

Summary
Contact-dependent growth inhibition (CDI) is a phenomenon by which bacterial cell growth is regulated by direct cell-to-cell contact via the CdiA/CdiB two-partner secretion system. Characterization of mutants resistant to CDI allowed us to identify BamA (YaeT) as the outer membrane receptor for CDI and AcrB as a potential downstream target. Notably, both BamA and AcrB are part of distinct multi-component machines. The Bam machine assembles outer membrane β-barrel proteins (OMPs) into the outer membrane and the Acr machine exports small molecules into the extracellular milieu. We discovered that a mutation that reduces expression of BamA decreased binding of CDI+ inhibitor cells, measured by flow cytometry with fluorescently-labeled bacteria. In addition, α-BamA antibodies, which recognized extracellular epitopes of BamA based on immunofluorescence, specifically blocked inhibitor-target cell binding and CDI. A second class of CDI-resistant mutants identified carried null mutations in the acrB gene. AcrB is an inner membrane component of a multidrug efflux pump that normally forms a cell envelope-spanning complex with the membrane-fusion protein AcrA and the outer membrane protein TolC. Strikingly, the requirement for the BamA and AcrB proteins in CDI is independent of their multi-component machines, and thus their role in the CDI pathway may reflect novel, import-related functions.
Introduction
Bacteria have developed sophisticated and highly efficient mechanisms to communicate with each other from a distance using diffusible chemical signals (Bassler et al., 1993; Bassler, 1999; Ji et al., 1997; Tomasz, 1965). Bacteria use these signals to coordinate cellular processes including uptake of DNA, cannibalism during spore formation, and toxin secretion (Bassler and Losick, 2006). In some cases communication occurs more intimately by direct cell-to-cell contact. In Myxococcus xanthus, a 17 kDa protein known as C-signal is required for coordinated cellular movement occurring during fruiting body formation. C-signal transmission appears to occur via end-to-end contact at the cell poles, although the C-signal receptor has not been identified (Kaiser, 2006). Recently, we identified a different type of cellular communication in Escherichia coli denoted as contact-dependent growth inhibition or CDI (Aoki et al., 2005). Like C-signaling in M. xanthus, CDI requires direct cell-to-cell contact between CDI+ inhibitor cells and CDI− target cells, but instead of coordinating motility, CDI regulates cell growth (Aoki et al., 2005).
CDI is dependent upon the growth state of inhibitory cells: logarithmic phase but not stationary phase CDI+ cells are growth inhibitory. In contrast, target cells are inhibited regardless of growth state (Aoki et al., 2005). In addition, CDI+ cells must synthesize proteins for growth inhibitory activity since CDI+ cells treated with chloramphenicol lose their growth inhibitory phenotype (Aoki et al., 2005). Binding between CDI+ inhibitory cells expressing green fluorescent protein (GFP) and target cells expressing Discosoma Red (DsRed) can be measured by fluorescence-activated cell sorting (FACS). However, the binding that occurs between cells appears to be transient (under conditions of rapid mixing), since one CDI+ inhibitor cell inhibits greater than 100 target cells over the period of two hours (Aoki et al., 2005). Together, these results indicate that CDI is a dynamic process involving the attachment of CDI+ cells to target cells, presumably via a cell surface receptor, and active protein synthesis occurring in the inhibitory cell, blocking growth of target cells by an unknown mechanism.
CDI requires two proteins, CdiB and CdiA, which are members of the TpsB and TpsA two-partner secretion (TPS) group of proteins (Mazar and Cotter, 2007; Tommassen, 2007). TpsB proteins form β-barrel channels in the outer membrane, facilitating transport of their TpsA counterpart through the outer membrane. Recently the structure of the TpsB protein FhaC from Bordetella pertussis was solved (Clantin et al., 2007). Two periplasmically located polypeptide-transport-associated domains (POTRA) (Sanchez-Pulido et al., 2003) located at the amino-terminus were shown to be important for secretion of filamentous hemagglutinin (FHA), its TpsA counterpart (Clantin et al., 2007). FHA binds to POTRA-1 via its amino-terminus and initiates secretion through the β-barrel channel of FhaC, possibly in an extended conformation facilitated by a chaperone function of the POTRA domain (Bos et al., 2007). Folding of FHA likely occurs at the cell surface after transit through the FhaC pore. Cell-surface FHA has adhesin activity, mediating binding of B. pertussis to eukaryotic target cells which plays an important role in the pathogenesis of whooping cough (Jacob-Dubuisson et al., 2004). FHA is also released into the medium, but the possible function(s) of unbound FHA is not clear (Mazar and Cotter, 2006). Notably, CdiB shares significant homology with FhaC, including conserved glycine residues within the POTRA regions that form a “POTRA signature” (see Fig. S1) (Clantin et al., 2007). Based on this homology it seems likely that CdiB is an outer membrane protein that facilitates export and assembly of CdiA.
CdiA shares a number of features with FHA. CdiA contains an N-terminal region homologous to FHA including the TPS domain required for interaction with the TpsB partner, CdiB and FhaC, respectively (note that FhaC was named prior to the TpsA/TpsB classification scheme). Like FHA, CdiA is both cell surface-associated and released into the extracellular milieu (Aoki et al., 2005). FHA is derived from processing of the FhaB precursor, which requires protease SphB1 (Coutte et al., 2003), generating mature FHA lacking the C-terminal one-third of the FhaB precursor. Release of FHA from the cell surface occurs in the absence of SphB1, however (Mazar and Cotter, 2006). Recent results indicate that the C-terminus of FHA is exposed on the cell surface, where it mediates adherence to epithelial cells (Mazar and Cotter, 2007). Analysis of CdiA containing different FLAG epitopes indicated that proteolytic cleavage occurs in at least three sites: one near the N-terminus, likely the signal peptide cleavage site, and two within the C-terminal portion of CdiA (see Fig. S1). Based on the sizes of CdiA peptides and ability to detect different FLAG epitopes at the cell surface, it appears that the mature form of CdiA at the cell surface lacks the C-terminal one-third of the protein, analogous to FHA, although it is not known if the C-terminus of CdiA is oriented outward. (Aoki et al., 2005).
Because CDI+ E. coli bind to target cells and growth inhibition requires cell-to-cell contact, it seems likely that CdiA, which localizes to the cell surface, binds to a cell surface receptor on target bacterial cells. Furthermore, it seems likely that once properly directed to a target cell, the CDI pathway must somehow interfere with an essential cellular process of the target cell in order to elicit growth inhibition. To probe the mechanisms of CDI-mediated growth inhibition, we took a genetic approach, isolating E. coli target cell mutants that are resistant to CDI-mediated growth inhibition. This genetic analysis identified an essential outer membrane protein, BamA (formerly known as YaeT but renamed as β-barrel assembly machine protein A, see http://ecocyc.org), which we demonstrate is the CDI receptor, and the inner membrane transport protein AcrB, which may be the target or the inner membrane portal for CDI-mediated growth inhibition.
Results
Genetic identification of AcrB and BamA in the CDI pathway
Previously we identified two open reading frames (ORFs), cdiA and cdiB, derived from E. coli strain EC93 that are required for CDI activity, and a third ORF, cdiI, that is necessary and sufficient to confer immunity to growth inhibition (Aoki et al., 2005) (Fig. S1). Because cell-to-cell contact is required for growth inhibition (Aoki et al., 2005), it seemed likely that CDI+ cells engage a cell surface receptor(s) on target cells. In addition CdiA must attack a cellular target, which may or may not be the receptor to elicit growth inhibition. We took a genetic approach to identify gene products in the target cell that are required for CDI-mediated growth inhibition by selecting for resistant mutants.
We mutagenized target E. coli EPI100 cells with a Tn5-based transposon EZ-Tn5<KAN-2> (Epicentre) to isolate CDI-resistant mutants (CDIR). Pools of CDIR mutants were selected after three rounds of challenge with CDI+ inhibitor cells (DH5α carrying the cdiB+A+I+ cosmid clone pDAL660Δ1–39, Fig S1). P1 lysates were prepared on each of the mutants obtained and these were used to transduce the transposon insertion from each mutant into MC4100 to verify linkage between the transposon insertion and CDI resistance.
The first round of mutagenesis yielded ten CDIR mutants that had insertions within the acrB gene coding for the inner membrane multidrug transport protein AcrB (Fig. 1A). Our acrB insertion mutations, exemplified by acrB11 and acrB18, as well as a known non-polar acrB deletion mutation conferred strong CDIR phenotypes (Fig. 1B). As expected, the acrB insertions are null mutations. Like known acrB mutations they conferred sensitivity to acridine orange and bile salts (data not shown). Furthermore, we demonstrated that CDI sensitivity is complemented by a plasmid carrying acrB+ (data not shown), showing that these mutations were recessive. Thus, it is the loss of acrB that is responsible for CDIR.
Figure 1. Mutations in acrB predominate amongst CDIR mutants.
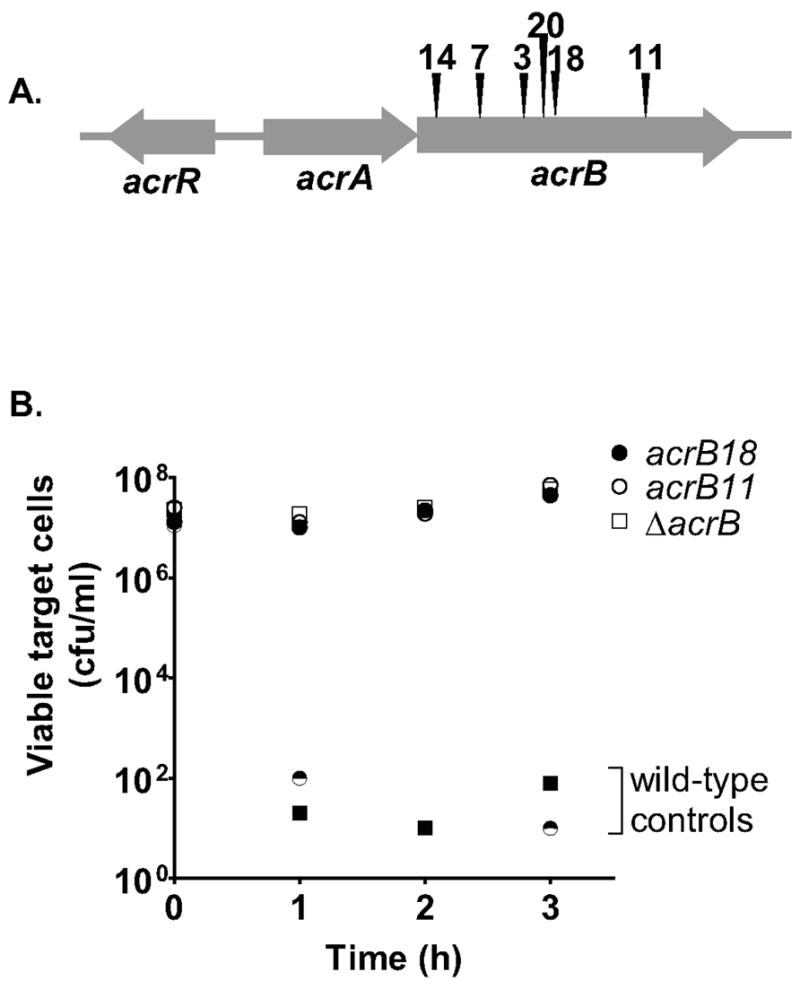
(A). The transposon locations in ten CDIR mutants are shown, with six unique insertion sites identified. (B). After transduction into E. coli MC4100, transposon mutants (two of ten are shown) and a ΔacrB mutant (RAM1279) were incubated with DL4608 CDI+ inhibitor cells at a 20:1 inhibitor to target ratio for 3 h, and cell numbers were determined by plating on LB-kan medium. Squares and circles represent data from two separate experiments, each with a wild-type control. The time course in Fig. 1B demonstrates that near maximal inhibition is achieved by two hours, therefore our remaining CDI experiments are limited to this time point.
The results shown in Fig. 1 demonstrate that AcrB plays either a direct or indirect role in the CDI pathway. However, since AcrB has an inner membrane location it did not seem likely that it could be the CDI receptor. Based on the fact that the mutagenesis strategy above yielded only mutations in the acrB gene, we performed a modified mutant hunt to enrich for other mutations that might lead to CDI resistance. A pool of transposon-mutagenized E. coli MC1061 target cells was subject to two rounds of selection against CDI+ inhibitor cells, then plated on MacConkey agar to inhibit growth of acrB mutant cells, which are sensitive to the bile salts in this medium. After transduction to move presumptive CDIR mutations into a fresh E. coli background, cells were reselected for CDI resistance. Four CDIR mutants that did not have an insertion in the acrB gene (tested by PCR analysis) and were not complemented by a plasmid expressing AcrB were picked and found to each contain an EZ-Tn5 insertion 23 bp upstream of the AUG start codon of the bamA gene, with a 9 bp chromosomal DNA sequence duplication (Experimental procedures). We will refer to these mutations as bamA101.
The bamA gene, formerly known as yaeT, encodes an essential outer membrane protein (Wu et al., 2005) and therefore bamA101 cannot be a null mutation. Given the position of the insertion, it was likely to affect bamA expression. The CDI phenotype of the bamA101 mutant was complemented by introduction of a low copy plasmid, pZS21-bamA+, expressing BamA constitutively from the tet promoter (Fig. 2A), demonstrating the recessive nature of the insertion mutation and suggesting that the bamA101 mutation caused decreased expression of BamA. To determine the effect of the bamA101 mutation on BamA protein levels, immunoblot analysis was performed using α-BamA antiserum. The results showed that BamA levels in the bamA101 mutant were significantly reduced compared to wild-type JCM158 cells. The presence of BamA degradation products complicates precise determination of expression levels, but we estimate that the bamA101 mutation reduces gene expression at least four to fivefold (Fig. 2B) (see Experimental procedures).
Figure 2. Analysis of the effects of the bamA101 mutation on CDI resistance and BamA expression.

(A). Complementation of the bamA101 mutant with bamA. E. coli MC1061 with the indicated genotypes were assessed for CDI resistance as described in Experimental procedures. Cell numbers were quantitated prior to addition of CDI+ inhibitor cells DL4577 (0 h), and after 2 h of incubation (2 h). Both the MC1061bamA101 and MC1061bamA+ isolates contained plasmid pZS21amp whereas bamA101(bamA+) contained plasmid pZS21amp-bamA+. (B). Effect of bamA101 on expression of BamA and other OMPs. Expression of BamA and partly degraded BamA (BamA*) (upper panel), and DegP, LamB, OmpA, BamD in E. coli JCM158 and mutant derivatives (lower panel) was determined by immunoblot analysis. Lanes (1), wild-type; (2), Δwzb; (3), bamA101; (4), bamA101 Δwzb; (5), bamD5; (6), bamD5 Δwzb. (C). AcrA and TolC are not required for CDI. MC4100 ΔacrA (RAM1277), ΔacrB (RAM1279), and tolC-(CAG12184) target cells were incubated with CDI+ inhibitory E. coli (DL4608) at a 20:1 inhibitor to target cell ratio. Viable target cell counts were determined after 3 h. MC4100 (“wild-type”) was included as a positive control for sensitivity.
BamA is an outer membrane protein that is essential for the biogenesis of outer membrane β-barrel proteins (OMPs), including LamB and OmpA (Wu et al., 2005). Immunoblot analysis indicated that the OmpA and LamB levels were decreased in the bamA101 mutant compared to wild-type cells (Fig. 2B). Correspondingly, the level of DegP, a periplasmic protease that is upregulated in response to defects in OMP assembly (Raivio, 2005), was increased in the bamA101 mutant (Fig. 2B). Thus, although the reduced levels of BamA in the bamA101 mutant are sufficient to support growth, they appear to be inadequate for efficient OMP assembly.
The cellular location of BamA suggests that it may be the receptor for CDI. In this case, the bamA101 mutation reduces receptor levels to a degree that confers resistance to CDI. Given that our screen for the CDIR phenotype identified four mutants with altered BamA levels, but no other potential receptor mutants, this suggests that BamA might function directly in CDI or that the “true” receptor is also essential. Additional experiments that support the conclusion that BamA is the CDI receptor are presented below.
In both of the mutant hunts described above, in addition to mutations in acrB and bamA, we found CDIR mutant colonies that were highly mucoid because of increased colanic acid capsule expression. Capsule could provide a physical barrier that prevents the direct cell-to-cell contact required for CDI. Further analysis suggested that some of these mucoid CDIR mutants had reduced expression of the histone-like nucleoid structuring protein (H-NS), since they fermented salicin (Dole et al., 2004), whereas other salicin− CDIR mutants (e.g. mutant 1S12) likely had other mutation(s) causing a mucoid phenotype. H-NS represses colanic acid synthesis by silencing the rcsA gene coding for a positive regulator of capsule expression (Gottesman et al., 1985; Sledjeski and Gottesman, 1995). Indeed, analysis of E. coli containing a known mutation in hns, which had a mucoid colony phenotype, showed that it was resistant to CDI as expected (Fig. S2). To determine if the CDI resistance of the hns mutant required colanic acid synthesis, a mutation in the wzb gene coding for a phosphotyrosine phosphatase required in the colanic acid biosynthesis pathway (Lescop et al., 2006; Vincent et al., 2000) was introduced. E. coli hns651 wzb− lost both its mucoid colony phenotype and CDI resistance, becoming about 104-fold more sensitive to CDI (Fig. S2). Similar results were obtained after introduction of the wzb mutation into the 1S12 CDIR mutant (Fig. S2), indicating that the resistance of mucoid mutants to CDI is due to production of the colanic acid capsule. In contrast, the CDI resistance of the acrB (not shown) and bamA101 mutants (see below) was not caused by capsule up-regulation since the CDIR phenotype was not affected by a wzb mutation. Together, these results indicate that AcrB and BamA play roles, either direct or indirect, in the CDI pathway. As noted above, colanic acid capsule production also confers resistance to CDI, likely by blocking the cell-to-cell contact necessary for growth inhibition (Aoki et al., 2005).
The AcrB and BamA multi-component machines are not required for CDI
It is striking that both AcrB and BamA are parts of multi-component complexes that function as an efflux pump and a β-barrel assembly machine, respectively (Kim et al., 2007; Seeger et al., 2006; Tikhonova and Zgurskaya, 2004; Wu et al., 2005). This raises the question of whether the functions of these two important cellular machines are required for CDI, or if AcrB and BamA act alone in the CDI pathway.
Trimeric AcrB interacts with AcrA in the periplasm and TolC in the outer membrane to form an efflux pump that spans the entire cell envelope. This pump transports small molecules including certain drugs from the cell using the proton gradient as an energy source (Nikaido and Zgurskaya, 2001). Deletion of the acrA gene did not confer significant resistance to CDI. After 3 h of mixing with CDI+ inhibitor cells, acrA mutant cells were over 105-fold more sensitive to CDI than acrB mutant cells (Fig. 2C). Similar results were observed using a tolC210::Tn10 null mutant (Fig. 2C). These results indicate that AcrA and TolC are not required for CDI. Thus, the function(s) of AcrB that plays a critical role in CDI is independent of both AcrA and TolC.
BamA interacts with four lipoproteins, BamB, BamC, BamD, and BamE (formerly known as YfgL, NlpB, YfiO, and SmpA respectively), that together form the Bam complex in the outer membrane, and with the periplasmic SurA chaperone, to assemble β-barrel OMPs (Jain and Goldberg, 2007; Kim et al., 2007; Malinverni et al., 2006; Ruiz et al., 2006; Sklar et al., 2007a; Werner and Misra, 2005). Derivatives of the MC4100 E. coli strain JCM158 containing mutations in genes coding for members of the Bam complex were analyzed for susceptibility to CDI and by immunoblotting to assess BamA and OMP levels. The results showed that bamB, bamD, and surA mutants had a low and somewhat variable level of resistance to CDI (based on multiple experiments), about 100-fold less than the bamA101 mutant (Table 1). As expected, the bamD4 (formerly yfiO4) mutant showed somewhat less resistance than bamD5 (formerly yfiO5), an allele causing a greater functional defect of this essential gene (Malinverni et al., 2006). Mutations in bamC and bamE, which both code for proteins in the Bam complex (Malinverni et al., 2006; Sklar et al., 2007a), did not have any effect on CDI (Table 1). The low resistance of the bamB, bamD, and surA mutations could be due to a number of possible factors including reduction in BamA level. However, immunoblot analysis indicated that the BamA level was reduced minimally or not at all by the bamD5 mutation and other mutations of genes coding for Bam complex members (Fig. 2B and 3A). In addition, the immunofluorescence analysis presented below indicated that the bamD5 mutant expresses a normal level of surface BamA, significantly higher than E. coli containing bamA101. These results rule out any effect of bamD5 on BamA level as the cause of CDI resistance.
Table 1.
Effect of mutations in the Bam complex on contact-dependent growth inhibition
| Strain | Relevant Genotype | Mean CDI Resistance1 | Range of CDI resistance |
|---|---|---|---|
| DL5503 | bamA101 | 30,950 | 21,100 – 40,800 |
| JCM344 | bamD4 | 3.6 | 1.3 – 7.6 |
| JCM345 | bamD5 | 25.5 | 4 – 65.3 |
| JCM175 | bamB | 31.9 | 3.3 – 80.5 |
| JCM647 | SurA | 38.6 | 2.8 – 105 |
| JCM304 | bamC | 0.4 | 0.3 – 0.5 |
| JCM376 | bamE | 0.6 | 0.1 – 1.2 |
CDI resistance was calculated as [cfu of mutant cells/cfu of wild-type cells] after a 2 h incubation with DL4577 CDI+ inhibitor cells. The mean of at least two independent experiments is shown in column 3, and the range of values is shown in column 4.
Fig. 3. Evidence that BamD, SurA, and BamB are not required for CDI.

(A). BamA levels were determined by immunoblot for E. coli samples containing mutations in genes coding for different outer membrane proteins. E. coli JCM158 with the indicated mutations were mixed with CDI+ E. coli DL4577 (B) or CDI− DL4956 mock inhibitor cells (C) for 2 h, and target cell number were determined (Experimental procedures). The first bar for each E. coli isolate shows target cell number prior to addition of inhibitor cells and the second bar shows cell number after 2 h incubation.
We next tested the possibility that mutations in bamB, bamD, and surA might indirectly affect CDI through induction of colanic acid capsule synthesis, since our results discussed above indicated that mutations that increase colanic acid production confer CDI resistance (Fig. S2). We found that the CDI resistance of the bamD5, surA, and bamB mutants was negated with the addition of a wzb mutation (Fig. 3B). This effect of the wzb mutation on CDI resistance was not due to a non-specific effect on cell fitness since each of the Bam complex mutants containing the wzb mutation grew well in mixed culture with CDI-minus control cells (Fig. 3C). These results indicate that the weak CDI resistance phenotypes of bamD5, surA, and bamB mutants are an indirect effect of up-regulation of capsule synthesis, possibly in response to the cellular stress caused by diminished OMP biogenesis (see Fig. 2B). Notably, there was no correlation between the OMP biogenesis phenotype and CDI resistance. A bamA101Δwzb double mutant is CDI-resistant (see below), and yet has a more normal OMP biogenesis phenotype than the bamD5Δwzb double mutant (see Fig. 2B), which is fully sensitive to CDI (Fig. 3B). This result supports the hypothesis that the CDI resistance of the bamA101 mutant is not caused by an indirect effect on Bam-regulated OMP levels.
BamA forms a β-barrel in the outer membrane and also has five POTRA domains located in the periplasm, designated P1 to P5, that interact with the BamA-associated outer membrane lipoproteins (Kim et al., 2007). The P3 domain appears to be required for an essential function of BamA in OMP biogenesis since a bamA mutant lacking P3 is non-viable (Kim et al., 2007). To determine if the BamAΔP3 mutant could function as a CDI receptor, we constructed bamA101/bamAΔP3 merodiploids. JCM158 bamA101 expressing bamAΔP3 was over 100-fold more sensitive to CDI compared to JCM158 bamA101 and within 10-fold of the CDI sensitivity of JCM158 control cells (Fig. 4A). These results indicated that BamAΔP3 retains the function of BamA required for CDI sensitivity.
Fig. 4. The POTRA-3 domain of BamA is not required for CDI.

(A). E. coli JCM158 with the indicated mutations (shown above the bars) and plasmids (shown below the x-axis) were mixed with CDI+ E. coli DL4577 and target cell numbers were determined after 2 h. The E. coli isolates used in (A) were analyzed in parallel by immunoblotting for BamA (panel B) and other outer membrane proteins (panel C) as described (Experimental procedures). Lane numbers in panels B and C correspond in order from left to right to the samples analyzed in panel A.
Note that the level of complementation observed with bamA101/bamAΔP3 merodipoid was significantly less than that observed for the bamA101/bamA+ merodiploid, even though the levels of the mutant BamAΔP3 and the wild-type BamA proteins produced in trans were similar based on immunoblot analysis (Fig. 4B, lanes 4, 5) and immunofluorescence (Fig. S3, compare with Fig. 5). Immunoblot analyses indicated a slight, but reproducible defect in OMP biogenesis in the bamA101/bamAΔP3 merodiploid relative to the bamA101/bamA+ merodiploid or the bamA101/pZS21 vector control based on a reduction of OMP levels and an increase in DegP levels (Fig. 4C). Therefore, we considered the possibility that the discrepancy in complementation could be due to increased colanic acid capsule production in the bamA101/bamAΔP3 merodiploid. This possibility was tested by measuring the effect of a Δwzb mutation on CDI complementation. Introduction of the Δwzb mutation into JCM158bamA101 did not affect its CDI resistance (Fig. 4A). Notably, in the Δwzb background, pZS21amp-bamAΔP3 complemented JCM158 bamA101 as well or better than pZS21amp-bamA+ (Fig. 4A), even though the Δwzb mutation did not significantly affect bamA expression or OMP biogenesis (Figs. 4B, 4C, compare lanes 3–5 with lanes 6–8). Thus, BamAΔP3 retains the function(s) of BamA required for CDI sensitivity. These results show that the POTRA-3 domain of BamA, which is essential for the BamA OMP biogenesis machine, is not required for CDI. Taken together, these data indicate that the functions of AcrB and BamA required for CDI can be separated from the functions of these two proteins in drug efflux and OMP assembly, respectively.
Fig. 5. Analysis of the effects of bamA101 on expression of BamA at the cell surface.

To detect expression of BamA at the cell surface, E. coli were fixed in formalin, which blocks antibody entry into cells, or methanol, which permeabilizes cells to antibodies. Antibodies to full-length BamA (α–BamA) or to the POTRA domains of BamA (α–POTRA) (indicated at left), were incubated with either formalin- or methanol-fixed cells (indicated at right) followed by an AlexaFluor 488-labeled secondary antibody as described in Experimental procedures. Left panels are phase contrast images, right panels are fluorescence images. (A). JCM158 bamA+ control, (B). DL5503 bamA101.
BamA mediates CDI-dependent inhibitor-target cells binding
The cellular locations of BamA in the outer membrane and AcrB in the inner membrane, suggest that BamA may function upstream of AcrB, possibly as a receptor and conduit for CdiA or a CdiA-derived peptide. For BamA to be a CDI receptor, a portion of BamA must be exposed at the cell surface. We tested this possibility by raising antisera against full-length BamA (α-BamA) and, as a control, to POTRA domains 1–5 (α–POTRA). Both antibody preparations, purified from whole sera, had similar activity against BamA in whole cell extracts, whereas antibodies from pre-immune sera did not react with BamA (Fig. S4).
To determine if α-BamA antibodies recognized a portion(s) of BamA exposed at the cell surface, we examined labeling of E. coli using immunofluorescence microscopy. Cells were fixed in methanol, which permeabilizes E. coli to antibodies, or formalin, which prevents antibody penetration. As shown in Fig. 5A, α–POTRA antibodies labeled the methanol-fixed cells but not cells fixed in formalin, consistent with the periplasmic location of the POTRA domain. In contrast, both formalin- and methanol-fixed E. coli were labeled with the α-BamA antibodies, indicating that some of the α-BamA antibodies recognize BamA epitopes exposed at the cell surface (Fig. 5A). Analysis of the bamA101 mutant showed that significantly less BamA was present at the cell surface (Fig. 5B), consistent with immunoblot results above (Figs. 2B, 3A) again showing that the BamA level in JCM158 bamA101 is about fivefold less than wild-type cells. In contrast, JCM158 bamD5 expresses a normal level of BamA (Fig. S5). These results are consistent with the hypothesis that the bamA101 mutant is resistant to CDI due to reduced levels of the cell surface receptor.
To more rigorously test the hypothesis that BamA is the CDI receptor, we determined if the α-BamA antibody, which binds to cell surface-exposed BamA (Fig. 5A), blocks CDI-mediated inhibitor-target cell binding. We used a FACS-based assay developed previously which measures CDI-dependent cell-to-cell binding (Aoki et al., 2005). CDI+ inhibitor cells (“I”), labeled with GFP (Fig. 6B), and wild-type target cells (“T”) labeled using DsRed (Fig. 6C), were mixed at a ratio of 1:2 (inhibitor:target) for 15 min, then analyzed by FACS to identify an “aggregate” (“A”) population, each of which contains at least one green inhibitor cell and one red target cell based on fluorescent intensity in both green and red channels (Fig. 6D). Quantitation showed that about 14% of wild-type JCM158 target cells were in aggregates with inhibitor cells compared to 1.5% of bamA101 mutant cells (Fig. 6E, Table 2). Thus, inhibitor-target binding was reduced about 9-fold by the bamA101 mutation. These results demonstrate that the bamA101 mutation affects CDI-dependent cell-to-cell adhesion, likely by reducing receptor levels.
Fig. 6. Analysis of the effects of target cell CDIR mutations on inhibitor-target cell binding.
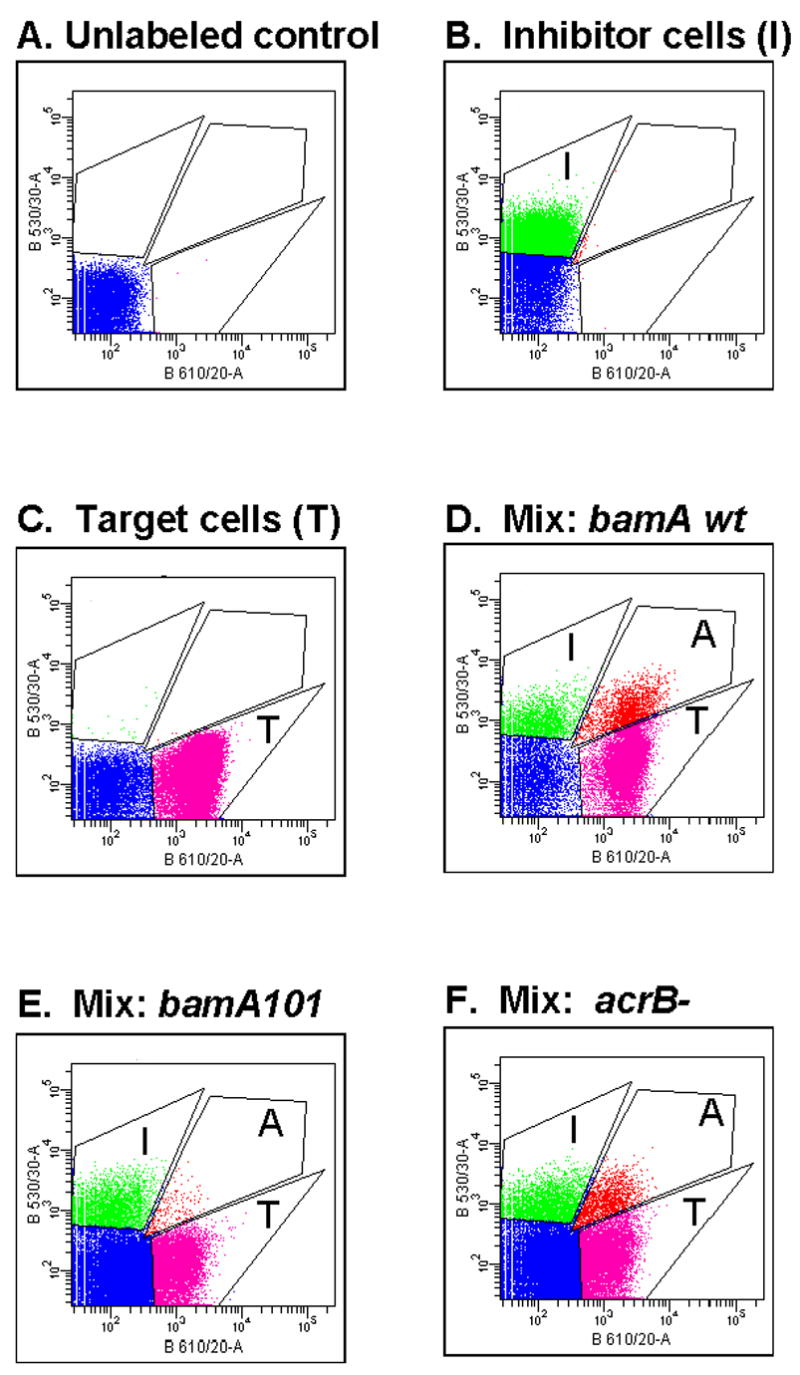
The effects of target cell acrB and bamA mutations on binding to CDI+ inhibitor cells was assessed by mixing GFP-labeled DL4905 CDI+ inhibitor cells with DsRed-labeled target cells (1 inhibitor with 4 targets) for 15 min, then scanning 105 bacteria by FACS (Experimental procedures). Boxed areas depicted show gating boundaries for GFP+ inhibitory cells (“I”), DsRed-labeled target cells (“T”), and cell aggregates (“A”) containing at least one inhibitory and one target cell. See Table II for data quantitation. (A). JCM158, (B). DL4905 GFP+ CDI+ inhibitor cells, (C). DL5529 (JCM158 DsRed+), (D). DL4095 + DL5529 (bamA wild-type), (E). DL4905 + DL5530 (bamA101), (F). DL4905 + DL5542 (acrB::kan).
Table 2.
FACS analysis of BamA-Mediated cell-to-cell adhesion
| Target Strain | Relevant Genotype of Target Cells1 | Inhibitor Strain | Antibody | Inhibitor to target ratio | % Aggregated Targets2 |
|---|---|---|---|---|---|
| JCM158 | Wild-type bamA CDIS | DL4577 CDI+ | None | 0.43 | 14.1% |
| DL5503 | bamA101 CDIR | “ “ | None | 0.48 | 1.5% |
| DL5542 | acrB::Kan CDIR | “ “ | None | 0.43 | 9.8% |
| JCM158 | bamA wt CDIS | “ ” | Pre-immune | 0.77 | 9.8% |
| JCM158 | bamA wt CDIS | “ “ | α-BamA POTRA | 0.78 | 10.1% |
| JCM158 | bamA wt CDIS | “ “ | α-BamA Whole | 0.79 | 0.8% |
CDIS = sensitive to contact-dependent growth inhibition, CDIR = resistant to contact-dependent growth inhibition
Calculated as [#aggregated cells]/[#target cells + # inhibitor cells] with data acquisition from 100,000 total cells for each datapoint
We next examined the effects of α-BamA antibodies on inhibitor-target cell binding using the FACS assay (see Fig. 6). About 10% of target cells were in complex with CDI+ inhibitor cells using pre-immune control or α-POTRA antibodies, similar to the aggregation levels obtained in the absence of antibody (Figs. 7A, 7B, Table 2). In contrast, less than 1% of target cells were aggregated in the presence of α-BamA antibodies (Fig. 7C, Table 2). Thus, α–BamA, which recognizes surface exposed BamA epitopes (Fig. 5A), blocks adhesion between inhibitor and target cells (Fig. 7C). However, α–POTRA antibodies, which do not react with BamA sequences at the cell surface (Fig. 5A), do not block inhibitor-target cell interaction (Fig. 7B). These data indicate that BamA is required for binding between inhibitor cells and target cells.
Fig. 7. α-BamA antibodies block inhibitor-target cell binding.
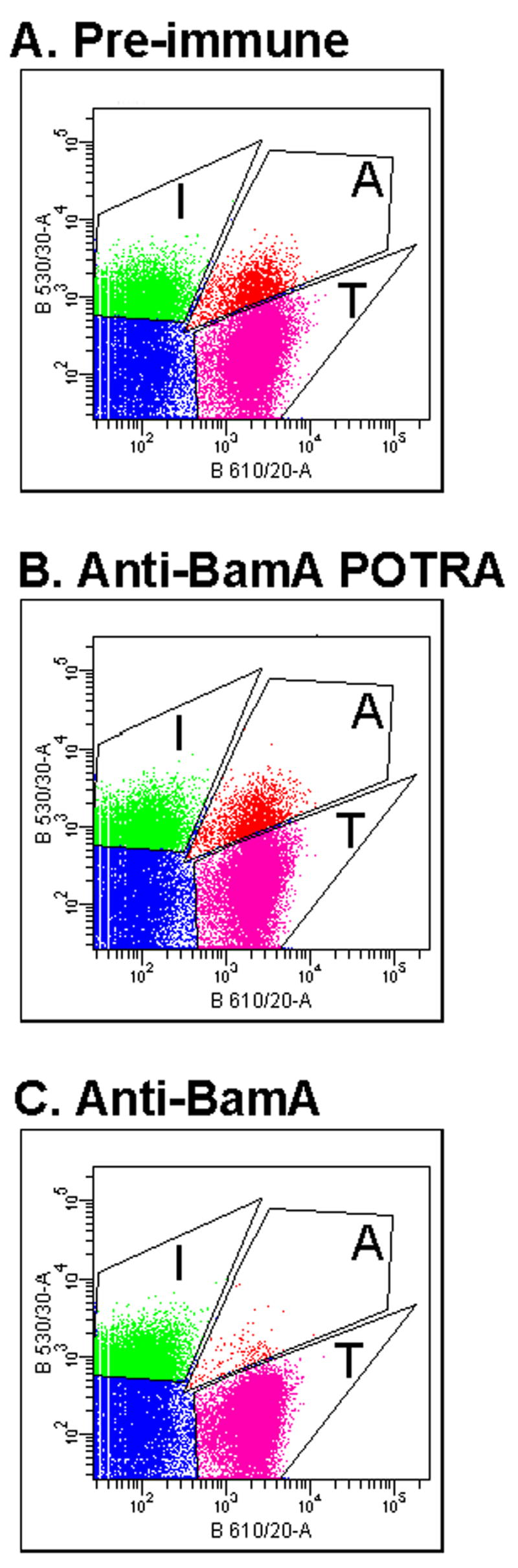
E. coli target cells (BamA+) labeled with DsRed (DL5529) were incubated with purified antibodies from (A) pre-immune, (B) α-BamA POTRA, and (C) α-BamA sera, then mixed for 15 min with GFP-tagged CDI+ inhibitor cells (DL4905) and analyzed by FACS (Experimental procedures). Cell gating boundaries are the same as for Fig. 6.
In contrast to results obtained with the bamA101 mutant, the acrB mutation did not significantly affect CDI-dependent cell-to-cell binding as measured by FACS (compare Figs. 6D, 6F, see Table 2). We conclude that AcrB does not mediate binding between CDI+ inhibitor cells and target cells. Taken together, these results strongly indicate that BamA is the CDI receptor, mediating cell to cell binding, and that AcrB functions downstream of BamA in the CDI-mediated growth inhibition pathway.
BamA sequences at the cell surface are required for CDI-mediated growth inhibition
Previously we sorted target cells that were in aggregates with CDI+ inhibitor cells using FACS and showed that they became growth inhibited more rapidly than free target cells (Aoki et al., 2005). These results showed a link between inhibitor-target cell binding, measured by FACS, and CDI-mediated growth inhibition. To determine if BamA plays a specific and direct role in CDI, the effect of α-BamA antibodies on CDI was measured (see Experimental procedures). Incubation of target cells with α-BamA antibodies significantly blocked CDI when compared with pre-immune antibodies (Fig. 8A, expressed as fold protection over buffer control). Notably, antibodies to the POTRA domains of BamA did not block CDI (Fig. 8A), consistent with results showing that the POTRA domain is not exposed at the cell surface (Fig. 5A), and that α-POTRA antibodies do not block inhibitor-target cell binding (Fig, 7B). These results indicate that BamA is required for CDI, and are consistent with results above showing that BamA mediates cell-to-cell binding.
Fig. 8. α-BamA antibodies inhibit CDI.
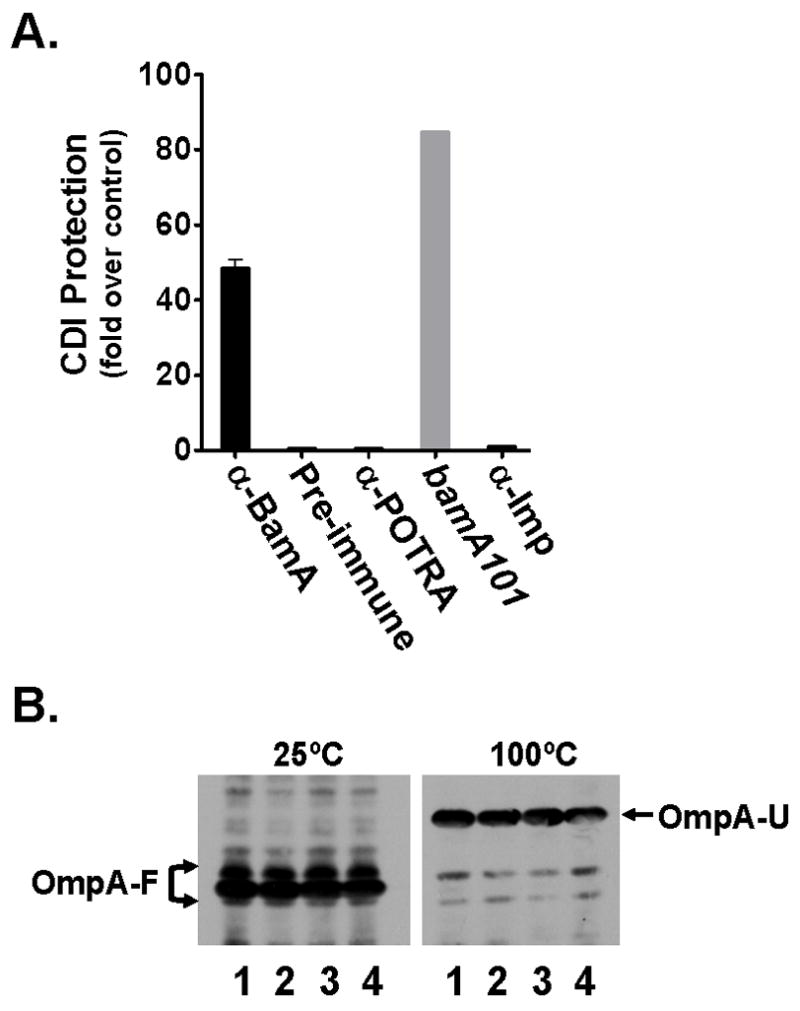
(A). α-BamA antibodies block CDI. Antibodies against whole BamA (α-BamA), the POTRA domains of BamA (α-POTRA), Imp, (α-Imp), and pre-immune serum (Preimmune) were purified and used to determine if they protect E. coli DL5422 from CDI as described in Experimental procedures. A bamA101 strain (DL5430) was included as a resistant control. CDI protection (y-axis) is expressed as the fold protection of DL5422 against CDI measured for each antibody preparation and the bamA101 resistant control compared with a no antibody addition control. (B). α-BamA antibodies do not induce OmpA misfolding. Samples were pre-incubated with either 1 = PBS, 2 = Pre-Imm, 3 = α–POTRA, or 4 = α–BamA for two hours and loaded onto an SDS-PAGE gel either with or without heat denaturation. Western blots were incubated with α–OmpA antiserum. In the unheated samples (25°C), OmpA is in a folded conformation (OmpA-F). Folded OmpA migrates faster than its unfolded conformation (OmpA-U), visible upon heat denaturation (100°C).
Three possible complications must be considered when interpreting the results of the experiment shown in Fig. 8A. First, binding of antibodies to BamA at the cell surface might inhibit BamA function and this could affect CDI indirectly, perhaps by killing the cells. This possibility was tested by determining if α-BamA antibodies altered the assembly of outer membrane proteins. Even after a 2 h incubation with α-BamA antibodies at the same concentration shown to block CDI (Fig. 8A), OmpA remained folded, migrating at the same position as cells pre-treated with pre-immune antibodies, α-POTRA antibodies, and a buffer control [Fig. 8B, compare folded OmpA (OmpA-F) at 25ºC with unfolded OmpA (OmpA-U) (100ºC)]. Further analysis showed that LamB assembly was also not affected by α–BamA antibodies (not shown), supporting the conclusion that αBamA antibodies did not affect OMP folding at the concentration used in this study.
A second complication arises since it is possible that binding of antibodies to any OMP epitopes exposed at the cell surface block CDI, in which case the CDI inhibition observed with α-BamA (Fig. 8A) was likely due to a nonspecific effect. This possibility was tested using purified polyclonal antibodies directed against the Imp protein, which is located in the outer membrane in similar abundance to BamA (Wu et al., 2005). Immunoblotting of an outer membrane fraction of our wild-type strain showed that α-Imp reacted with Imp as well as additional OMPs including OmpA; reactivity of α-Imp with BamA was not observed (Fig. 9A). We then tested if α-Imp antibodies could bind to surface exposed OMP epitopes of whole cells. α-Imp-treated cells were incubated with with AlexaFluor secondary antibody to monitor immunofluoresence. Polyclonal α-Imp recognized cell surface-exposed OMP epitopes with similar or greater reactivity compared with α-BamA (Fig. 9B), yet unlike α-BamA, the α-Imp antibody did not block CDI (Fig. 8A). These results indicate that the inhibitory effect of α-BamA antibodies on CDI (Fig. 8A) is due to a specific effect on blocking ligand binding to BamA.
Fig. 9. Evidence that the inhibition of CDI by α-BamA is specific.
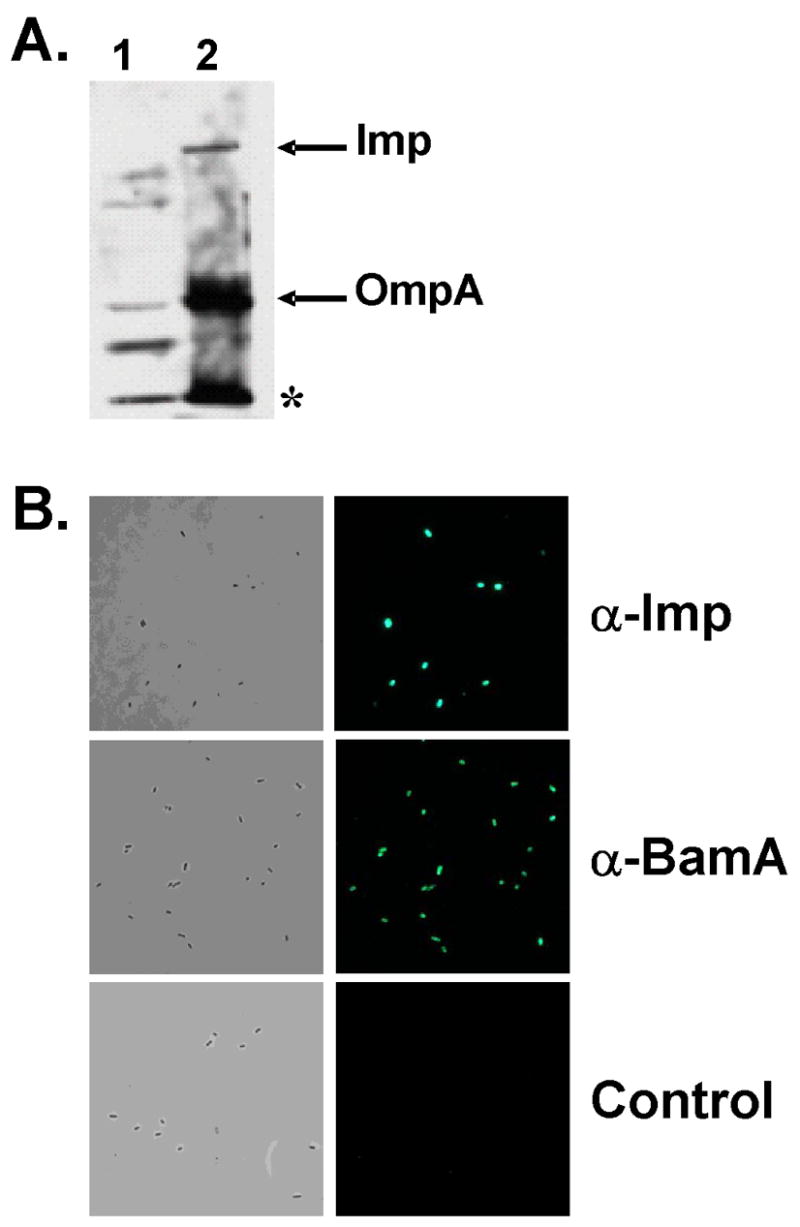
(A). Immunoblot analysis of E. coli DL5432 with α-Imp antibodies. Analysis was performed as described in Fig. S3/Experimental procedures using inner membrane plus soluble protein (lane 1) and outer membrane (lane 2) fractions. α-Imp is reactive with Imp and OmpA (arrows), and an unknown protein (asterisk). (B). Immunofluorescence was carried out using formalin fixed DL5432 cells incubated with α-Imp and α-BamA antibodies under the same conditions as described in Fig. 8A. The control was treated identically except that primary antibody was not added. Left panels are phase contrast images and right panels are fluorescence images.
A third possible complication arises because of the presence of BamA on both inhibitor cells as well as target cells. Perhaps the binding of α–BamA antibodies to BamA present on the surface of inhibitor cells might also block CDI. This could occur, for example, if CDI+ inhibitor cells require BamA for their growth inhibitory activity. This possibility was tested by transduction of the bamA101 mutation into CDI+ inhibitor cells and determining if their effectiveness at blocking target cell growth was altered. The results in Fig. 10 showed that E. coli bamA101 CDI+ cells inhibit target cell growth about 10-fold less than bamA+ CDI+ cells do. In contrast, target cells containing the bamA101 mutation were 1,000-fold more resistant to growth inhibition than bamA+ cells (Fig. 2). Thus, the bamA101 mutation had about 100-fold less effect on inhibitor cells compared to its effect on target cells. The 10-fold reduction in CDI activity of bamA101 inhibitor cells compared to bamA+ cells may be due to decreased export of CdiB into the outer membrane, since CdiB is predicted to be a β-barrel protein and BamA is essential for export of all β-barrel proteins tested to date (Jain and Goldberg, 2007; Kim et al., 2007; Ruiz et al., 2006; Werner and Misra, 2005). Taken together, these results strongly support the conclusion that BamA is the receptor on target cells that mediates binding with CDI+ inhibitor cells and subsequent growth inhibition.
Figure 10. Effect of the bamA101 allele on CDI+ inhibitor cells.
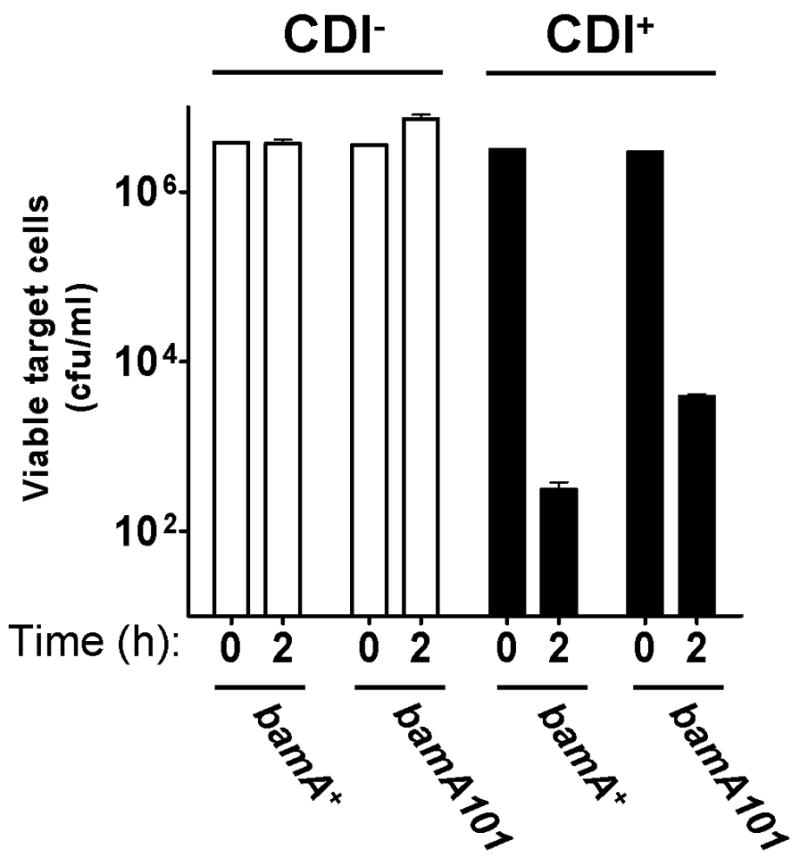
E. coli with the indicated genotypes (x-axis), carrying either cosmid vector pWEB::TNC (CDI−) or cosmid pDAL660Δ1–39 (CDI+) (see Fig. S1), were used as inhibitor cells against E. coli DL4372 targets as described in Experimental procedures. Viable cell target numbers were measured at “0” time and after 2 h incubation for each sample.
Discussion
In this study we used a genetic approach to identify components in target cells that are required for contact-dependent growth inhibition. We found that mutations in the genes coding for BamA and AcrB conferred CDI resistance, indicating that these two proteins play roles in the CDI pathway (Figs. 1, 2). Our results show that BamA is the receptor for CDI (Figs. 2, 6, 7,8, 9) and that AcrB functions downstream of BamA in the CDI pathway (Fig. 6).
BamA is a highly conserved member of the YaeT/Omp85 family of proteins (Gentle et al., 2005), required for the biogenesis of β–barrel OMPs (Jain and Goldberg, 2007; Kim et al., 2007; Werner and Misra, 2005). The C-terminal portion of BamA forms a β-barrel structure and inserts into the outer membrane, with the N-terminal portion extending into the periplasm. Five POTRA domains are present at the N-terminus, which mediate interactions between BamA and four lipoproteins, BamB, BamC, BamD, and BamE (Malinverni et al., 2006; Sklar et al., 2007a; Wu et al., 2005). In addition, the periplasmic protein SurA appears to chaperone OMPs to the Bam complex (formerly referred to as the YaeT complex) and is found associated with BamA (Sklar et al., 2007b). In concert, these proteins form a multi-component assembly machine that is required for OMP biogenesis (Wu et al., 2005). We determined if these Bam complex proteins play a role in CDI using mutants that either lack the proteins altogether or produce proteins with compromised function. Although some of these mutations confer low-level resistance by the stress-induced production of a protective colanic acid capsule, it is clear that none of these components play a direct role in CDI. Moreover our data also indicate that BamAΔP3, which cannot function in OMP biogenesis (Kim et al., 2007), is fully functional for CDI (Fig. 4A).
The results presented here show that the essential outer membrane protein BamA is the receptor for CDI. Indeed, antibodies directed against surface epitopes of BamA blocked binding between CDI+ inhibitor cells and target cells (Fig. 7C), and blocked CDI-dependent growth inhibition (Fig. 8A). This inhibitory effect of α-BamA on CDI was shown to be specific since α-Imp antibodies, which bind to cell surface exposed epitopes equally or better compared with α-BamA (Fig. 9B), did not block CDI (Fig. 8A). In this regard the function of BamA is conceptually similar to its proposed function as a receptor for short-tailed Stx phages, which may bind to BamA within an extracellular loop (Smith et al., 2007).
AcrB is one of three components of a resistance nodulation division (RND)-type multi-drug efflux pump. It is an inner membrane protein with 12 transmembrane segments and functions as a drug/proton antiporter. AcrA is a periplasmic membrane fusion protein that couples AcrB to the outer membrane, channel-forming protein TolC. Together, these three components form a complex that spans the entire cell envelope allowing the transport of drugs into the medium.
Mutants lacking AcrB are insensitive to CDI even though these mutants produce a functional CDI receptor (BamA) and form contacts with CDI+ cells (Fig. 6F). These results demonstrate that AcrB functions downstream of BamA in the CDI growth-inhibition pathway, and it seems likely that this protein is the target of the CDI growth-inhibitory signal. Since mutants that lack AcrA or TolC are fully sensitive to CDI, AcrB must have some activity that is evidenced in the absence of a functioning efflux pump. Moreover, AcrB containing a carboxyl-terminal histidine tag (Tikhonova and Zgurskaya, 2004) does not complement the CDIR phenotype of an acrB null mutant, even though it retains a normal drug transport phenotype (unpublished data). These results suggest that CDI might involve an additional function of AcrB not required for drug transport. Alternatively, these data might be explained by a difference in thresholds for the CDI and drug efflux phenotypes.
A model for contact-dependent growth inhibition
The ligand for BamA has not yet been identified, but it is most likely CdiA and not CdiB, which by analogy with other TPS systems is an outer membrane protein required for CdiA export (Jacob-Dubuisson et al., 2004). Based on sequence homology of the TPS protein FHA (the prototype for the TpsA family), CdiA is predicted to have an extended structure formed by stacking of repeated β-strands (Makhov et al., 1994). Our previous work showed that, like FHA, CdiA is located at the cell surface and is also released into the extracellular milieu (Aoki et al., 2005). It is not known if either FHA or CdiA extends out perpendicular to the cell surface, lies flat against the outer membrane, or is at some angle between these states.
Assuming that CdiA is the ligand that binds to BamA, what is the growth-inhibitory signal transmitted to target cells? Previous results showed that CdiA is processed at two sites besides the putative signal sequence (Aoki et al., 2005). CDI-mediated growth inhibition requires continuous protein synthesis by cdiA+B+I+ inhibitory cells, and a single CDI+ inhibitor cell can inhibit hundreds of target cells over a two hour time course (Aoki et al., 2005). These observations could be explained if the growth inhibitory signal is generated by autoproteolysis of CdiA, since CdiA would be used up in the process, and multiple peptides might be required for growth inhibition. A CdiA peptide could be released after binding to BamA and be transported through the BamA β-barrel or through a central pore of a BamA oligomer (Kim et al., 2007; Tommassen, 2007). Although there is no evidence that protein transport can occur through the BamA β-barrel, we note, based on analogy to other two-partner secretion systems (Clantin et al., 2007; Mazar and Cotter, 2006), that export of CdiA is thought to occur through the CdiB β-barrel.
Another possibility is that contact between CdiA and BamA generates a signal, for example a conformational change that alters a downstream effector. We think this explanation less likely. We showed previously that CdiA containing a FLAG epitope at its C-terminus [FLAG 1, (Aoki et al., 2005)] is no longer active in growth inhibition (CDI−) but maintains intercellular binding based on FACS analysis. Thus, binding to BamA is clearly necessary but not sufficient for growth inhibition.
The most likely target of the postulated CdiA peptide or signal transduction pathway is the AcrB antiporter. Mutants lacking this inner membrane protein are fully resistant to CDI (Fig. 1) even though AcrB clearly functions downstream of the BamA receptor in the CDI growth inhibitory pathway. AcrB is not an essential protein under normal laboratory conditions, and thus CDI cannot occur by inhibition of AcrB function. However, it is possible that CDI might inhibit growth by altering this antiporter to cause an ion leakage that disrupts the proton-motive force. Alternatively, in analogy to the mannose transporter (Erni, 2006), AcrB might function to transport a CdiA peptide into the cell where it might interact with an unidentified cellular target.
Other human pathogens including Yersinia pestis and Burkholderia pseudomallei contain possible CdiA and CdiB homologues in their genomes (Aoki et al., 2005). The identification of the highly conserved and essential BamA protein as the CDI receptor opens the possibility that the CDI systems of uropathogenic E. coli isolates and possibly other pathogens also bind to BamA, which can be readily tested.
Experimental procedures
Strains, plasmids, and growth conditions
E. coli strains and plasmids used in this study are listed in Table 3. Cells were grown in Luria-Bertani (LB) (1% tryptone, 0.5% yeast extract, 1% NaCl) or Tryptone (TB) (1% tryptone, 1% NaCl) broth or agar supplemented with antibiotics at the following concentrations (unless otherwise noted): ampicillin (amp), 100 μg/ml; chloramphenicol (cam), 34 μg/ml; kanamycin (kan), 25 μg/ml; nalidixic acid (nal), 10 μg/ml; streptomycin (str), 100 μg/ml; tetracycline (tet), 12.5 μg/ml; rifampicin (rif), 150 μg/ml. Cultures were incubated at 37°C in an environmental shaker apparatus (New Brunswick Series 25) at 225 rpm unless otherwise indicated.
Table 3.
Strains, plasmids, and oligonucleotides used in this study.
| Strain | Relevant characteristic(s) | Reference or source |
|---|---|---|
| CAG12095 | MG1655 zac-3051::Tn10 | (Singer et al., 1989) |
| CAG12184 | MG1655 tolC210:: Tn10 | (Singer et al., 1989) |
| DH5α | F− supE44 hsdR17(rK-mK+) recA1 φ80dlacZΔM15 ΔlacU169 gyrA96 endA1 thi-1 relA1 deoR λ- | (Hanahan, 1983) |
| EC100/EPI100 | F− mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ− rpsL nupG | Epicentre |
| EC100D pir+ | F− mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ− rpsL nupG pir+(DHFR) | Epicentre |
| FB23374 | MG1655 wzb::Tn5KAN-I-SceI | Blattner Laboratory |
| JCM158 | MC4100 araR (spontaneous arabinose resistance) | (Wu et al., 2005) |
| JCM175 | JCM158 bamB::kan | (Sklar et al., 2007a) |
| JCM304 | JCM158 bamC::kan | (Malinverni et al., 2006) |
| JCM344 | JCM158 bamD4::cam | (Malinverni et al., 2006) |
| JCM345 | JCM158 bamD5::cam | (Malinverni et al., 2006) |
| JCM376 | JCM158 bamE::kan (Keio collection) | This study |
| JCM647 | JCM158 surA::kan (Keio collection) | This study |
| MC4100 | F− araD139 Δ(argF-lac)U169 rpsL relA1 flb5301 deoC1 ptsF25 RbsR | (Casadaban, 1976) |
| RAM1277 | MC4100 Δara ΔacrA | (Augustus et al., 2004) |
| RAM1279 | MC4100 Δara ΔacrB | (Augustus et al., 2004) |
| MC4100 acrB3 | EZ-Tn5 <KAN-2> Tnp chromosomal transposon insertion at 988 bp downstream of the acrB ATG start | This study |
| MC4100 acrB7 | EZ-Tn5 <KAN-2> Tnp chromosomal transposon insertion at 609 bp downstream of the acrB ATG start | This study |
| MC4100 acrB11 | EZ-Tn5 <KAN-2> Tnp chromosomal transposon insertion at 2571 bp downstream of the acrB ATG start | This study |
| MC4100 acrB14 | EZ-Tn5 <KAN-2> Tnp chromosomal transposon insertion at 199 bp downstream of the acrB ATG start | This study |
| MC4100 acrB18 | EZ-Tn5 <KAN-2> Tnp chromosomal transposon insertion at 1114 bp downstream of the acrB ATG start | This study |
| MC4100 acrB20 | EZ-Tn5 <KAN-2> Tnp chromosomal transposon insertion at 1062 bp downstream of the acrB ATG start | This study |
| DL1976 | MC4100 hns651 | (White-Ziegler et al., 1998) |
| DL4372 | MC4100 araD+ (zac-3051::Tn10) | This study |
| DL4577 | EPI100 pDAL660Δ1-39 | (Aoki et al., 2005) |
| DL4608 | DH5α pDAL660Δ1-39 | (Aoki et al., 2005) |
| DL4905 | MC4100 λ640-13 pDAL660Δ1-39 | (Aoki et al., 2005) |
| DL4956 | EPI100 pDAL660Δ2-63 | This study |
| DL5311 | MC4100 pacrA+B+ | This study |
| DL5311 1S12 | MC4100 pacrA+B+ mucoid mutant | This study |
| DL5311 1S12 wzb− | MC4100 pacrA+B+ wzb::Tn5KAN-1-SceI | This study |
| DL5413 | MC1061 bamA101 | This study |
| DL5422 | MC1061 pZS21 | This study |
| DL5428 | MC1061 pZS21amp | This study |
| DL5430 | MC1061 bamA101 pZS21amp | This study |
| DL5490 | MC1061 pWEB::TNC | This study |
| DL5491 | MC1061 pDAL660Δ1-39 | This study |
| DL5492 | MC1061 bamA101 pWEB::TNC | This study |
| DL5493 | MC1061 bamA101 pDAL660Δ1-39 | This study |
| DL5431 | MC1061 pZS21amp-bamA+ | This study |
| DL5503 | JCM158 bamA101 | This study |
| DL5525 | JCM158 bamA101 pZS21amp-bamAΔP3 | This study |
| DL5526 | JCM158 bamA101 pZS21amp-bamA+ | This study |
| DL5527 | JCM158 bamA101 pZS21amp | This study |
| DL5529 | JCM158 pDAL672 | This study |
| DL5530 | JCM158 bamA101 pDAL672 | This study |
| DL5541 | JCM158 acrB3::kan | This study |
| DL5542 | JCM158 acrB3::kan pDAL672 | This study |
| DL5561 | JCM158 Δwzb bamD5::cam | This study |
| DL5562 | JCM158 wzb::kan | This study |
| DL5564 | JCM158 Δwzb | This study |
| DL5576 | JCM158 Δwzb bamA101 | This study |
| DL5580 | JCM158 pZS21 | This study |
| DL5584 | JCM158 Δwzb surA::kan | This study |
| DL5585 | JCM158 Δwzb bamB::kan | This study |
| DL5632 | JCM158 Δwzb bamA101 pZS21amp | This study |
| DL5633 | JCM158 Δwzb bamA101 pZS21amp-bamA+ | This study |
| DL5634 | JCM158 Δwzb bamA101 pZS21amp-bamAΔP3 | This study |
| DL5650 | MC4100 hns651 wzb::Tn5KAN-I-SceI | This study |
| Plasmid | Relevant characteristic(s) | Reference or source |
| pacrAB+ | pACYC184 constitutively expressing acrAB | (Augustus et al., 2004) |
| pCP20 | Temperature-sensitive plasmid that shows thermal induction of FLP recombinase AmpR CamR | (Cherepanov and Wackernagel, 1995) |
| pDAL660Δ1-39 | pDAL660 deletion, cdiA+B+I+ AmpR | (Aoki et al., 2005) |
| pDAL660Δ2-63 | pDAL660 deletion, cdiABI AmpR | This study |
| pDAL672 | plac-DsRed AmpR | (Aoki et al., 2005) |
| pUHE-1 Pzl-1 | ColE1 replicon with an AmpR cassette interchangeable with pZ plasmids | (Lutz and Bujard, 1997) |
| pWEB::TNC | Cosmid vector AmpR, CamR | Epicentre |
| pZS21 | PLtetO plasmid KanR | (Lutz and Bujard, 1997) |
| pZS21-bamA+ | pZS21 expressing BamA KanR | (Kim et al., 2007) |
| pZS21-bamAΔP3 | pZS21 expressing BamAΔP3 KanR | (Kim et al., 2007) |
| pZS21amp | pZS21 converted from KanR to AmpR | This study |
| pZS21amp-bamA+ | pZS21-BamA converted from KanR to AmpR | This study |
| pZS21amp-bamAΔP3 | pZS21-bamAΔP3 converted from KanR to AmpR | This study |
| pZS21-His-bamA+ | pZS21 expressing N-terminally His-tagged BamA KanR | (Kim et al., 2007) |
| Oligonucleotide | Sequence | Reference or source |
| 885 | 5′ TTGGTTGTAACACTGGCAGAGC 3′ | This study |
| 886 | 5′ GCGAGCCCATTTATACCCAT 3′ | This study |
| 889 | 5′ CAACAAGGATCCACGTCTGGAAGAAGGGCTT 3′ (BamHI tag is underlined) | This study |
| 890 | 5′ CAACAAACTAGTGACGTAATAACCGAGGAATG 3′ (SpeI tag is underlined) | This study |
| 1085 | 5′ GATGCCAATCACCCACCAG 3′ | This study |
| 1086 | 5′ GCTGGATATGTCCGATGCC 3′ | This study |
| KAN-2 FP-1 | 5′ ACCTACAACAAAGCTCTCATCAACC 3′ | Epicentre |
| R6KAN-2 RP-1 | 5′ CTACCCTGTGGAACACCTACATCT 3′ | Epicentre |
Plasmids pZS21amp and pZS21amp-bamA+ were constructed by excising the kanR cassette from pZS21 and pZS21-BamA with restriction enzymes SacI and AatI and replacing it with the ampicillin-resistance cassette from pUHE-1 Pzl-1 (Lutz and Bujard, 1997). The majority of bamA on pZS21amp-bamA+ was then exchanged with bamAΔP3 from pZS21-bamAΔP3 between restriction sites EcoRI and KpnI resulting in pZS21amp-bamAΔP3.
The bamE::kan and surA::kan alleles originate from the Keio deletion collection (Baba et al., 2006), and were moved by P1 transduction into our wild type JCM158 background to create JCM376 and JCM647, respectively.
CDI Competition Assay
E. coli EPI100 cells were transformed with cdiA+B+I+ plasmid pDAL660Δ1–39 to construct CDI+ isolate DL4577, or with cdiA−B−I− plasmid pDAL660Δ2–63 (see Fig. S1) to construct CDI− “mock” inhibitor isolate DL4956. E. coli inhibitor (DL4577) or mock inhibitor (DL4956) cells were grown overnight in LB medium containing ampicillin, diluted 1/100 the next day in fresh LB lacking antibiotics, and grown to an OD600 between 0.2 and 0.5. Target E. coli isolates to be tested for CDI sensitivity were grown overnight in LB containing appropriate antibiotics, diluted 1/50 the next day in antibiotic-free LB, and used at an OD600 between 0.6 and 1.2. 1 ml of DL4577 or DL4956 cells were mixed in 15 ml loose-capped glass tubes with a volume of target cells calculated to provide five to ten inhibitor cells per target cell. Cell mixtures were incubated upright in an environmental shaking incubator (New Brunswick) at 37°C with shaking at 250 rpm. Samples were taken initially, prior to mixing, and at various times, depending upon the experiment, diluted 1:10 serially in M9 minimal medium salts (Sigma Co.) and plated on LB medium with appropriate antibiotics at 37°C to measure viable target cell numbers.
Mutation analysis
Transposon mutagenesis
Identification of acrB
Random transposon insertions were generated in the chromosome of strain EPI100 using an Epicentre EZ-Tn5 <KAN-2> Tnp Transposome kit. Briefly, 1 μl of EZ-Tn5<KAN-2> Tnp Transposome was electroporated into 50 μL of electrocompetent EPI100 cells using a MicroPulser Electroporator (Bio-Rad) according to the manufacturer’s instructions. Transformants were selected by plating on LB-kan, pooled together and stored at −70°C in LB containing 15% glycerol. To enrich for EPI100 target cell transposon mutants resistant to inhibition by CDI+ E. coli (DL4608), frozen EZ-Tn5 <KAN-2>Tnp mutant library cells were used to inoculate LB-kan, grown overnight, then mixed with log phase DL4608 cells at an inhibitor to target ratio of 20 to 1 (based on OD600). The cell mixture was incubated at 37°C with shaking at 225 rpm for 3 h, diluted 10-fold serially and plated on LB-kan. All colonies growing on LB-kan (survivors) were pooled and used as target culture to repeat the experiment. This process was repeated 3 times before individual colonies were screened for resistance to growth inhibition. Phage T4GT7 lysates were prepared on CDI-resistant mutants and transduced into MC4100. Transductants were then tested for CDI resistance. Transposon insertion positions were determined using inverse PCR as previously described (Ochman et al., 1988). Briefly, genomic DNA was digested with Hpy99I (which cuts once within the transposon) and ligated. Oligonucleotide primers 885 and 886 were then used to amplify the transposon/chromosome junction for sequencing.
Identification of bamA
Electroporation of E. coli isolate EPI100 was carried out using a EZ-Tn5<R6Kγori/KAN-2>Tnp transposome (Epicentre) to generate a library of transposon insertions. The EPI100 transposon library was subjected to selection for CDI-resistant mutants by challenge with DL4577 at a 10:1 inhibitor to target ratio, plating on MacConkey-kan agar to select for carriage of the transposon and to select against acrB mutants, which are inhibited by the bile salts in MacConkey agar. Surviving colonies were pooled, grown to an OD600 of 1, and the selection was repeated. A phage T4GT7 lysate was prepared on pooled colonies, transduced into E. coli strain MC1061 with selection on MacConkey-kan agar, and pooled transductants were subject to one additional round of selection by challenge with DL4577 as above. Presumptive CDI-resistant (CDIR) colonies were tested individually for CDI resistance, and tested by PCR for insertions within acrB using oligonucleotide primers 889 and 890. Four CDIR mutants that did not have transposon insertions within acrB maintained CDI resistance after introduction of plasmid pacrAB+ (Augustus et al., 2004) by electroporation. Chromosomal DNA from each of these CDIR mutants was digested with restriction enzyme Hpy99I, which does not cut within the transposon, ligated with Fast-Link Ligase (Epicentre), and introduced into E. coli EC100D pir+ by electroporation with selection on LB-kan plates. DNA sequencing was carried out using KAN-2 FP-1 and R6KAN-2 RP-1 primers from Epicentre. All four mutants contained an insertion 14 bp upstream of the AUG start site of bamA and contained a 9 bp duplication, 5′-GTTAGGAAG-3′, flanking the transposon. This mutation was designated bamA101.
Chemical mutagenesis
Chemical mutagenesis of E. coli DL5311 was carried out using diethyl sulfate (DES, Sigma) with slight modification of the method described by John Roth (University of California, Davis: http://rothlab.ucdavis.edu/protocols/des-mutagenesis.html). Briefly, 50 μl of DES was added to a 10 ml screw-capped tube containing 5 ml of M9 minimal medium lacking a carbon source. The tube was mixed, incubated 10 min at 37°C, and 0.1 ml of a logarithmic phase culture in LB (109 cells/ml) was added to the aqueous phase. After a 45 min incubation at 37°C, 0.1 ml aliquots were transferred to LB (5 ml) and incubated overnight on a tube roller at 37°C. Mutant pools were subjected to two rounds of CDI selection using DL4577 (10:1 inhibitor to target), plating on MacConkey-salicin-cam plates to select for acrB+, hns+ (salicin−phenotype), CDIR mutants. A wzb mutation was transduced into CDIR mutant 1S-12 by phage P1 transduction using E. coli FB23374 wzb::Tn5KAN-I-SceI. The presence of the wzb::Tn5 was confirmed by PCR using oligonucleotides 1085 and 1086.
Construction of Δwzb double mutants
Transduction of wzb::kan into JCM158
wzb::kan (wzb gene replaced with a FRT-flanked kanamycin-resistance cassette) was transduced from E. coli JW2046 into JCM158 cells. To construct JCM158 Δwzb (DL5564), the antibiotic resistance gene was then removed by FLP-mediated recombination as previously described (Cherepanov and Wackernagel, 1995). Briefly, FLP recombinase plasmid pCP20 was transformed into JCM158 wzb::kan (DL5562) and transformants were selected for on LB-amp at 30°C. Individual transformants were then colony-purified on LB (no antibiotics) at 42°C. Cells were then tested for kanamycin, ampicillin, and chloramphenicol sensitivity. Loss of the kanamycin-resistance cassette was verified by PCR using oligonucleotide primers 1085 and 1086. Phage P1 or phage T4GT7 transduction was then used to move various mutations (bamA101, bamB::kan, bamD5::cam, and surA::kan) into JCM158 Δwzb to create the double mutants.
OMP analysis and antibody blocking
Preparation of purified α-Bam and α-Imp antisera
Two different BamA rabbit polyclonal antisera were generated in this study. For the preparation of α-POTRA BamA antibody, a DNA fragment containing codons 21–810 was amplified from MC4100 template with primers 5′-ACACCATATGGCTGAAGGGTTCGTAGTGAA-3′ and 5′-ATATGCGGCCGCTTACTCTTTTACCTTGTAGACGACATC-3′, and digested and inserted into pET28b (Novagen) using NdeI and NotI restriction sites. The plasmid was introduced into BL21(DE3) (Promega) for overexpression of the peptide, which contained five POTRA domains with an N-terminal His-tag and thrombin cleavage site. The protein was purified using a Ni-nitrilotriacetic acid (Ni-NTA) superflow resin (Qiagen), with subsequent removal of the N-terminal His-tag by digestion with thrombin. The peptide was dialyzed against 20 mM Tris pH 8.0 and 150 mM NaCl (TBS). Preparation of the full-length a-BamA antibody proceeded as described above with the following modifications. The primers used to amplify full-length BamA were 5′-ACACCATATGGCTGAAGGGTTCGTAGTGAA-3′ and 5′-ACACGCGGCCGCTTACCAGGTTTTACCGATGTTAAACTG-3′, which was subsequently cloned into pET28b and introduced into BL21(DE3) as above. The protein was expressed as inclusion bodies, which was extracted in 8M urea. The refolding was performed by 20-fold dilution of urea solution into the buffer, 50 mM Tris (pH 8), 1 mM Tris[2-carboxyethyl] phosphine (TCEP), 0.1 mM EDTA, and 0.5% (NN-dimethyldodecylamine N-oxide) (LDAO), and incubation at room temperature overnight. The refolded protein was purified and cleaved with thrombin as above. The gel-filtration was carried out with TBS + 1% OG, and the collected protein solution was dialyzed against TBS. The resulting protein preparations were used to generate rabbit polyclonal antisera (Princeton Animal Facility, Princeton University). Whole serum immunoglobins were purified from the pre-immune and BamA antigen-containing sera using the PURE1A Protein A Antibody Purification kit (Sigma), and subsequently dialyzed against two changes of TBS buffer (1 L) and filter sterilized. For immunoblotting experiments both antibodies were used at a concentration of 1:10,000.
The α-Imp antibody has been previously described (Braun and Silhavy, 2002). Whole serum immunoglobins were purified and filter-sterilized as described above for the BamA antibody preparation. We verified the ability of purified α-Imp to recognize the Imp protein, however our particular batch of antibody detected several more outer membrane proteins than the one previously described (Braun and Silhavy, 2002). This was visualized by immunoblot analysis against fractionated JCM158 samples. The protocol for the fractionation is essentially as described by Osborn et al. (Osborn et al., 1972) with the following modifications. E. coli JCM158 was grown to mid-log in LB medium at 37°C and resuspended in 10 mM Tris (pH 7.5), 5 mM EDTA (pH 7.5), 1 mM PMSF, 0.05 mg/mL DNAse, and 0.1 mg/ml lysozyme. Lysis was achieved by passing the suspension twice through a French-press (14,000 p.s.i.). Unlysed cells were pelleted at 3500 × g for 10 min at 4°C. The lysed sample was transferred to new tube and brought to a concentration of 25% sucrose. A 55–30% step sucrose gradient was poured, and sample was loaded on top of the gradient. All sucrose solutions contained 10 mM Tris and 5 mM EDTA, pH.7.5. Tubes were ultracentrifuged at 25,000 rpm in an SW28 rotor (Beckman Coulter, Inc, Fullerton, CA) for 20 hrs at 4°C (Osborn et al., 1972). The gradient was disassembled from the top in 4 ml fractions, and the sucrose content of each fraction was determined using a refractometer. From these percentages, the density of each fraction was estimated and used to identify the fractions enriched in soluble (ρ = 1.113 g/cc), inner membrane ρ = 1.146 g/cc), and OM proteins (ρ = 1.220 g/cc).
Western blot analysis
Cultures were grown in LB medium overnight at 37°C, with aeration. Cultures were diluted 1:100 in the same medium and grown to mid-exponential phase (OD600 ~0.5). For each sample, 0.5 ml culture was pelleted and resuspended in a volume of sample buffer (70 mM Tris-HCl (pH 6.8), 3% SDS, 10% glycerol, and 5% β-mercaptoethanol) equivalent to OD600/12 (in ml) and boiled for 10 minutes unless otherwise specified. Samples (15 μl each) were loaded onto 10% polyacrylamide gels. Gels were electrophoresed in a Protean II xi cell (Bio-Rad, Hercules, CA) at 110V, with the exception of the Western shown in Fig. 8B (see below), and transferred onto nitrocellulose membranes for 2 hours at 10 volts using a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad) according to protocol. Membranes were hybridized against BamD, DegP and LamB/OmpA antbodies as previously described (Sklar et al., 2007a; Sklar et al., 2007b). Purified α–Imp was used at a dilution of 1:8000, and BamA was detected using purified α–BamA (described above) at a dilution of 1:10,000.
In Fig. 8B, samples were processed using a gentle lysis procedure as previously described (Misra et al., 1991; Wu et al., 2005). Samples were treated with or without boiling for 10 minutes (25°C vs. 100°C) and loaded onto a 10% polyacrylamide gel. OmpA, like many β-barrel proteins, exhibits a heat-modifiable migration pattern in SDS-PAGE experiments. Therefore, samples were electrophoresed at low voltage (50V) at room temperature in order to prevent further thermal denaturation of OmpA in the unheated samples. Both folded and unfolded forms of OmpA were detected as cross-reacting bands of the αLamB monomer antibody (Sklar et al., 2007b; Wu et al., 2005), used at a dilution of 1:15,000.
Membranes hybridized against the indicated primary antibodies were washed and incubated with donkey anti-rabbit IgG horse radish peroxidase conjugate (Amersham) at a 1:8000 dilution. Protein bands were visualized using ECL (Amersham) and exposed onto Hyblot CL autoradiography film (Denville Scientific, Metuchen, NJ).
Antibody blocking experiments
The antibody blocking experiment (Fig. 8A) was carried out as follows. E. coli DL5422 (10 μl of an OD600 = 1.0) was incubated with 20 μl of the purified antibodies indicated and 70 μl of 1× M9 minimal salts (100 μl total volume) using 2 ml conical microcentrifuge tubes. In addition, a DL5430bamA101 CDI-resistant control was included (10 μl DL5430 cells + 90 μl 1× M9 salts) as well as a control (M9 control) receiving 10 μl DL5422 cells and 90 μl 1× M9 salts. After 45 min of incubation at 23°C with rotation at 100 rpm, DL4577 CDI+ inhibitor cells (900 μl of an LB medium culture at OD600 = 0.5) were added, cells were transferred to 16 mm × 150 mm glass tubes (1 ml total volume) and incubated upright at 37°C with rotational shaking at 260 rpm. After 30 min, cells were diluted in 1× M9 minimal salts and plated as described above to select for target cells (CDI Competition Assay). The CDI protection indicated on the y-axis was calculated as [#viable cells after 30 min incubation with DL4577 inhibitor cells/#viable cells at zero time (just prior to addition of inhibitor cells)] divided by the above quotient obtained with the M9 control. This value represents the fold increase in protection against CDI compared to the M9 control.
Immunofluorescence microscopy
Strains to be analyzed were grown in LB medium with appropriate antibiotics to an OD600 of 1.0. 0.2 ml of culture was centrifuged for 1 min at 16,000 × g in 0.6 ml microcentrifuge tubes (Robbins Scientific) and supernatant fluid was removed by vacuum suction using a drawn glass pipette tip. Cell pellets were resuspended by vortexing in either 0.2 ml of 4% formalin in phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate, 2 mM potassium phosphate, pH 7.4), or 0.2 ml of 100% methanol, for 15 min at 23°C. Cells were then washed 2 times in 0.2 ml PBS and then resuspended in 50 μl PBS containing 1% bovine serum albumin (BSA, Fraction V, Sigma). Purified antibodies (10 μl) were added for 30 min at 23°C before washing cells 2 times in PBS and then resuspended in 0.2 ml PBS with 1% BSA. Secondary Alexa Fluor 488 goat α–rabbit antibodies (Invitrogen) were added at a 1:700 to 1:1,000 dilution for 30 min on ice. Cells were washed 2 times in 1% BSA in PBS, and then resuspended in 40 μl PBS. 10 μl to 20 μl of cells were added to the centers of coverslips coated with poly-D-lysine (prepared by spreading a 0.1% poly-D-lysine solution in water onto coverslips for 15 min, rinsing and drying). After 10 min, coverslips were rinsed in water, inverted onto glass slides containing 15–20 μl of SlowFade Gold solution (Invitrogen), and examined by microscopy using a GFP filter (Omega Optical XF116-2). Fluorescence exposures were all taken at a gain of 1.0 with a 0.9 s exposure.
Fluorescence-activated cell analysis (FACS)
FACS analysis was carried out as previously described (Aoki et al., 2005) with the following modifications. CDI+ E. coli DL4905 (GFPmut3+) was mixed with E. coli JCM158 target cells containing plasmid pDAL672 (DsRed+), pre-induced with 0.8 mM IPTG in TB broth (overnight incubation) at a 1 to 4 inhibitor to target ratio. Cells were mixed by shaking at 225 rpm (37°C), and green and red fluorescence quantitated using a FACS Aria (Becton Dickinson) with a 488 nm blue laser and 530 nm and 610 nm filters, respectively.
Supplementary Material
Acknowledgments
We thank F. Blattner, H. Nikaido, R. Misra, and H. Zgurskaya for strains and plasmids. We also thank Seokhee Kim and Daniel Kahne for providing the BamA peptides used for the generation of BamA antibodies. We are very grateful to the National Science Foundation for their generous support of this project (N.S.F. grant 0642052 to D.A.L. and R.E.U. grant to K.J. and B.T), the Department of Homeland Security (DOE contract number DE-AC05-00OR22750 to J.W.) and the National Institutes of General Medical Sciences (GM34821 grant to T.J.S).
References
- Aoki SK, Pamma R, Hernday AD, Bickham JE, Braaten BA, Low DA. Contact-dependent inhibition of growth in Escherichia coli. Science. 2005;309:1245–1248. doi: 10.1126/science.1115109. [DOI] [PubMed] [Google Scholar]
- Augustus AM, Celaya T, Husain F, Humbard M, Misra R. Antibiotic-sensitive TolC mutants and their suppressors. J Bacteriol. 2004;186:1851–1860. doi: 10.1128/JB.186.6.1851-1860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006–0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassler BL, Wright M, Showalter RE, Silverman MR. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol Microbiol. 1993;9:773–786. doi: 10.1111/j.1365-2958.1993.tb01737.x. [DOI] [PubMed] [Google Scholar]
- Bassler BL. How bacteria talk to each other: regulation of gene expression by quorum sensing. Curr Opin Microbiol. 1999;2:582–587. doi: 10.1016/s1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- Bassler BL, Losick R. Bacterially speaking. Cell. 2006;125:237–246. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Bos MP, Robert V, Tommassen J. Functioning of outer membrane protein assembly factor Omp85 requires a single POTRA domain. EMBO Rep. 2007;8:1149–1154. doi: 10.1038/sj.embor.7401092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, Silhavy TJ. Imp/OstA is required for cell envelope biogenesis in Escherichia coli. Mol Microbiol. 2002;45:1289–1302. doi: 10.1046/j.1365-2958.2002.03091.x. [DOI] [PubMed] [Google Scholar]
- Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Clantin B, Delattre AS, Rucktooa P, Saint N, Meli AC, Locht C, Jacob-Dubuisson F, Villeret V. Structure of the membrane protein FhaC: a member of the Omp85-TpsB transporter superfamily. Science. 2007;317:957–961. doi: 10.1126/science.1143860. [DOI] [PubMed] [Google Scholar]
- Coutte L, Willery E, Antoine R, Drobecq H, Locht C, Jacob-Dubuisson F. Surface anchoring of bacterial subtilisin important for maturation function. Mol Microbiol. 2003;49:529–539. doi: 10.1046/j.1365-2958.2003.03573.x. [DOI] [PubMed] [Google Scholar]
- Dole S, Nagarajavel V, Schnetz K. The histone-like nucleoid structuring protein H-NS represses the Escherichia coli bgl operon downstream of the promoter. Mol Microbiol. 2004;52:589–600. doi: 10.1111/j.1365-2958.2004.04001.x. [DOI] [PubMed] [Google Scholar]
- Erni B. The mannose transporter complex: an open door for the macromolecular invasion of bacteria. J Bacteriol. 2006;188:7036–7038. doi: 10.1128/JB.01074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentle IE, Burri L, Lithgow T. Molecular architecture and function of the Omp85 family of proteins. Mol Microbiol. 2005;58:1216–1225. doi: 10.1111/j.1365-2958.2005.04906.x. [DOI] [PubMed] [Google Scholar]
- Gottesman S, Trisler P, Torres-Cabassa A. Regulation of capsular polysaccharide synthesis in Escherichia coli K-12: characterization of three regulatory genes. J Bacteriol. 1985;162:1111–1119. doi: 10.1128/jb.162.3.1111-1119.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166:557–580. doi: 10.1016/s0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- Jacob-Dubuisson F, Fernandez R, Coutte L. Protein secretion through autotransporter and two-partner pathways. Biochim Biophys Acta. 2004;1694:235–257. doi: 10.1016/j.bbamcr.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Jain S, Goldberg MB. Requirement for YaeT in the outer membrane assembly of autotransporter proteins. J Bacteriol. 2007;189:5393–5398. doi: 10.1128/JB.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Beavis R, Novick RP. Bacterial interference caused by autoinducing peptide variants. Science. 1997;276:2027–2030. doi: 10.1126/science.276.5321.2027. [DOI] [PubMed] [Google Scholar]
- Kaiser D. A microbial genetic journey. Annu Rev Microbiol. 2006;60:1–25. doi: 10.1146/annurev.micro.60.080805.142209. [DOI] [PubMed] [Google Scholar]
- Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, Kahne D. Structure and function of an essential component of the outer membrane protein assembly machine. Science. 2007;317:961–964. doi: 10.1126/science.1143993. [DOI] [PubMed] [Google Scholar]
- Lescop E, Hu Y, Xu H, Hu W, Chen J, Xia B, Jin C. The solution structure of Escherichia coli Wzb reveals a novel substrate recognition mechanism of prokaryotic low molecular weight protein-tyrosine phosphatases. J Biol Chem. 2006;281:19570–19577. doi: 10.1074/jbc.M601263200. [DOI] [PubMed] [Google Scholar]
- Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhov AM, Hannah JH, Brennan MJ, Trus BL, Kocsis E, Conway JF, Wingfield PT, Simon MN, Steven AC. Filamentous hemagglutinin of Bordetella pertussis. A bacterial adhesin formed as a 50-nm monomeric rigid rod based on a 19-residue repeat motif rich in beta strands and turns. J Mol Biol. 1994;241:110–124. doi: 10.1006/jmbi.1994.1478. [DOI] [PubMed] [Google Scholar]
- Malinverni JC, Werner J, Kim S, Sklar JG, Kahne D, Misra R, Silhavy TJ. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol. 2006;61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- Mazar J, Cotter PA. Topology and maturation of filamentous haemagglutinin suggest a new model for two-partner secretion. Mol Microbiol. 2006;62:641–654. doi: 10.1111/j.1365-2958.2006.05392.x. [DOI] [PubMed] [Google Scholar]
- Mazar J, Cotter PA. New insight into the molecular mechanisms of two-partner secretion. Trends Microbiol. 2007;15:508–515. doi: 10.1016/j.tim.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Misra R, Peterson A, Ferenci T, Silhavy TJ. A genetic approach for analyzing the pathway of LamB assembly into the outer membrane of Escherichia coli. J Biol Chem. 1991;266:13592–13597. [PubMed] [Google Scholar]
- Nikaido H, Zgurskaya HI. AcrAB and related multidrug efflux pumps of Escherichia coli. J Mol Microbiol Biotechnol. 2001;3:215–218. [PubMed] [Google Scholar]
- Ochman H, Gerber AS, Hartl DL. Genetic applications of an inverse polymerase chain reaction. Genetics. 1988;120:621–623. doi: 10.1093/genetics/120.3.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, Gander JE, Parisi E, Carson J. Mechinism of assembly of the outer membrane of Salmonella typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J Biol Chem. 1972;247:3962–3972. [PubMed] [Google Scholar]
- Raivio TL. Envelope stress responses and Gram-negative bacterial pathogenesis. Mol Microbiol. 2005;56:1119–1128. doi: 10.1111/j.1365-2958.2005.04625.x. [DOI] [PubMed] [Google Scholar]
- Ruiz N, Wu T, Kahne D, Silhavy TJ. Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem Biol. 2006;1:385–395. doi: 10.1021/cb600128v. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pulido L, Devos D, Genevrois S, Vicente M, Valencia A. POTRA: a conserved domain in the FtsQ family and a class of beta-barrel outer membrane proteins. Trends Biochem Sci. 2003;28:523–526. doi: 10.1016/j.tibs.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–1298. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev. 1989;53:1–24. doi: 10.1128/mr.53.1.1-24.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar JG, Wu T, Gronenberg LS, Malinverni JC, Kahne D, Silhavy TJ. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc Natl Acad Sci U S A. 2007a;104:6400–6405. doi: 10.1073/pnas.0701579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar JG, Wu T, Kahne D, Silhavy TJ. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 2007b;21:2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledjeski D, Gottesman S. A small RNA acts as an antisilencer of the H-NS-silenced rcsA gene of Escherichia coli. Proc Natl Acad Sci U S A. 1995;92:2003–2007. doi: 10.1073/pnas.92.6.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DL, James CE, Sergeant MJ, Yaxian Y, Saunders JR, McCarthy AJ, Allison HE. Short-tailed stx phages exploit the conserved YaeT protein to disseminate Shiga toxin genes among enterobacteria. J Bacteriol. 2007;189:7223–7233. doi: 10.1128/JB.00824-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli Form a Stable Intermembrane Multidrug Efflux Complex. J Biol Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- Tomasz A. Control of the competent state in Pneumococcus by a hormone-like cell product: an example for a new type of regulatory mechanism in bacteria. Nature. 1965;208:155–159. doi: 10.1038/208155a0. [DOI] [PubMed] [Google Scholar]
- Tommassen J. Getting into and through the outer membrane. Science. 2007;317:903–904. doi: 10.1126/science.1146518. [DOI] [PubMed] [Google Scholar]
- Vincent C, Duclos B, Grangeasse C, Vaganay E, Riberty M, Cozzone AJ, Doublet P. Relationship between exopolysaccharide production and protein-tyrosine phosphorylation in gram-negative bacteria. J Mol Biol. 2000;304:311–321. doi: 10.1006/jmbi.2000.4217. [DOI] [PubMed] [Google Scholar]
- Werner J, Misra R. YaeT (Omp85) affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol Microbiol. 2005;57:1450–1459. doi: 10.1111/j.1365-2958.2005.04775.x. [DOI] [PubMed] [Google Scholar]
- White-Ziegler CA, Angus Hill ML, Braaten BA, van der Woude MW, Low DA. Thermoregulation of Escherichia coli pap transcription: H-NS is a temperature-dependent DNA methylation blocking factor. Mol Microbiol. 1998;28:1121–1137. doi: 10.1046/j.1365-2958.1998.00872.x. [DOI] [PubMed] [Google Scholar]
- Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell. 2005;121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.