
- 34.Yang CC, Askanas V, Engel WK, Alvarez RB. Immunolocalization of transcription factor NF-kappaB in inclusion-body myositis muscle and at normal human neuromuscular junctions. Neurosci. Lett. 1998;254:77–80. doi: 10.1016/s0304-3940(98)00657-0. [DOI] [PubMed] [Google Scholar]
- 35.Nogalska A, Wojcik S, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress induce myostatin precursor protein and NF-kappaB in cultured human muscle fibers: relevance to inclusion body myositis. Exp. Neurol. 2007;204:610–618. doi: 10.1016/j.expneurol.2006.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tateyama M, Takeda A, Onodera Y, Matsuzaki M, Hasegawa T, Nunomura A, Hirai K, Perry G, Smith MA, Itoyama Y. Oxidative stress and predominant Abeta42(43) deposition in myopathies with rimmed vacuoles. Acta Neuropathol. 2003;105:581–585. doi: 10.1007/s00401-003-0685-2. [DOI] [PubMed] [Google Scholar]
- 37.Li HM, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic Res. 39:1091–1099. doi: 10.1080/10715760500260348. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto M, Rockenstein E, Crews L, Masliah E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer's and Parkinson's diseases. Neuromolecular Med. 2003;4:21–36. doi: 10.1385/NMM:4:1-2:21. [DOI] [PubMed] [Google Scholar]
- 39.Schapira AHV. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 40.Santorelli FM, Sciacco M, Tanji K, Shanske S, Vu TH, Golzi V, Griggs RC, Mendell JR, Hays AP, Bertorini TE, Pestronk A, Bonilla E, DiMauro S. Multiple mitochondrial DNA deletions in sporadic inclusion body myositis: a study of 56 patients. Ann Neurol. 1996;39:789–795. doi: 10.1002/ana.410390615. [DOI] [PubMed] [Google Scholar]
- 41.Engel WK. “Ragged-red fibers” in ophthalmoplegia syndromes and their differential diagnosis (abstract). Abstracts 2nd Intl Cong Muscle Diseases, Perth, Australia. Excerpta Med Inter Cong Series. 1971;237:28. [Google Scholar]
- 42.Hong WK, Han EH, Kim DG, Ahn JY, Park JS, Han BG. Amyloid-beta peptide reduces the expression level of mitochondrial cytochrome oxidase subunits. Neurochem Res. 2007;32:1483–1488. doi: 10.1007/s11064-007-9336-7. [DOI] [PubMed] [Google Scholar]
- 43.Askanas V, McFerrin J, Baqué S, Alvarez RB, Sarkozi E, Engel WK. Transfer of beta-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc Natl Acad Sci U S A. 1996;93:1314–1319. doi: 10.1073/pnas.93.3.1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]