

Abstract
Motivation: Gene-set enrichment analysis (GSEA) can be greatly enhanced by linear model (regression) diagnostic techniques. Diagnostics can be used to identify outlying or influential samples, and also to evaluate model fit and explore model expansion.
Results: We demonstrate this methodology on an adult acute lymphoblastic leukemia (ALL) dataset, using GSEA based on chromosome-band mapping of genes. Individual residuals, grouped or aggregated by chromosomal loci, indicate problematic samples and potential data-entry errors, and help identify hyperdiploidy as a factor playing a key role in expression for this dataset. Subsequent analysis pinpoints suspected DNA copy number abnormalities of specific samples and chromosomes (most prevalent are chromosomes X, 21 and 14), and also reveals significant expression differences between the hyperdiploid and diploid groups on other chromosomes (most prominently 19, 22, 3 and 13)—differences which are apparently not associated with copy number.
Availability: Software for the statistical tools demonstrated in this article is available as Bioconductor package GSEAlm.
Contact: assaf.oron@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Gene set enrichment analysis (GSEA, Mootha et al., 2003; Subramanian et al., 2005) is an important new approach to the analysis of gene expression data, and it has already been extended and generalized in a number of ways (Hummel et al., 2008; Jiang and Gentleman, 2007; Kim and Volsky, 2005; Tian et al., 2005). Expression analysis in general and GSEA in particular can be viewed as a cascade of successive data reductions: first, biochemical hybridization information is reduced to a set of pixel images (typically one or two per sample). Second, the images are preprocessed to produce probe-level summaries, which are then further summarized to a G × n matrix of normalized average expression estimates (G genes, n samples). This matrix is then filtered to remove redundant probesets and genes identified as unexpressed or otherwise uninformative (non-specific filtering). Next, dataset-level differential expression statistics are calculated for each gene. Finally, these statistics are used to calculate gene-set (GS) level statistics, which help identify differentially expressed or otherwise interesting GSs. This data-reduction process is essential. It helps bring the amount of information generated by the microarray experiment down to a manageable level, while retaining its core features. However, the quality of such massive data reduction can and should be monitored. Monitoring the last stages of this process is where linear model tools may prove beneficial.
Several studies (e.g. Goeman et al., 2004; Hummel et al., 2008; Jiang and Gentleman, 2007; Kim and Volsky, 2005; Kong et al., 2006) have demonstrated the potential of using a linear model (regression) framework for GSEA. In particular, with linear models one can adjust for important explanatory covariates, such as sex and estrogen receptor (ER) status for breast cancer. These studies focus mostly on the use of linear models to evaluate covariate effects upon GS expression, averaged over the relevant genes and samples. This averaging aspect of linear models is complemented by diagnostics, in particular residuals—which examine the model's adequacy in describing the original data patterns, and also individual deviations from the average effects. In this article, we demonstrate the application of linear model diagnostics to GSEA.
2 METHODS
2.1 Linear models and diagnostics
A linear (regression) model assumes that the mean of the response variable has a linear relationship with the explanatory covariate(s). In gene expression terminology, a simple generic model could be written as:
| (1) |
where
ygi, g=1, …, G, i=1, …, n is the gene expression value of gene g in sample i;
p is the number of explanatory covariates in the model;
Xij is the value of the j-th covariate for the i-th sample. For dichotomous covariates, such as phenotype, one typically sets X to zero or one (e.g. NEG will be zero and BCR/ABL one);
βgj is the magnitude of the effect of covariate j upon the expression of gene g (βg0 is the intercept, or baseline expression for gene g); and
ɛgi is a random error (‘noise’), here assumed to follow a Normal distribution with mean zero and variance σg2.
The data and model are used to calculate a fitted value for each observation, denoted as 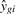 , an estimate for each effect's magnitude, denoted as
, an estimate for each effect's magnitude, denoted as 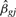 , and a t-statistic for each covariate quantifying the strength of evidence for its effect, denoted as tgj. Applying linear models to gene expression in the way outlined here, involves fitting the same model form to all genes independently and simultaneously [a more general formulation allowing for explicit gene–gene dependence can be found in Hummel et al. (2008)]. Note also that a simple gene-by-gene two-sample t-test is identical to a linear model with p = 1 and with the sole covariate taking on only two values: zero or one.
, and a t-statistic for each covariate quantifying the strength of evidence for its effect, denoted as tgj. Applying linear models to gene expression in the way outlined here, involves fitting the same model form to all genes independently and simultaneously [a more general formulation allowing for explicit gene–gene dependence can be found in Hummel et al. (2008)]. Note also that a simple gene-by-gene two-sample t-test is identical to a linear model with p = 1 and with the sole covariate taking on only two values: zero or one.
The regression residuals, 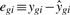 , are used to estimate the residual standard error needed for inference on model effects—but they also play a key role in diagnostics. While model estimates summarize the information about mean tendencies, residuals convey information about deviations or discrepancies from these tendencies. Residuals can help to identify outlying observations, examine model assumptions and evaluate whether there are missing terms in the model (Neter et al., 1996). For example, outlying residuals indicate suspect observations that need to be more carefully inspected and accounted for. Grouping of residuals, or a trend in residuals as a function of fitted values, may indicate a poor model fit, which may be improved by adding terms to the model or by modifying its assumptions. In the gene expression case, where we run a large number of identical models in parallel, we will show that residuals can also be used to identify genes or samples with discrepant or unusual residual patterns across the entire dataset.
, are used to estimate the residual standard error needed for inference on model effects—but they also play a key role in diagnostics. While model estimates summarize the information about mean tendencies, residuals convey information about deviations or discrepancies from these tendencies. Residuals can help to identify outlying observations, examine model assumptions and evaluate whether there are missing terms in the model (Neter et al., 1996). For example, outlying residuals indicate suspect observations that need to be more carefully inspected and accounted for. Grouping of residuals, or a trend in residuals as a function of fitted values, may indicate a poor model fit, which may be improved by adding terms to the model or by modifying its assumptions. In the gene expression case, where we run a large number of identical models in parallel, we will show that residuals can also be used to identify genes or samples with discrepant or unusual residual patterns across the entire dataset.
There also exist diagnostic tools designed to test a single observation's impact, or influence upon the calculated mean tendencies. Typically, to be influential an observation has to display some combination of a large residual and off-center or rare Xij (i.e. covariate) values. One of these measures is Cook's D (Cook and Weisberg, 1982), representing the squared distance by which the observation in question ‘moves’ the fitted model's parameter estimates. This distance is measured in p-dimensional parameter space and normalized by the standard error of parameter estimates.1
2.2 GSEA and diagnostics in a linear model framework
GSEA involves using the gene-level statistics (usually, t-statistics) to produce summary statistics for each GS. As mentioned above, there already exist several ways to achieve this. Here, we choose the statistic of (Jiang and Gentleman, 2007) (hereafter, ‘the J–G statistic’), as it enables the easy implementation of diagnostic analysis.
The J–G statistic for a GS indexed k can be defined as
| (2) |
where tg is the regression t-statistic for the effect of our covariate of interest upon gene g expression, and |Sk| is the size of GS Sk. Under independence between genes and under the null hypothesis that GS Sk's expression is not affected by the covariate in question, τk→N(0,1) as |Sk|→∞. However, in microarray experiments where all genes in a given sample come from the same organism, we expect their expression levels to be correlated. Even mild gene–gene correlations can induce a size effect on τk; methods to account for these correlations are a subject of ongoing research (Efron, 2007; Hummel et al., 2008). Here, we address correlations by calculating GS P-values via sample (‘column’) label permutations rather than by comparing τk to standard Normal or t-distributions. Thus, a GS would be considered interesting vis-a-vis a specific covariate, if its J–G statistic for this covariate is very extreme, compared with a large ensemble of analogous statistics calculated on the same dataset via the same linear model, but with sample labels repeatedly scrambled (see e.g. Ernst, 2004). The use of permutation tests also relieves us of the need to make sure τk's behavior is close enough to Normal, and thus we can examine relatively small GSs.
Just as we aggregate gene-level t-statistics to calculate the GS effect statistic τk, we can aggregate gene-level residuals to calculate GS-level residuals. When aggregating residuals from different regression models fitted in parallel, the residuals should first be normalized to prevent some genes from dominating the rest. There exist several normalization approaches (Cook and Weisberg, 1982; see Supplementary Material A). In this article, we mainly use externally-Studentized residuals, which (if model assumptions hold) are t-distributed with n−p−2 degrees of freedom. The resulting formula for normalized, aggregated GS residuals is
| (3) |
where rgi is the normalized residual from sample i and gene g. Note that we have n GS residuals per GS. GS residuals can be used in the same manner as an individual gene residuals, with the advantage of being averages: if a sample or group of samples does not really deviate in its expression for a given GS, then we expect its GS residuals to roughly average out—even if some individual gene residuals may be large. When this does not happen, we have evidence that expression patterns of the sample in question are poorly explained by the model. Similarly, we can also identify discrepant GSs via their GS residual patterns.
Finally, we can also aggregate Cook's D values within a GS. Since Cook's D is not symmetric around zero, the aggregation takes a somewhat different form:
| (4) |
Δki, the GS root-mean Cook's D, provides a measure of the typical amount by which the sample in question affects t-statistics for genes in the GS.
2.3 Chromosomal loci as GSs
A hallmark of most cancers is gene disregulation, which is often associated with certain chromosomal loci, due to deletion, amplification or epigenetic events (Pollack et al., 2002). For that reason, examining gene loci for evidence of disregulation is of potential benefit. One can attempt to model disregulation as a function of continuous chromosomal coordinates (Nilsson et al., 2008), or use the more traditional, hierarchical structure of chromosome bands and sub-bands. We chose the latter, being compatible with GSEA methodology. Chromosomal loci (chromosomes, bands, sub-bands, etc.) are modeled as GSs. This GS structure forms a tree graph: the trunk is the organism, the first branches are complete chromosomes, and so forth—down to the lowest resolution sub-bands, which are known in graph theory as the tree's leaves. We impose a cutoff of at least five genes, for a chromosome sub-band to be included as a GS in our analysis.
2.4 Dataset: acute lymphoblastic Leukemia
We demonstrate the use of diagnostics on an adult acute lymphoblastic leukemia (ALL) clinical trial dataset (Chiaretti et al., 2004, hereafter: ‘the ALL dataset’). It contains 128 samples, each hybridized to an Affymetrix HGU95-Av2 chip containing probes associated with 12 625 genes. One question of interest is finding chromosomal locations with differential expression between the B-cell BCR/ABL and NEG phenotypes of the disease. Non-specific filtering was performed (Jiang and Gentleman, 2007), and multiple probes targeting the same gene were filtered out as well. The filtered dataset contains 79 samples and 4502 unique genes. We mapped the chromosomal location of these genes, using tools available in R package Category. In the filtered dataset, 4495 genes mapped to 524 chromosome bands or sub-bands containing at least five genes each. This mapped subset of genes was used for the analysis described below.
3 IMPLEMENTATION ON THE ‘ALL’ DATASET
3.1 GSEA for the phenotype effect only
3.1.1 Simple diagnostics
We fitted the expression data of each gene to the generic model (1) with a single covariate denoting phenotype (BCR/ABL or NEG). Before continuing to the next GSEA step—calculating GS statistics—we pause and examine residuals at the individual gene level.
Figure 1 summarizes all externally Studentized residuals by sample arranged by phenotype. Even though there is no single overwhelmingly outlying sample, several samples do catch the eye. For example, residuals from samples 28001 and 68001 (NEG phenotype, top left) are predominantly negative, and also exhibit relatively high variability. Residuals from sample 84004 display high variability combined with a positive tendency (BCR/ABL phenotype, bottom right). If a sample's expression levels are systematically higher or lower across the board, it is impossible to tell whether this is due to real biological differences or due to a normalization offset; we suspect that the latter case is more common. It is interesting to note that the dataset had already been normalized during preprocessing with all 12 625 features present. Apparently, the 4495 features shown on Figure 1 are different enough from the rest to somewhat disrupt the early normalization. Moreover, removal of the average per-gene baseline via regression, and Studentization of the residuals, seem to improve our sensitivity to normalization offsets. In any case, whether corrective normalization action is warranted—and also whether a phenotype-only model fits the data well—becomes clearer upon observing GS-level residuals.
Fig. 1.

Raw externally Studentized residuals from the linear model of gene expression on phenotype for the ALL dataset, grouped by sample, arranged by phenotype (NEG on left, BCR/ABL on right) and sorted by each sample's median residual.
3.1.2 GSEA diagnostics
Figure 2 displays a heatmap of GS residuals, with chromosome bands in rows and samples in columns. Red indicates positive values and blue negative values. In order to avoid overlaps, only the leaves of the chromosome-location tree are shown. Both rows and columns are simultaneously re-ordered according to correlation, allowing us to detect patterns deviating from model fit—whether they occur by sample or by GS.
Fig. 2.

GS residuals from the linear model of gene expression on phenotype, for each lowest level chromosome band (row) and sample (column). Residuals in each row were standardized to have mean 0 and SD 1. Heatmap colors change in increments of 0.8 (on the normalized scale), with reds positive and blues negative. The horizonal band at the top indicates the value of the kinet variable: red for hyperdiploid, gray for diploid and white for unknown.
One of the samples identified above as having low residuals, 28001, is visible as a narrow predominantly blue vertical strip (Fig. 2, somewhat right of center). This indicates no association between chromosomal loci and low expression levels for this sample; unless we realign expression levels on the filtered dataset (most simply by removal of sample-specific medians), sample 28001—and quite possibly others with smaller offsets—are likely to appear as outliers during more detailed analysis. More interesting from a modeling perspective is the apparent block or checkerboard pattern of the heatmap. This pattern indicates a potential association between groups of samples and overall expression levels at certain chromosomal locations; an association not explained by the phenotype-only model. In particular, there is a relatively tight cluster of 20 samples (left-hand side of map), whose expression pattern is roughly the opposite of most other samples. Among the dataset's 21 descriptive variables, we identified the ‘kinet’ variable to be most strongly associated with the pattern-induced grouping of samples (χ2 P-value conditional on the clustering: <0.001). This variable indicates whether the sample is classified as hyperdiploid. The association between hyperdiploidy and gene expression of chromosomal loci or complete chromosomes among pediatric ALL patients, has been well documented in research (Ross et al., 2003; Teixeira and Heim, 2005), and we can plausibly assume it holds for adult patients as well. The kinet variable is illustrated as a colored band at the top of Figure 2, with red indicating hyperdiploid samples, gray diploid samples and white samples of unknown status. Even though only 19 of 79 samples are hyperdiploid, they form a clear majority in the 20-sample cluster described above, and are further differentiated from diploid samples within that cluster as well. We concluded that it may be useful to add kinet to the model.
Another variable that is known with certainty to be associated with chromosome-level expression differences is sex. Females do not have the Y chromosome, and therefore observed expression differences for non-autosomal Y chromosome genes can serve several functions at once: a test of microarray technology, a test of GSEA methodology and a test for data-entry errors. Since the Y chromosome has relatively few genes, it is represented in Figure 2 by two rows only, making its effect hard to detect at this level. A direct inspection of GS residuals, with the GS defined as the 11 non-autosomal Y chromosome genes in our filtered dataset, reveals the expected strong sex-related pattern—albeit with some noise (Fig. 3). In fact, several samples’ GS residuals deviate from their sex baseline so strongly towards the other sex, as to suggest a possible sex mis-assignment in the dataset annotation. A more careful analysis led us to conclude beyond reasonable doubt, that two females have been mis-assigned as males. Additionally, up to three males have apparently been mis-assigned in the opposite direction, though the evidence is somewhat weaker.2 For subsequent analysis in this article, we have reassigned two samples to female and one sample to male. An additional sample with a missing sex entry was identified as male by its Y chromosome expression patterns.
Fig. 3.

Boxplot of GS residuals, calculated on the set of non-autosomal genes on the Y chromosome, obtained from a linear model with only phenotype as predictor and grouped by sex.
3.2 GSEA using the expanded model
3.2.1 Chromosome-level patterns
The GSEA procedure was repeated with the changes indicated above—adding sex and hyperdiploidy to the model, relabeling the sex entries of three samples and recentering each sample's expression values by its median to diminish the impact of outlying samples. Four samples with missing data for hyperdiploidy were dropped from the analysis, leaving us with n=75. It is of interest to compare the evaluation of the phenotype effect before and after model expansion. There are minor changes: the correlation between phenotype-effect t-statistics generated by the two models is 0.99. We performed GS-level inference to see if the minor variations between the two models are localized to certain GSs. Inference was obtained via sample-label permutation as explained above. For the expanded model, care must be taken to permute sample labels only within groups that have the same sex and hyperdiploidy status. The test was performed only for the leaves of the chromosomal-loci tree, using 5000 permutations. Overall, the 3-covariate model's inference is somewhat more conservative, and less tilted towards over-expressed bands. However, there is substantial agreement between the significant chromosomal-loci lists generated via the two models.3
At the other end of the chromosomal-loci hierarchy, Figure 4 shows complete-chromosome mean expression trends calculated using the 3-covariate model. Even for normal samples (black line) there are marked inter-chromosome differences, as is known from literature (Caron et al., 2001). BCR/ABL's trend (red dots) is almost indistinguishable from the normal group, with the biggest gap observed at chromosome 22, which is directly affected by that phenotype's anomaly. The hyperdiploid trend (blue dashes), though following the normal group's general trend, exhibits much larger deviations from it—with chromosomes 19, 21, 22 and X most strongly over-expressed and chromosomes 3 and 13 most strongly under-expressed. All these effects are statistically significant at the 0.05 false discovery rate (FDR) level (Benjamini and Hochberg, 1995; Benjamini and Yekutieli, 2001). The sex covariate (trend not shown) has negligible effect, except for the Y chromosome.
Fig. 4.

Complete-chromosome mean expression levels relative to the median gene, as found by the 3-covariate model. Shown are the baseline mean (NEG-diploid, black and ‘+’ signs), the BCR/ABL-diploid mean (red, dots and ‘B’) and the NEG-hyperdiploid mean (blue, dashes and ‘H’). All estimates are for male samples; the female-sample pattern was nearly identical, except for the Y chromosome.
These hyperdiploidy-related differences raise the question whether they are the result of individual hyperdiploid samples exhibiting aneuploidy while others have normal expression levels, or of a subtle expression shift across the entire hyperdiploid group. In the former case, chromosome-level GS residuals of samples with abnormal DNA copy number should be flagged as gross outliers. Figure 5 displays a map of these outliers, using GS residuals from an intercept-only model. Outliers were identified via standard robust location and scale methods (Huber, 1981), using a numerically generated outlier-free reference distribution (Wisnowski et al., 2001), and FDR thresholds of 0.05, 0.1 and 0.2. We imposed the additional constraint that the sample's average expression for the chromosome in question must differ from the median of all samples by a relative amount of at least 1:6 [similarly to Hertzberg et al.'s, (2007) approach which was tested against verified aneuploidies].4
Fig. 5.

Map of suspected aneuploidies in the ALL dataset, by chromosome (rows) and sample (columns). Red–brown hues correspond to extra copies, and blue hues to missing copies. Dark, medium and light shades correspond to FDR levels of 0.05, 0.1 and 0.2, respectively. The top bar indicates hyperdiploidy, as in Figure 2. Samples and chromosomes with no flags have been omitted.
Most hyperdiploid samples, and about a dozen diploid samples, are flagged for at least one aneuploidy. Observing Figure 5 from the perspective of chromosomes, chromosome X is by far the most prevalent, with 12 samples flagged as potential multisomies at the 0.2 FDR level. The next most prevalent multisomies are of chromosomes 21 and 14, respectively. Equally interesting are some chromosomes absent from Figure 5, because they have no flagged samples. These include chromosomes 19 and 22, identified in Figure 4 as over-expressed by the hyperdiploid group, and chromosomes 3 and 13, identified as under-expressed. Sample-level inspection reveals that these chromosomes are mildly over- or under-expressed across the board, i.e. the second of the two potential explanations suggested above seems to hold for them.
3.2.2 Influence analysis
Beside identifying outliers, res-earchers may need to answer the practical question: how strongly does a specific outlying sample affect model estimates? This is where Cook's D, mentioned above, can be useful. For the phenotype-only model, which splits the dataset into two roughly equal-sized groups of 42 and 37, no sample is influential enough to cause concern—not even 28001. The story is somewhat different under the 3-covariate model, where both the female and hyperdiploid groups are much smaller. Figure 6 summarizes all Δki values for lowest level chromosome bands, by sample. Two samples belonging to hyperdiploid female subjects (far right) have much larger overall influence than most other samples. However, even they are not dominant to the point of questioning the validity of hyperdiploidy or sex effect inference.
Fig. 6.

Chromosome-band root-mean Cook's D values, summarized by sample, for the 3-covariate model. Samples are ordered by mean.
4 DISCUSSION
Diagnostics, an indispensable and versatile component of regression analysis, are especially useful for finding unexpected data patterns. On the single dataset used here for demonstration, diagnostics have helped us recognize the need to realign expression values; decide whether the sex covariate has been entered in error for certain samples; explore model expansion and pinpoint suspected individual aneuploidies.5 Some of the uses of diagnostics can be formalized and even automated (see Supplementary Materials B and D); others, such as recognizing that there may be a Y-chromosome problem or interpreting Figure 2, are more exploratory and intuition driven.
Software tools used to produce the analysis reported here are publicly available as Bioconductor package GSEAlm.6 Researchers wishing to perform the main regression analysis using a package of their choice, can still take advantage of GSEAlm's diagnostic features by extracting residuals using lmPerGene followed by getResidPerGene. Detailed information appears in the package's vignette and manual pages. The ALL dataset is available as Bioconductor package ALL.
Funding
United States National Institutes of Health [grant numbers NHGRI-1-P41-HG004059, P50-CA-083636 (Ovarian SPORE)].
Conflict of Interest: none declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank S. Chiaretti and J. Ritz for making the ALL dataset available, and C. Lottaz for a summarized and preprocessed version of the St Jude dataset used in Supplementary Material D. We also thank the anonymous referees for a timely review that helped improve this article.
Footnotes
1More detailed information on residuals and influence measures is available in Supplementary Material A.
2Details can be found in Supplementary Material B.
3See Supplementary Material C for a significant loci list according to the 3-covariate model.
4More details can be found in Supplementary Material D.
5Regarding aneuploidies, following a referee's suggestion we applied our residuals method to the St Jude pediatric ALL dataset (Ross et al., 2003), on which Hertzberg et al.'s (2007) expression-based aneuploidy detection method was optimized. For that dataset, cytogenetic information on chromosome 21 is available. Our method, lifted ‘as is’ from the ALL dataset and applied to the St Jude dataset with no further optimization, exhibits somewhat weaker sensitivity but somewhat better specificity than (Hertzberg et al.'s, 2007). More details can be found in Supplementary Material D.
6Included in this package is a function to test a single covariate's effect at the GS level, while adjusting for other covariates (gsealmPerm). Package GlobalAncova offers a wider variety of such tests; that package uses the F-test, while gsealmPerm uses the permutation analogue to the t or Wald test.
REFERENCES
- Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001;29:1165–1188. [Google Scholar]
- Caron H, et al. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- Chiaretti S, et al. Gene expression profile of adult T-cell acute lymphocytic leukemia identifies distinct subsets of patients with different response to therapy and survival. Blood. 2004;103:2771–2778. doi: 10.1182/blood-2003-09-3243. [DOI] [PubMed] [Google Scholar]
- Cook R, Weisberg S. Monographs on Statistics and Applied Probability. New York: Chapman and Hall; 1982. Residuals and Influence in Regression. [Google Scholar]
- Efron B. Correlation and large-scale simultaneous significance testing. J. Am. Stat. Assoc. 2007;102:93–103. [Google Scholar]
- Ernst M. Permutation methods: a basis for exact inference. Stat. Sci. 2004;19:686–696. [Google Scholar]
- Goeman J, et al. A global test for groups of genes: testing association with a clinical outcome. Bioinformatics. 2004;20:93–99. doi: 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- Hertzberg L, et al. Prediction of chromosomal aneuploidy from gene expression data. Genes Chromosome Cancer. 2007;46:75–86. doi: 10.1002/gcc.20391. [DOI] [PubMed] [Google Scholar]
- Huber PJ. Wiley Series in Probability and Mathematical Statistics. New York: John Wiley & Sons Inc.; 1981. Robust statistics. [Google Scholar]
- Hummel M, et al. GlobalANCOVA: exploration and assessment of gene group effects. Bioinformatics. 2008;24:78–85. doi: 10.1093/bioinformatics/btm531. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Gentleman R. Extensions to gene set enrichment analysis. Bioinformatics. 2007;23:306–313. doi: 10.1093/bioinformatics/btl599. [DOI] [PubMed] [Google Scholar]
- Kim S.-Y, Volsky D. Page: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong S, et al. A multivariate approach for integrating genome-wide expression data and biological knowledge. Bioinformatics. 2006;22:2373–2380. doi: 10.1093/bioinformatics/btl401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Neter J, et al. Applied Linear Statistical Models. Boston: McGraw-Hill Companies, Inc.; 1996. [Google Scholar]
- Nilsson B, et al. An improved method for detecting and delineating genomic regions with altered gene expression in cancer. Genome Biol. 2008;9:R13. doi: 10.1186/gb-2008-9-1-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack J, et al. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl Acad. Sci. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross M, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102:2951–2959. doi: 10.1182/blood-2003-01-0338. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira M, Heim S. Multiple numerical chromosome aberrations in cancer: what are their causes and what are their consequences? Sem. Canc. Biol. 2005;15:3–12. doi: 10.1016/j.semcancer.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Tian L, et al. Discovering statistically significant pathways in expression profiling studies. Proc. Natl Acad. Sci. 2005;102:13544–13549. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnowski J, et al. A comparative analysis of multiple outlier detection procedures in the linear regression model. Comp. Stat. Data Anal. 2001;36:351–382. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.