

Abstract
The delivery of platelet-derived growth factor (PDGF) for tissue engineering of skin and periodontal wounds has become an active area of interest. However, little is known regarding the extended effects of PDGF on cell signaling via gene therapy and how such an approach facilitates the exiting of cells from growth arrest and entry to competence required for cell cycling. We show in vitro expression and secretion of PDGF-AA by recombinant adenovirus encoding the PDGF-A gene (Ad-PDGF-A). The bioactive PDGF-AA protein released induces sustained downregulation of PDGFαR that is encoded by a growth arrest-specific (gas) gene. Ad-PDGF-A induces sustained phosphorylation of PDGFαR as well as prolonged phosphorylation of downstream extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Furthermore, the phosphorylation of PDGFαR is abolished by cotransducing cells with adenovirus encoding a dominant negative mutant of the PDGF-A gene that disrupts PDGF bioactivity. These findings demonstrate the prolonged effects of adenoviral delivery of PDGF and aid in the better understanding of sustained PDGF signaling.
Keywords: platelet-derived growth factor, growth arrest, extracellular signal-regulated kinase, Akt/protein kinase B
PLATELET-DERIVED GROWTH FACTOR (PDGF) is a potent mitogen for cells of mesenchymal origin and possesses pluripotential effects on wound healing (15, 17). Recently, multiple investigators have studied the promotion of tissue repair using plasmid DNA and adenoviral approaches for PDGF gene delivery (5, 9, 10, 12). In addition to the well-described PDGF-A and -B chains, two new members of this family, PDGF-C and PDGF-D, were recently identified (2, 23, 24). Two PDGF receptors (PDGFRs) also have been comprehensively described, PDGFαR and PDGFβR (6, 15). In this work we have focused on PDGF-AA, which selectively binds to and activates PDGFαR receptor homodimerization and the triggering of several signaling cascades (15, 17).
PDGFαR protein binds to PDGF-AA, -AB, -BB, and -CC isoforms and has a central role in mediating the state of competence induced by PDGF (6, 7). In addition to its traditional role as a receptor tyrosine kinase, PDGFαR was later identified as being encoded by a growth arrest-specific (gas) gene (25). Gas genes are preferentially expressed when cell division in culture is prevented by serum deprivation. In principle, gas genes encode products that cause growth arrest or, conversely, can be expressed persistently during the growth-arrested state (G0) because factors required for cell cycling are lacking (3, 28). It has been shown that expression of PDGFαR, which is important in cellular mitogenic responses and early stage embryogenesis following growth arrest (27), may facilitate the cell cycling that occurs following the addition of PDGF (25).
In regard to cell signaling, it is traditionally believed that growth factor-stimulated signaling events including PDGF signal transduction occur transiently. However, it is difficult to reconcile how PDGF as a mitogen contributes to cell cycle entry, which occurs many hours later. Harrington et al. (14) first demonstrated the possibility of “late” PDGF receptor phosphorylation following PDGF exposure under specific experimental conditions. More recent cumulative evidence suggests that growth factor-dependent signaling is not restricted to the 1- to 2-h time frame subsequent to stimulation as has been generally thought for many years. (1, 19, 20). PDGF triggers a second wave of “late” phosphorylation (i.e., phosphatidylinositol 3-kinase and protein kinase C activities) that occur ˜ 2-7 h after PDGF exposure. The second phase of phosphorylation events appears critical for cellular proliferation (32). In our study, we observed time periods beyond the late phase of phosphorylation for up to 96 h after PDGF treatment. We utilized gene transfer of adenovirus encoding PDGF-A (Ad-PDGF-A) to continuously produce PDGF-AA protein to study the signaling events and PDGFαR expression as a measure of traversing cell cycle growth arrest. We have shown that Ad-PDGF-A prolongs phosphorylation of extracellular signal-regulated kinase (ERK), Akt, and the correlative sustained downregulation of PDGFαR, a gas gene product.
MATERIALS AND METHODS
Adenovirus construction
We have previously described the construction of adenoviruses encoding the PDGF-A and PDGF-1308 (dominant negative mutant of PDGF-A) genes (37). In brief, the full-length murine PDGF-A or PDGF-1308 cDNA (gifts of Dr. C. D. Stiles, Boston, MA) was subcloned into a shuttle plasmid (obtained from Genzyme, Cambridge, MA) under the control of the cytomegalovirus promoter. The precut viral backbone DNA Ad2/EGFP (encoding green fluorescent protein) and the shuttle plasmid containing either PDGF-A or PDGF-1308 cDNA were linearized by restriction enzyme digestion. The linearized shuttle plasmid and viral backbone DNA were cotransfected into 293 packaging cells. Recombination between the shuttle plasmid and the GFP viral backbone resulted in replacement of the GFP cDNA with PDGF-A or PDGF-1308 cDNA. Subsequent recombinant viral plaques were identified, picked, and purified. Titers of the virus stocks were determined on 293 cells by plaque assay and expressed as the number of plaque-forming units per ml.
Cell culture and gene transfer
Primary cultures of rat dermal fibroblasts (DFs; kindly donated by Dr. R. B. Rutherford, University of Michigan) were utilized as previously described for ex vivo gene transfer (22). Low-passage cells were plated in six-well culture dishes containing DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin/streptomycin, and 2 mM glutamine. Subconfluent cultures were then brought to a stage of quiescence by washing with PBS and reducing the serum concentration to 0.1% for 48 h prior to adenovirus transduction or recombinant human PDGF-AA (rhPDGF-AA; Upstate Biotechnology, Lake Placid, NY) treatment. In all experiments, cells were treated either with recombinant adenoviruses (Ad-GFP, Ad-PDGF-A or Ad-PDGF-1308) for 4-5 h at a multiplicity of infection (MOI) of 100 or with 20 ng/ml rhPDGF-AA. Cells were incubated for various time periods (see below) depending on the experiment and were harvested at 8, 24, 48, and 96 h.
Measurement of PDGF-AA after transduction with Ad-PDGF-A
To determine the production of PDGF-AA protein following gene transfer, we infected cells with Ad-PDGF-A as described in Cell culture and gene transfer. The conditioned cell culture medium was harvested 24 and 96 h after Ad-PDGF-A transduction and was centrifuged to remove cell debris. Phenylmethylsulfonyl fluoride (PMSF; 1 mM; Sigma Chemical, St. Louis, MO) was added to the conditioned medium. The medium was then transferred to molecular porous membrane tubing (Spectrum Laboratories, Rancho Dominguez, CA) and dialyzed at 4°C overnight while deionized H2O containing 0.1 mM PMSF was gently stirred in. The samples were lyophilized and then suspended in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA) containing β-mercaptoethanol for subsequent analysis by Western blotting techniques as previously described (11). In brief, samples were resolved on SDS-polyacrylamide gels and electrophoretically transferred to polyvinylidene difluoride membranes. Membranes were blocked for 1 h in PBS containing 5% nonfat dry milk, followed by incubation for 1 h with polyclonal anti-PDGF-A as the primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Donkey anti-rabbit Ighorseradish peroxidase conjugate (Amersham Life Science, Amersham, UK) was added as the secondary antibody. In experiments in which a monoclonal antibody was used as primary antibody, sheep anti-mouse Ig-horseradish peroxidase conjugate (Amersham Life Science) was added as the secondary antibody. Specific immunoreactive protein bands were detected by using an enhanced chemiluminescence reagent (ECL; Amersham Pharmacia Biotech, Piscataway, NJ).
Measurement of PDGFαR expression
Cells were washed twice with ice-cold PBS and then lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris·HCl, pH 7.3, 150 mM NaCl, 0.25 mM EDTA, pH 8.0, 1% Triton X-100, 1% C24H39O4Na, 0.2% NaF, 0.1% Na3VO4, 1 mM PMSF, 5 µg/ml aprotinin, and 20 µM leupeptin). Protein concentrations of cell lysates were normalized using a protein assay kit (Bio-Rad). Equal amounts of proteins were processed for Western blotting analysis with PDGFαR (Santa Cruz Biotechnology) as primary antibody. In addition, α-tubulin (Sigma) was measured to show the corresponding equal loading of cell lysates. Densitometric scanning was performed by normalizing PDGFαR expression to α-tubulin to demonstrate relative changes in PDGFαR expression following PDGF-AA exposure, using NIH Image analysis software (National Institutes of Health, Bethesda, MD).
Measurement of phosphorylation of PDGFαR, ERK1/2, and Akt
For phosphotyrosine analysis of immunoprecipitated PDGFαR, 200 µg of total protein from cell lysates were incubated with 1 µg of PDGFαR antibody (Upstate) for 2 h at 4°C. Subsequently, 30 µl of protein A-agarose suspension beads were added to capture the immunocomplexes for 1 h at 4°C. Agarose immunocomplexes were then centrifuged and resuspended for phosphotyrosine Western blot analysis. Anti-phosphotyrosine antibodies PY20 (Santa Cruz Biotechnology) and 4G10 (Upstate) were combined and used to detect the presence of tyrosine phosphorylated PDGFαR. The activated form of ERK1/2 was tested by using a specific anti-phospho-ERK1/2 antibody (Cell Signaling Technology, Beverly, MA) that recognized only the activated forms phosphorylated on Thr202 and Tyr204, and the activated form of protein kinase B (PKB)α/Akt was tested by using a specific anti-phospho-Akt antibody (Cell Signaling) that recognized only the activated forms phosphorylated on Ser473. ERK1, PKBα/Akt (BD Transduction Laboratories, Lexington, KY), and α-tubulin proteins were measured to show the relative protein loading for each of the lanes.
Cell cycle regulation by Ad-PDGF-A: flow cytometry and assessment of cyclin D1 and cyclin E
To determine cell cycle distribution, DF cell monolayers were harvested by trypsinization, washed once with cold PBS, stained and hypotonically treated with 500 µl of propidium iodide hypotonic lysis buffer [0.1% sodium citrate, 0.1% Triton X, 100 µg/ml RNase A (Sigma), and 50 µg/ml propidium iodide (Sigma)], and analyzed by flow cytometry as previously described by using 488-nm excitation (19). In addition, cell lysates in RIPA buffer were tested for the expression of cyclin D1 and cyclin E with anti-cyclin D1 or anti-cyclin E antibodies (Santa Cruz Biotechnology) by using Western blotting analysis.
Coinfection of cells with Ad-PDGF-A and Ad-PDGF-1308
To determine the reversibility and specificity of Ad-PDGF-A on signal transduction, we coinfected DFs with Ad-GFP or Ad-PDGF-1308 with Ad-PDGF-A. The cells were initially transduced with Ad-PDGF-1308 or Ad-GFP (MOI = 300) 24 h before infection with Ad-PDGF-A or Ad-GFP (MOI = 100) for an additional 24 h. Cell lysates were harvested, and PDGFαR expression and PDGFαR tyrosine phosphorylation were assessed by immunoprecipitation using PDGFαR antibody.
RESULTS
Expression of PDGF-AA by Ad-PDGF-A gene transfer
To examine whether Ad-PDGF-A-transduced cells produce and secrete bioactive PDGF-A protein, we collected conditioned media from transduced cells and then immunoprobed for PDGF-A by Western blot analysis. To choose the appropriate media collection time for maximal protein release, we performed a fluorescence-activated cell sorting (FACS) time course using the control virus Ad-GFP to measure protein expression kinetics. FACS analysis revealed initial GFP fluorescence as early as 6-8 h after Ad-GFP exposure, with > 95% of the cells exhibiting GFP protein by 20-24 h after transduction with a MOI of 100 (data not shown). Therefore, conditioned media were analyzed at 24 and 96 h for PDGF-AA protein secretion.
Figure 1 demonstrates a Western blot of protein that was concentrated and lyophilized from medium that was probed with a monospecific PDGF-A antibody (Santa Cruz Biotechnology). Ad-PDGF-A-transduced cells produced immunoreactive protein that exhibited the same electrophoretic mobility as purified PDGF-A peptide on β-mercaptoethanol-reduced samples. Using these Western blot data and correcting for sample concentration after lyophilization and resuspension, we estimate that DFs transduced with Ad-PDGF-A (MOI = 100) produced ˜200 ng of PDGF-AA protein per 106 cells in serum-free conditions.
Fig. 1.
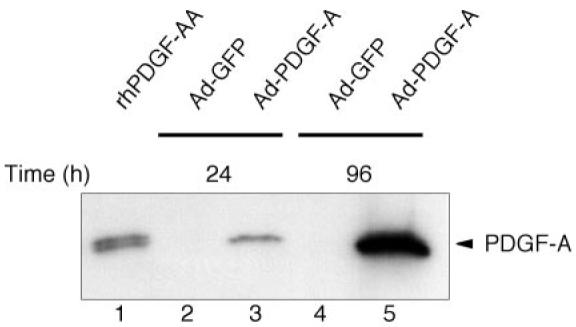
In vitro production of platelet-derived growth factor (PDGF)-AA by adenovirus encoding PDGF-A (Ad-PDGF-A). After 24 h of serum starvation, subconfluent dermal fibroblasts (DFs) were transduced with either Ad-PDGF-A or Ad-GFP (green fluorescent protein). Conditioned medium was collected after 24 and 96 h of culture followed by dialysis, concentration, and lyophilization. The lyophilized samples were resuspended in reducing sample buffer containing β-mercaptoethanol and were analyzed by Western blotting with anti-PDGF-A polyclonal antibody. Recombinant human PDGF-AA (rhPDGF-AA; 5 ng) was loaded in lane 1 as control. Note the continued accumulation of PDGF-AA in cultures from 1 to 4 days after PDGF-A gene transfer (lanes 2-5).
Prolonged downregulation of PDGFαR by Ad-PDGF-A gene transfer
To determine whether continuous treatment with PDGF via adenovirus results in prolonged effects, we examined the downregulation of PDGFαR that results from sustained ligand binding and activation. Cells treated with the control virus Ad-GFP (Fig. 2, lanes 1-6), serum-free medium (data not shown) or Ad-PDGF-1308 (data not shown) did not demonstrate any measurable changes in PDGFαR expression over the 96-h observation period. PDGF-AA treatment revealed a potent reduction in PDGFαR protein levels within 2-3 h after treatment (data not shown), and this diminution was sustained for at least 24 h before it returned to baseline levels at 48 h (Fig. 2, lanes 7-12). On the other hand, cells treated with Ad-PDGF-A resulted in prolonged downregulation of PDGFαR for at least 96 h (Fig. 2, lanes 13-18). This reduction of PDGFαR was first noted at 24 h and was sustained for 96 h. The lag in PDGFαR downregulation by Ad-PDGF-A was presumably due to the time required for PDGF-AA protein expression (Fig. 2, lanes 13-18). These results suggest that the brief exposure of cells to PDGF-AA results in a transient (∼24 h) reduction in PDGFαR levels, whereas adenoviral delivery results in constant downregulation of the PDGFαR for at least 96 h.
Fig. 2.

Gene delivery of Ad-PDGF-A promotes downregulation of the growth arrest-specific (gas) gene product PDGF α-receptor (PDGFαR). A: DFs were transduced with either Ad-GFP [multiplicity of infection (MOI) = 100] or Ad-PDGF-A (MOI = 100) or exposed to 20 ng/ml rhPDGF-AA for 0, 8, 24, 48, and 96 h. Cells were lysed, and 200 µg of total protein from cell lysates were subjected to anti-αPDGFR Western blotting in which a monospecific antibody to PDGFαR was used. The upper band represents the mature isoform of the receptor, whereas the weaker lower band is consistent with the immature, nonglycosylated receptor. Western blots were then stripped and reprobed with anti-α-tubulin to determine the relative equal loading of protein. B: relative protein expression levels were normalized to α-tubulin expression and are depicted by densitometric scanning using NIH Image analysis software. There is little relative change in PDGFαR expression with Ad-GFP treatment (lanes 1-6), whereas rhPDGF-AA treatment caused a transient reduction in PDGFαR for 8-24 h (lanes 8 and 9) followed by a rebound in baseline receptor levels (lanes 10-12). Ad-PDGF-A treatment resulted in a slightly delayed but sustained downregulation of PDGFαR for up to 96 h (lanes 13-18).
Coordination of sustained phosphorylation of ERK and Akt pathways with the sustained tyrosine phosphorylation of PDGFαR
The goal of these experiments was to identify the effects on signaling elicited by continuous PDGF-AA treatment. Binding of ligand to tyrosine receptor kinase results in an elevation of activity of a receptor kinase and a triggering of downstream signal transduction pathways. Because the prolonged adenoviral protein expression was shown previously, we sought to determine whether the adenoviral PDGF-A protein is as biologically active as rhPDGF-AA and whether adenoviral delivery could extend intracellular signaling events; we therefore evaluated the phosphorylation of PDGFαR and several key pathways involved in PDGF signaling. Our results show that Ad-PDGF-A induces the phosphorylation of PDGFαR by 8 h after gene delivery and that the phosphorylation increases gradually and is maintained over 96 h (Fig. 3A, lanes 7-12). The phosphorylation induced by PDGF-AA peptide was sustained and weakened gradually by 72 h (Fig. 3A, lanes 1-6).
Fig. 3.

Phosphorylation of PDGFαR by Ad-PDGF-A leads to sustained downstream activation of extracellular signal-regulated kinase (ERK) and Akt. Cell lysates treated with either 20 ng/ml of PDGF-AA (lanes 1-6) or Ad-PDGF-A (lanes 7-12) for 8-96 h were immunoprecipitated with anti-PDGFαR (A) and subjected to SDS-PAGE gel electrophoresis followed by phosphotyrosine assessment (p-PDGFαR) with Western blot analysis. The blots were probed with both α-PY20 (1:1,000) and 4G10 (1:1,000) antibodies. The same cell lysates also were probed with phospho-ERK1/2 (p-ERK1/2; B) and phospho-Akt (p-Akt; C). Relative levels of ERK, Akt, and α-tubulin were determined by normalization for relative expression levels. Note the extended phosphorylation of cells treated with Ad-PDGF-A compared with rhPDGF-AA exposure. Also, observe the corresponding minimal alteration in relative levels of α-tubulin, ERK1/2, and Akt to respective phosphorylated proteins.
Multiple signaling pathways might cooperate in promoting cell growth. To test this hypothesis, we examined the activation of ERK1/2 and Akt pathways. Our results showed that in cells exposed to rhPDGF-AA, phosphorylation of ERK1/2 and Akt increased by 8 h compared with basal levels and was sustained for a period of 48-72 h (Fig. 3, B and C, lanes 1-6). The phosphorylation of ERK1/2, which peaked at 72 h, was apparently more sustained than that of PDGFαR, which peaked at 24 h. In DFs treated with Ad-PDGF-A, the phosphorylation of ERK1/2 and Akt has phosphorylation curve patterns similar to that of PDGFαR itself, which remained elevated for at least 96 h and to a greater relative extent (Fig. 3, B and C, lanes 7-12).
Our cell cycle study by propidium iodide hypotonic lysis method failed to demonstrate cell progression to S phase after induction with rhPDGF-AA for up to 16 h, whereas the majority of quiescent cells entered S phase after induction with 10% FBS (data not shown). Also, addition of 100 ng/ml insulin-like growth factor-I (IGF-I) protein induced cells treated with PDGF-AA or Ad-PDGF-A to enter into S phase at a level about twofold greater than that of PDGF alone (data not shown). Moreover, we found that there was enhancement of cyclin D1 and cyclin E protein levels as early as 8 h after either infection by Ad-PDGF-A or induction with rhPDGF-AA. However, no alteration of cyclin D1 and E levels was observed during the time frame from 8 to 96 h for either Ad-PDGF-A or rhPDGF-AA treatment (data not shown). These findings suggest no difference between pulse (protein delivery) and sustained (adenovirus delivery) treatment on cyclins D1 and E levels over 4 days.
Competition between PDGF-1308 and PDGF-A on receptor tyrosine phosphorylation
To investigate the reversibility of PDGF-A signaling events, we performed a competition study on receptor tyrosine phosphorylation between PDGF-1308 and PDGF-A. In this experiment, DFs were infected with Ad-PDGF-1308 24 h before Ad-PDGF-A or Ad-GFP transduction to “prime” the cells with PDGF-1308 protein. Figure 4 demonstrates that PDGF-1308 (MOI = 300) completely abolished the phosphorylation of PDGFαR induced by Ad-PDGF-A (MOI = 100), whereas Ad-PDGF-1308 (MOI = 100-200) only partially inhibited PDGFαR tyrosine phosphorylation (data not shown). Interestingly, coinfection of Ad-PDGF-1308 (MOI = 100-300) and Ad-PDGF-A (MOI = 100) could not affect PDGFαR protein levels elicited by Ad-PDGF-A (data not shown). This datum suggests that the mechanisms of PDGF-1308 antagonism with native PDGF-A chains may not prevent dimer association with the PDGFαR to alter receptor expression but does disrupt the ability of PDGF-A isoforms to elicit functional PDGFαR tyrosine phosphorylation.
Fig. 4.

Dominant negative mutant PDGF-1308 abolishes Ad-PDGFA-mediated PDGFαR tyrosine phosphorylation. A: DFs were initially transduced with Ad-PDGF-1308 for 24 h followed by infection with Ad-PDGF-A or Ad-GFP for an additional 24 h. Cell lysates were immunoprecipitated with anti-PDGFαR and Western blotted with combined 4G10 and PY20 anti-phosphotyrosine antibodies. B: note the near complete abolishment of PDGFαR tyrosine phosphorylation by comparing Ad-PDGF-A (lane 2) and coinfection with Ad-PDGF-A and Ad-PDGF-1308 (lane 4). The ratio of Ad-PDGF-A to Ad-PDGF-1308 was 3:1 as shown in lane 4. Ad-GFP was used as a negative control. All samples were infected with a total adenovirus MOI of 400.
DISCUSSION
Despite the active interest in utilizing long-term PDGF delivery to stimulate tissue repair, there is minimal information available regarding the signal transduction mechanisms involved following PDGF gene transfer. Our observations have provided evidence suggesting that prolonged PDGF-A expression by adenovirus allows for continued downregulation of PDGFαR, which results from sustained activation by ligand binding. Our findings also illustrate the sustained stimulation of several signaling pathways; notably, activation of ERK1/2 and Akt, which coincides with prolonged activation of PDGFαR, may contribute to the mechanisms of growth-arrest exiting. In addition, this is the first study to demonstrate the sustained effects of PDGF-AA on signal transduction by Ad-PDGF-A and its antagonist, Ad-PDGF-1308.
In serum-free conditions, cells are induced to leave the cell cycle and enter a state of growth arrest, where they remain unless triggered to reenter the cycle by mitogenic signals. Certain genes are known to be preferentially or selectively expressed during growth arrest and are called gas genes (3, 28). PDGFαR was shown to be a gas gene by use of the lacZ gene to identify chromosomal loci that are transcriptionally active during the growth arrest of NIH/3T3 cells (25) or cell cycle repression genes by microarray (8). Lih et al. (25) demonstrated that PDGFαR is not induced in NIH/3T3 cells cultured in serum-deprived medium when transformed with erb2, src, or raf, all of which block growth arrest under conditions of serum deprivation. Their results indicated that regulation of PDGFαR expression is governed by cell cycling, rather than the presence or absence of serum growth factors per se. The addition of PDGF-AA protein to growth-arrested cells enables progression in the cell cycle. While such PDGF-stimulated cells become “competent” for cycling, progression through G1 and entry into S phase require additional factors usually present in serum or in platelet-poor plasma in “traditional” cell types such as BALB/c 3T3 cells (15). However, recently it was found that in certain cell types such as AKR-2B (32) and NIH/3T3 (1, 19, 20), PDGF-BB alone could function not only as a competence factor but also as a progression factor when it was continuously present in the medium. Our studies have shown that the DF is more like a traditional cell type, where PDGF-AA functions solely as competence factor. Our data show that adenovirus produces PDGF-AA dimers continuously and that this prolonged expression of PDGF activates and downregulates PDGFαR for extended time periods. Nevertheless, this sustained PDGF expression by adenovirus cannot drive cells to progress beyond the G1/S boundary without progression factors. Using propidium iodide staining, we failed to demonstrate cell cycle progression into S phase following treatment with PDGF-AA for up to 16 h unless either serum or IGF-I was added to the medium (data not shown). Although PDGF alone did not elicit DNA synthesis, we believe these data support the potential efficacy of PDGF gene delivery for tissue repair. In these studies serum deprivation conditions were used to specifically examine effects of long-term PDGF delivery in vitro (which avoids the effects of other growth factors or cytokines in serum). However, in vivo gene therapy approaches would result in much different results under physiological conditions, where there are various other growth factors, cytokines, etc., that collaborate with PDGF in cell cycle progression. Furthermore, this study focused on mitogenic signal transduction events elicited by PDGF. PDGF is known to be not only a strong mitogen but also a chemoattractant, a potent stimulator of matrix biosynthesis and granulation tissue formation (17). Therefore, the pleotropic effects of PDGF highlight its efficacy as a strong promoter of wound repair in vivo using gene therapy (5, 9, 10).
It is well documented that sustained receptor activation by ligand induces receptor downregulation. This phenomenon results mainly from receptor internalization such as receptor endocytosis and intracellular degradation process as effects of a ligand autocrine system. The downregulation may provide a mechanism for desensitizing the cells to subsequent stimulation by that ligand (4, 16, 18, 21, 33). The sustained downregulation of PDGFαR by Ad-PDGF-A in our study is therefore an expected result of sustained activation of PDGFαR by continuous PDGF-AA production and binding. However, several unexpected findings show that sustained stimulation of cells by mitogenic growth factors or certain transforming oncogenes induce receptor downregulation not only at the protein level but also at the mRNA level via reduction of mRNA transcription (25, 26, 34, 36). It has been suggested that the state of growth arrest (G0) is associated with enhanced expression of PDGFαR and/or PDGFβR with corresponding repression of these receptors at both the mRNA and protein levels in normal cells after growth factor stimulation or in oncogene-transformed cells (25, 34). For instance, Vaziri and Faller (34) tested the regulation of PDGFαR in BALB/c 3T3 cells by using basic fibroblast growth factor (FGF)-2 instead of PDGF and found that both mRNA and protein of PDGFβR were downregulated. We also noted similar results on the downregulation of PDGFαR by FGF-2 in our primary DFs by semiquantitative PCR (data not shown). Together, these findings are important to elucidate the role of PDGFαR in cell cycling and better understand the phenomenon of PDGFαR downregulation.
We and others have shown that PDGF stimulates sustained ERK activity (35). Induction of activated MEK (mitogen-activated protein kinase/ERK kinase) also has been revealed to elevate cyclin D1 levels (35). Whether activated MEK alone is sufficient to trigger DNA synthesis needs further investigation. Our results showed that there was the expected early induction of cyclin D1 and cyclin E protein levels as early as 8 h after treatment with either Ad-PDGF-A or PDGFAA; however, these levels remained relatively constant throughout the 96-h observation period (data not shown). Cyclin D1 and cyclin E are characterized as G1 proteins because they increase when reentering the cell cycle from G0. Cyclins D1 and E are further degraded after their active combination with their specific cyclin-dependent kinases (13, 31). Given the lack of significant alterations in cyclin D1 and E levels over time, we speculate that minimal DNA synthesis occurs when DFs are exposed to PDGF-AA or Ad-PDGF-A in the absence of other serum or plasma factors despite the fact that Ad-PDGF-A prolongs activation of ERK1/2 and Akt. Here we have shown that the extended ERK and Akt activities correlate with the sustained tyrosine phosphorylation of PDGFαR and the continued downregulation of PDGFαR in G1, suggesting a necessary link to mitogen-induced progression through G1. However, such signaling events are insufficient to commit cells beyond the G1/S boundary. Further study of other more downstream signaling molecules following PDGF exposure may aid in the better understanding of S phase entry in the cell cycle. Moreover, future gene delivery approaches using other vectors and evaluating additional growth factors would be important to a better understanding of sustained signaling using viral vectors or plasmid DNA. To date, little has been evaluated to determine what other effects these vector strategies may play in modulating signal transduction in vitro and in vivo.
Furthermore, we have shown that a dominant negative form of PDGF-A, PDGF-1308, can compete with the wild-type growth factor in PDGFαR activation. PDGF-1308, generated by site-directed mutagenesis (cysteine-129 to serine), reduces both homo- and heterodimer secretion of PDGF by forming inactive or unstable heterodimers with wild-type A and B subunits (29, 30). We used adenoviral infection to show the antagonistic effect of PDGF-1308 in terms of receptor activation. We found that coinfection of Ad-PDGF-A and PDGF-1308 failed to reverse the downregulation of the PDGFαR elicited by continuous PDGF-A exposure (data not shown). However, when we evaluated the perturbation in receptor tyrosine phosphorylation, we demonstrated that Ad-PDGF-A blocked the Ad-PDGF-A mediation of PDGFαR downregulation. Hence, PDGF-1308 acts by preventing the transduction of a signal while having no effect on altering PDGFαR protein levels.
In conclusion, the results from this study suggest that delivery of the PDGF-A gene by adenovirus elicits the production of PDGF-AA protein with extended effects on PDGFαR downregulation and tyrosine phosphorylation. Furthermore, Ad-PDGF-A also results in significant prolongation of phosphorylation of two key signaling molecules, ERK1/2 and Akt, over time.
Acknowledgments
This study was funded by National Institute of Dental and Craniofacial Research Grants DE-11960 and DE-13397 (to W. V. Giannobile). FACS experiments were performed at the University of Michigan Flow Cytometry Facility.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Balciunaite E, Jones S, Toker A, Kazlauskas A. PDGF initiates two distinct phases of protein kinase C activity that make unequal contributions to the G0 to S transition. Curr Biol. 2000;10:261–267. doi: 10.1016/s0960-9822(00)00358-4. [DOI] [PubMed] [Google Scholar]
- 2.Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, Alitalo K, Eriksson U. PDGF-D is a specific, protease-activated ligand for the PDGF β-receptor. Nat Cell Biol. 2001;3:512–516. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- 3.Brenner DG, Lin-Chao S, Cohen SN. Analysis of mammalian cell genetic regulation in situ by using retrovirus-derived “portable exons” carrying the Escherichia coli lacZ gene. Proc Natl Acad Sci USA. 1989;86:5517–5521. doi: 10.1073/pnas.86.14.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter G, Cohen S. 125I-labeled human epidermal growth factor. Binding, internalization, and degradation in human fibroblasts. J Cell Biol. 1976;71:159–171. doi: 10.1083/jcb.71.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandler LA, Doukas J, Gonzalez AM, Hoganson DK, Gu DL, Ma C, Nesbit M, Crombleholme TM, Herlyn M, Sosnowski BA, Pierce GF. FGF2-targeted adenovirus encoding platelet-derived growth factor-B enhances de novo tissue formation. Mol Ther. 2000;2:153–160. doi: 10.1006/mthe.2000.0102. [DOI] [PubMed] [Google Scholar]
- 6.Claesson-Welsh L. Cytokines. Karger; Basel: 1993. pp. 31–43. [Google Scholar]
- 7.Claesson-Welsh L. Platelet-derived growth factor receptor signals. J Biol Chem. 1994;269:32023–32026. [PubMed] [Google Scholar]
- 8.Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doukas J, Chandler LA, Gonzalez AM, Gu D, Hoganson DK, Ma C, Nguyen T, Printz MA, Nesbit M, Herlyn M, Crombleholme TM, Aukerman SL, Sosnowski BA, Pierce GF. Matrix immobilization enhances the tissue repair activity of growth factor gene therapy vectors. Hum Gene Ther. 2001;12:783–798. doi: 10.1089/104303401750148720. [DOI] [PubMed] [Google Scholar]
- 10.Eming SA, Whitsitt JS, He L, Krieg T, Morgan JR, Davidson JM. Particle-mediated gene transfer of PDGF isoforms promotes wound repair. J Invest Dermatol. 1999;112:297–302. doi: 10.1046/j.1523-1747.1999.00522.x. [DOI] [PubMed] [Google Scholar]
- 11.Franceschi RT, Wang D, Krebsbach PH, Rutherford RB. Gene therapy for bone formation: in vitro and in vivo osteogenic activity of an adenovirus expressing BMP7. J Cell Biochem. 2000;78:476–486. doi: 10.1002/1097-4644(20000901)78:3<476::aid-jcb12>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 12.Giannobile WV, Lee CS, Tomala MP, Tejeda KM, Zhu Z. Platelet-derived growth factor (PDGF) gene delivery for application in periodontal tissue engineering. J Periodontol. 2001;72:815–823. doi: 10.1902/jop.2001.72.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez Lahoz E, Liegeois NJ, Zhang P, Engelman JA, Horner J, Silverman A, Burde R, Roussel MF, Sherr CJ, Elledge SJ, DePinho RA. Cyclin D- and E-dependent kinases and the p57KIP2 inhibitor: cooperative interactions in vivo. Mol Cell Biol. 1999;19:353–363. doi: 10.1128/mcb.19.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrington MA, Estes JE, Leof E, Pledger WJ. PDGF stimulates transient phosphorylation of 180,000 Dalton protein. J Cell Biochem. 1985;27:67–81. doi: 10.1002/jcb.240270202. [DOI] [PubMed] [Google Scholar]
- 15.Heldin CH, Ostman A, Ronnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta. 1998;1378:F79–F113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 16.Heldin CH, Wasteson A, Westermark B. Interaction of platelet-derived growth factor with its fibroblast receptor. Demonstration of ligand degradation and receptor modulation. J Biol Chem. 1982;257:4216–4221. [PubMed] [Google Scholar]
- 17.Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- 18.Hicke L. Ubiquitin-dependent internalization and down-regulation of plasma membrane proteins. FASEB J. 1997;11:1215–1226. doi: 10.1096/fasebj.11.14.9409540. [DOI] [PubMed] [Google Scholar]
- 19.Jones SM, Kazlauskas A. Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat Cell Biol. 2001;3:165–172. doi: 10.1038/35055073. [DOI] [PubMed] [Google Scholar]
- 20.Jones SM, Kazlauskas A. Growth factor-dependent signaling and cell cycle progression. FEBS Lett. 2001;490:110–116. doi: 10.1016/s0014-5793(01)02113-5. [DOI] [PubMed] [Google Scholar]
- 21.Keating MT, Williams LT. Processing of the platelet-derived growth factor receptor. Biosynthetic and degradation studies using anti-receptor antibodies. J Biol Chem. 1987;262:7932–7937. [PubMed] [Google Scholar]
- 22.Krebsbach PH, Gu K, Franceschi RT, Rutherford RB. Gene therapy-directed osteogenesis: BMP-7-transduced human fibroblasts form bone in vivo. Hum Gene Ther. 2000;11:1201–1210. doi: 10.1089/10430340050015248. [DOI] [PubMed] [Google Scholar]
- 23.LaRochelle WJ, Jeffers M, McDonald WF, Chillakuru RA, Giese NA, Lokker NA, Sullivan C, Boldog FL, Yang M, Vernet C, Burgess CE, Fernandes E, Deegler LL, Rittman B, Shimkets J, Shimkets RA, Rothberg JM, Lichenstein HS. PDGF-D, a new protease-activated growth factor. Nat Cell Biol. 2001;3:517–521. doi: 10.1038/35074593. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, Backstrom G, Hellstrom M, Bostrom H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U. PDGF-C is a new protease-activated ligand for the PDGF α-receptor. Nat Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- 25.Lih CJ, Cohen SN, Wang C, Lin-Chao S. The platelet-derived growth factor α-receptor is encoded by a growth-arrest-specific (gas) gene. Proc Natl Acad Sci USA. 1996;93:4617–4622. doi: 10.1073/pnas.93.10.4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paasinen-Sohns A, Holtta E. Cells transformed by ODC, c-Ha-ras and v-src exhibit MAP kinase/Erk-independent constitutive phosphorylation of Sos, Raf and c-Jun activation domain, and reduced PDGF receptor expression. Oncogene. 1997;15:1953–1966. doi: 10.1038/sj.onc.1201366. [DOI] [PubMed] [Google Scholar]
- 27.Schatteman GC, Morrison-Graham K, van Koppen A, Weston JA, Bowen-Pope DF. Regulation and role of PDGF receptor alpha-subunit expression during embryogenesis. Development. 1992;115:123–131. doi: 10.1242/dev.115.1.123. [DOI] [PubMed] [Google Scholar]
- 28.Schneider C, King RM, Philipson L. Genes specifically expressed at growth arrest of mammalian cells. Cell. 1988;54:787–793. doi: 10.1016/s0092-8674(88)91065-3. [DOI] [PubMed] [Google Scholar]
- 29.Shamah SM, Stiles CD, Guha A. Dominant-negative mutants of platelet-derived growth factor revert the transformed phenotype of human astrocytoma cells. Mol Cell Biol. 1993;13:7203–7212. doi: 10.1128/mcb.13.12.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shao ZM, Nguyen M, Barsky SH. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene. 2000;19:4337–4345. doi: 10.1038/sj.onc.1203785. [DOI] [PubMed] [Google Scholar]
- 31.Sherr CJ. D-type cyclins. Trends Biochem Sci. 1995;20:187–190. doi: 10.1016/s0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 32.Simm A, Hoppe V, Karbach D, Leicht M, Fenn A, Hoppe J. Late signals from the PDGF receptors leading to the activation of the p70S6-kinase are necessary for the transition from G1 to S phase in AKR-2B cells. Exp Cell Res. 1998;244:379–393. doi: 10.1006/excr.1998.4200. [DOI] [PubMed] [Google Scholar]
- 33.Sorkin A, Waters CM. Endocytosis of growth factor receptors. Bioessays. 1993;15:375–382. doi: 10.1002/bies.950150603. [DOI] [PubMed] [Google Scholar]
- 34.Vaziri C, Faller DV. Repression of platelet-derived growth factor β-receptor expression by mitogenic growth factors and transforming oncogenes in murine 3T3 fibroblasts. Mol Cell Biol. 1995;15:1244–1253. doi: 10.1128/mcb.15.3.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J. 1997;326:61–68. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang QX, Walker F, Burgess AW, Baldwin GS. Reduction in platelet-derived growth factor receptor mRNA in v-src-transformed fibroblasts. Biochim Biophys Acta. 1995;1266:9–15. doi: 10.1016/0167-4889(94)00232-4. [DOI] [PubMed] [Google Scholar]
- 37.Zhu Z, Lee CS, Tejeda KM, Giannobile WV. Gene transfer and expression of platelet-derived growth factors modulate periodontal cellular activity. J Dent Res. 2001;80:892–897. doi: 10.1177/00220345010800030901. [DOI] [PMC free article] [PubMed] [Google Scholar]