

Abstract
Drugs developed for the treatment of conditions other than neoplasia can also show promise as potential antitumor agents. The fluoroquinolone antibiotic ciprofloxacin (CPFX) is known to modulate cycle cell progression and apoptosis in cancer cells, and is thought to induce DNA double-strand breaks (DSBs) via topoisomerase II (topo II) inhibition and stabilized cleavage complex (SCC) formation. DSBs trigger Ser-139 phosphorylation of histone H2AX (γH2AX) by PI-3-like kinases including ATM; γH2AX can serve as a marker of DNA damage when measured in situ using immunocytochemistry and flow cytometry. The aim of the present study was to investigate the relationship between CPFX-mediated DNA damage and induction of apoptosis in human lymphoblastoid cells and phytohaemagglutinin (PHA)-stimulated lymphocytes (Lymphs). Treatment of TK6 cells (wild-type p53) with 100 µg/ml CPFX for 2-10 h produced no increase in γH2AX; to the contrary, its level in S phase cells was reduced at 10 h compared to controls. Nevertheless, stabilization of topo IIα, ATM Ser-1981 phosphorylation and G2 arrest was observed in TK6 cells exposed to CPFX for ≥4 h. However, following 24 h treatment, γH2AX was dramatically increased in a sub-population of cells indicating the onset of apoptosis (confirmed by presence of activated caspase 3). CPFX had a similar lack of effect on induction of γH2AX at early time points in WTK1 and NH32 cells (devoid of functional p53) and proliferating Lymphs, however, induction of apoptosis was less pronounced than in TK6 cells. Formation of SCC and activation of ATM (but lack of γH2AX induction) indicates topo II-mediated chromatin or DNA changes in the absence of DSBs; ATM activation apparently triggers the G2M checkpoint leading to G2 arrest. The subsequent induction of apoptosis appears to be facilitated by functional p53. CPFX may therefore have a potential use as a chemotherapeutic agent in the treatment of lymphoblast-derived cancer.
Keywords: ciprofloxacin, topoisomerase II, DNA double-strand breaks, γH2AX, cell cycle, apoptosis
Introduction
In recent years, in vitro experiments with a number of drugs typically used for conditions other than neoplasia have shown promise in the potential treatment of leukemia and lymphoma. Acting through diverse mechanisms, these compounds usually retard or inhibit progression through the cell cycle and induce apoptosis. Examples of such compounds include fibrates and medroxyprogesterone acetate in Burkitt's lymphoma cells,1 and psychotropics in B-cell-derived lines.2,3 Thalidomide, an antiemetic which was withdrawn from the market in the 1960s, is thought to act by inducing cell differentiation and thereby preventing unrestrained proliferation of immature myelogenous cells.4 Hematopoietic-derived cancer cells are not the only target for such novel therapies; nonsteroidal anti-inflammatory drugs are known to inhibit proliferation and induce apoptosis in glioma and ovarian cancer cell lines.5,6 In experimental animals, acetaminophen has been shown to reduce carcinogen-induced cell proliferation and subsequent neoplasia.7 Furthermore, epidemiology studies have shown a decreased risk of various types of cancer in susceptible individuals regularly exposed to aspirin.8,9
Fluoroquinolone compounds (FQs) are a class of antibacterial agents that are commonly used in the treatment of human and animal infections.10 They are potent inhibitors of bacterial DNA gyrase enzymes, which regulate DNA supercoiling and play a key role in chromosome condensation, DNA replication, transcription and recombination.11 Inhibition of DNA gyrase enzymes results in modulation of these processes leading to DNA strand breakage and ultimately cell death.12 FQs also possess a lesser affinity towards the eukaryotic DNA gyrase homologue, topoisomerase II (topo II) and, at concentrations higher than normally achieved in blood, inhibition of topo II can occur resulting in the formation of stabilized cleavage complexes (SCC) and ultimately the production of DNA double-strand breaks (DSBs).13 Should inefficient DSB repair take place, mutagenesis or cell death by apoptosis can follow.14 An example of one such compound is ciprofloxacin (CPFX); previous studies have shown it to induce DNA strand breaks and be clastogenic in mammalian cells.15-18 CPFX is also known to induce G2M cell cycle arrest and apoptosis in a variety of cancer cell lines, without such pronounced effects in normal cells.19-22 Interestingly, the authors of these studies suggest CPFX may be used as a prophylactic and/or adjunct therapy in tumor management.
A promising technique available for the detection of DSBs is the use of cytometry to detect phosphorylated histone H2AX.23 Phosphorylation of Ser-139 on histone H2AX (defined as γH2AX) by PI-3-related kinases ATM, ATR and/or DNA-PK occurs rapidly following the generation of DSBs.24 γH2AX is thought to act as a molecular anchor by holding DSB ends in close proximity and therefore facilitating DNA repair.25 Radiation and chemotherapeutic agents known to cause DSBs, e.g., X-rays, UV-B and topo I and II inhibitors, have been shown to efficiently induce γH2AX foci in various cell types.26-29 Discrete γH2AX foci are easily detected in situ via immunocytochemistry and epifluorescence microscopy.30 Moreover, measurement of γH2AX by multiparameter flow cytometry offers the advantages of high-throughput screening, rapid and accurate analysis of individual cells, and the ability to correlate DNA damage with cellular DNA content, i.e., cell cycle phase, and apoptosis.31
In the present study, the genotoxic and apoptotic effects of CPFX in TK6 lymphoblastoid cells were studied simultaneously using γH2AX in a multiparameter flow cytometric approach. In addition, an upstream mediator of histone H2AX phosphorylation, Ser-1981 phosphorylated-ATM (ATMP1981),32 and a key enzyme in caspase-mediated apoptosis, activated caspase 3,33 were studied. Lymphoblast-derived WTK1 and NH32 cells with altered p53 status and phytohaemagglutinin (PHA)-stimulated human lymphocytes (Lymphs) were also used in some experiments for comparison.
Results
Lack of γH2AX induction and G2 cell cycle arrest
Treatment of TK6 cells, which possess wild-type p53, with either 50 or 100 µg/ml CPFX for 2-10 h produced no overall increase in expression of γH2AX compared to controls; to the contrary, a decrease (−18%) in the background level of γH2AX in S phase cells was observed in cells treated with 100 µg/ml (the highest concentration tested) at 10 h (Fig. 1A). In addition, after 10 h treatment with both 50 and 100 µg/ml CPFX, TK6 cells were arrested in G2 phase of the cell cycle (observation under fluorescence microscope showed exclusive G2 arrest and not M arrest; data not shown) and this arrest progressed in a time-dependent manner (Fig. 1B). Similar to TK6 cells, WTK1, NH32 cells and PHA-stimulated Lymphs showed no increase in γH2AX following 2 or 4 h treatment with 50 or 100 µg/ml CPFX (data not shown).
Figure 1.

Effects of 100 µg/ml CPFX treatment in TK6 cells. (A) Suppression of ‘constitutive’ γH2AX in S phase cells after 10 h, and dramatic induction in a sub-population of apoptotic cells after 24 h, compared to controls. (B) Time-dependent G2 accumulation (also evident in DNA histograms of part A)
Onset of apoptosis: induction of γH2AX and activation of caspase 3
Following 24 h treatment with 100 µg/ml CPFX, the level of γH2AX was dramatically increased in a sub-population of TK6 cells, indicating the onset of apoptosis,28 (34% in treated cells vs. 3% in controls) (Figs. 1A and 3A). Extensive nuclear fragmentation and formation of apoptotic bodies was also observed (Fig. 2). Interestingly, levels of γH2AX were still decreased compared to controls in the major proportion of cells at the same time point. An accumulation of TK6 cells in G2 (40% in treated cells vs. 20% in controls) was also observed (Figs. 1B and 5). To assess whether the dramatic increase in γH2AX in a sub-population of CPFX-treated TK6 cells was triggered by apoptosis-associated DNA fragmentation in these cells, caspase 3 activation was measured following treatment with CPFX for 6-48 h. While exposure of these cells to 100 µg/ml CPFX for 6, 8 and 10 h produced no detectable caspase 3 activation (data not shown), enhanced levels of activated caspase 3 were observed following 24 h (and also after 48 h; data not shown) treatment (Fig. 3B). No increase in γH2AX was detected in PHA-stimulated Lymphs treated with 50 µg/ml CPFX for 24 h and only a modest increase (6% in treated cells vs. 3% in controls) in cells treated with 100 µg/ml (Fig. 4). Exposure of PHA-stimulated Lymphs to 100 µg/ml CPFX for 48 h resulted in a increase in levels of activated caspase 3 in relatively few cells (data not shown). Treatment of WTK1 cells for 24 h with either 50 or 100 µg/ml CPFX failed to induce γH2AX (3% in treated cells vs. 2% in controls) which suggests a lack of apoptosis in these cells (Fig. 4). However, WTK1 cells were still arrested in G2 (50 µg/ml; 31% and 100 µg/ml; 40% vs. 20% in controls) at the same time point (Fig. 5). Following 24 h treatment with 100 µg/ml CPFX in NH32 cells, a rise in γH2AX was detected in some cells (17% in treated cells vs. 8% in controls), again indicative of apoptosis (Fig. 4). NH32 cells were also arrested in G2 (50 µg/ml; 18% and 100 µg/ml; 26% vs. 12% in controls) at the same time point (Fig. 5).
Figure 3.

Onset of apoptosis in TK6 cells. (A) High expression of γH2AX in a sub-population of cells treated with 100 µg/ml CPFX for 24 h (right panel) compared to controls (left panel). (B) Expression of activated caspase 3 in identically treated cells. Number in top left indicates % of apoptotic cells, and numbers in parentheses indicate % of nonapoptotic (lower) and apoptotic cells (upper) in G2 phase of the cell cycle
Figure 2.

Untreated (A and B) and TK6 cells treated with 100 µg/ml CPFX for 24 h (C-E) were stained with 7-AAD and examined by microscopy (Nikon Microphot FXA, 60×). The red fluorescence emission of DNA-bound 7-AAD (A and C) was induced by exposure to green wavelengths and the blue emission of CPFX (E) was induced by exposure to UV light. (B and D) show cell morphology under differential interference (Nomarski) contrast. Note extensive nuclear fragmentation and formation of apoptotic bodies (C-E), changes typical of apoptosis, in cells treated with CPFX. Interestingly, the blue fluorescence of CPFX is primarily associated with nuclei and fragments of chromatin
Figure 5.

Cell cycle phase distribution in CPFX-treated cells; concentration-dependent G2 accumulation in PHA-stimulated Lymphs, TK6, WTK1 and NH32 cells following 24 h exposure
Figure 4.

Limited frequency of apoptosis and modest induction of gH2AX (and G2 arrest) in PHA-stimulated Lymphs, and p53-deficient NH32 and WTK1 cells following 100 mg/ml CPFX treatment for 24 h, compared to controls. Number in top-right indicates % of apoptotic cells
Induction of ATM phosphorylation
Expression of ATMP1981 was studied in TK6 cells and PHA-stimulated Lymphs treated with CPFX for 2-48 h. Treatment of TK6 cells with 50 µg/ml CPFX for 2 and 4 h produced no detectable increase in ATMP1981(data not shown). TK6 cells exposed to 100 µg/ml CPFX also showed no significant increase in ATMP1981 between 2-6 h. A marked increase (all phases of the cell cycle were affected), however, was observed after 10 h (34%) and 24 h (29%) treatment, compared to controls (Fig. 6). CPFX had no effect on levels of ATMP1981 in PHA-stimulated Lymphs at any time point (2-48 h) assessed (data not shown).
Figure 6.

Time-dependent induction of ATMP1981 in TK6 cells following 100 µg/ml CPFX, compared to controls
CPFX-induced SCC formation
To determine whether formation of CPFX-induced SCC occurred prior to ATM activation, genomic DNA was probed for the presence of stabilized topo IIα with a specific antibody. Untreated TK6 showed no presence of stabilized topo IIα (Fig. 7). A concentration-dependent increase in stabilized topo IIα was detected in TK6 cells following treatment with 50 and 100 µg/ml CPFX for 4 h.
Figure 7.

Concentration-dependent formation of genomic DNA-topo IIα SCC in TK6 cells following CPFX treatment for 4 h
Lack of H3P induction
To confirm the findings of exclusive G2 arrest (and not M) as assessed by fluorescence microscopy, the mitotic marker H3P,35 was studied in TK6 cells treated with CPFX. As expected, no detectable increase in H3P was observed in TK6 cells treated with either 50 or 100 µg/ml CPFX for 24 h (Fig. 8). To the contrary, a decrease in frequency of cells with a G2M DNA content expressing H3P was seen, consistent with CPFX-induced G2 arrest.
Figure 8.
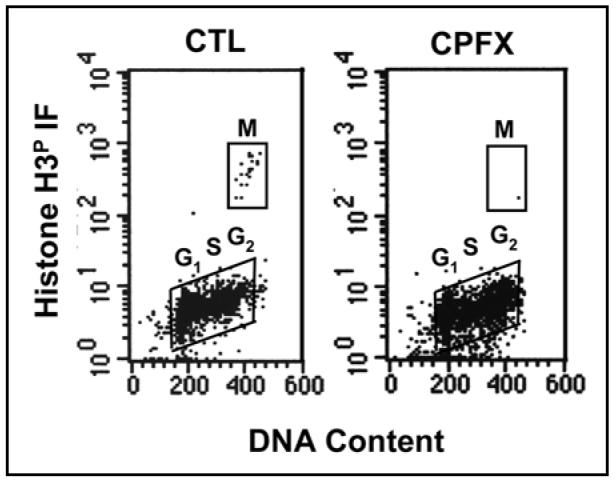
Lack of H3P induction in TK6 cells treated with 100 µg/ml CPFX for 24 h, indicating exclusive G2 arrest and not M
Discussion
The FQ antibiotic CPFX has been previously shown to induce DNA strand breakage in mammalian cell lines, ostensibly as a result of topo II inhibition.16,17 In addition to this, several other studies have described cell cycle and pro-apoptotic effects in cancer cells treated with CPFX.19-22 The main aim of the current study was to investigate the relationship between CPFX-mediated DNA damage and the induction of apoptosis in TK6 lymphoblastoid cells.
It is well known that topo II poisons, e.g., etoposide and mitoxantrone, indirectly generate DSBs and, if breaks are sufficient in numbers, they result in activation of the apoptosis pathway and cell death.28,29 Similar cellular effects are observed in X-irradiated cells, where DSBs are produced in DNA by reactive oxygen species (ROS).26 In the present study, TK6 cells were treated for 2-24 h with CPFX and the established DSB marker, γH2AX, was measured using multiparameter flow cytometry. γH2AX is generated early during a DNA damage response and is thought to facilitate DSB repair by holding broken DNA ends close together and recruiting other DNA repair factors to the damaged area.23-25 No induction of γH2AX was detected in cells treated with either 50 or 100 µg/ml CPFX for 2-10 h; to the contrary, a suppression in the ‘constitutive’ γH2AX level,36 was observed in S phase cells after 10 h treatment with 100 µg/ml. Furthermore, this effect was still evident at 24 h in the major proportion of treated TK6 cells compared to controls. A similar phenomenon was reported in TK6, A549 and bronchial epithelial cells treated with the ROS scavenger N-acetyl-L-cysteine (NAC), and the opposite occurred in A549 cells treated with buthionine sulphoximine, an inhibitor of glutathione (GSH) biosynthesis.36 While it is possible that CPFX operates like NAC and GSH as a ROS scavenger, acting to reduce the number of endogenous oxidant-induced DSBs, it is also likely that activation of ATM in TK6 cells leads to the recruitment of the Mre11/Rad50/Nbs1 complex, which engages the S phase checkpoint and reduces the rate of DNA replication. Because DSBs are formed in S phase as a result of collisions of replication forks with oxidative damage induced by endogenous ROS such as single-strand DNA lesions, suppression of the DNA replication rate is expected to reduce the frequency of DSBs and thereby expression of γH2AX. Studies are in progress to elucidate the mechanism(s) of CPFX-mediated attenuation of γH2AX.
Our findings differ from two early studies by Bredberg et al.,16,37 who reported a significant induction of DSBs in Raji lymphoblastoid cells treated for 2 h with 80 µg/ml CPFX. These authors used a limited DNA elution technique, whereas, we used histone γH2AX, a molecular marker that is closely associated with the induction of DSBs. In addition, in a recent study utilizing the comet assay and concentrations of CPFX up to 1000 µg/ml, treated WTK1 lymphoblastoid cells showed concentration-dependent increases in DNA single-strand breaks (SSBs), without any induction of DSBs.17 The present study does not rule out the possibility of other types of CPFX-mediated DNA damage, e.g., single-strand DNA lesions, but, taken together, recent evidence suggests DSBs are not induced by 0-100 µg/ml CPFX (0-10 h exposure) in human lymphoblastderived cells. It is therefore likely that CPFX-induced clastogenicity in such cells arises from DNA damage that is different to direct DSBs.
Although a lack of γH2AX induction was observed at 2-8 h, CPFX did induce both Ser-1981 ATM phosphorylation and cell cycle effects. Phosphorylation of ATM suggests the presence of modified DNA and/or altered chromatin structure. However, as mentioned earlier, one of its downstream targets, histone H2AX, was not phosphorylated, and DSBs were not detected by the neutral comet assay. Bakkenist and Kastan,38 elegantly describe a model in which the ATM activation can be actually a response to a change in chromatin structure and where ATM substrates are specifically phosphorylated depending on the type (or lack) of DNA damage present. We therefore speculate that CPFX induces changes in chromatin structure via topo II inhibition (as determined by presence of stabilized topo IIα after 4 h CPFX treatment), and these are sufficient to cause activation of ATM. However, as DSBs are not present, ATMP1981 does not target H2AX, but rather other downstream proteins that control cell cycle progression, e.g., checkpoint kinases and p53. Indeed, modulation of the cell cycle occurred after 8 h treatment with 100 µg/ml CPFX; TK6 cells started to arrest exclusively in G2 phase and this continued over time. Related effects were also observed in WTK1 and NH32 cells (and to a lesser extent in PHA-stimulated lymphocytes), however, G2 arrest was not apparent until 24 h. A number of different cell types including CC-531, SW-403 and HT-29 colon carcinoma cells, MC3T3-E1 and MG-63 osteoblast-like cells and HTB9 bladder carcinoma cells show time-dependent G2M cell cycle arrest following exposure to similar concentrations of CPFX.20,21,39 Aranha et al.,20 reported that CPFX treatment, at least in HTB9 cells, produced a downregulation in cyclins B and E, and dephosphorylation of cyclin-dependent kinase-2 and thereby causing G2M arrest. Although we provide limited evidence, we hypothesize that ATMP1981 triggers the signaling cascade, which is ultimately responsible for G2 arrest (and apoptosis) in these cancer cell lines. It is also possible that, through the recruitment of Mre11/Rad50/Nbs1 complex, activated ATM also engages the S phase checkpoint, which, as discussed earlier, may be reflected by the reduced level of constitutive γH2AX in S phase cells.
In addition to cell cycle arrest, CPFX is also known to induce apoptosis in cancer cell lines.20,39 Importantly, the pro-apoptotic effects appear to be cancer cell-specific, as illustrated in a study showing that nontumorigenic prostate epithelial cells (MLC8891) were not affected when treated in parallel to malignant PC3 prostate cells.19 It was suggested that CPFX could be a potential chemotherapeutic agent for treatment of a range of cancers, e.g., bladder and prostate.19,20 We studied the induction of γH2AX, which as well as being a marker for DSBs, can also be used as an indicator of apoptosis.29,40 The level of γH2AX is dramatically increased during the process of apoptosis as DNA is cleaved by nucleolytic enzymes and PI-3 kinases are activated; a sub-population of cells can be identified with levels of γH2AX orders of magnitude above nonapoptotic cells. Such a sub-population of TK6 cells was observed following exposure to CPFX for 24 h and this was further confirmed by a large increase in the level of activated caspase 3 at 24 and 48 h. Changes in γH2AX were absent in treated lymphoblast-derived WTK1 cells (mutant p53) and not as pronounced in NH32 lymphoblastoid cells (p53-null), indicating that CPFX-induced apoptosis is facilitated by p53. PHA-stimulated Lymphs showed no evidence of apoptosis (both in the level of γH2AX and activated caspase 3) after 24 h treatment with CPFX, however, a small increase in activated caspase 3 was observed after 48 h. These results are in concordance with the findings of Aranha et al.,19 as CPFX appears to induce high levels of apoptosis in TK6 leukemia cells, without having such effects in normal mitogen-stimulated Lymphs. A functional p53 signaling pathway also appears to be required for achieving high levels of CPFX-induced apoptosis in B-cell leukemia; p53 status should therefore be assessed before considering CPFX in future chemotherapy studies. However, it should be noted that p53 status may not be as important in T-cell leukemia, since apoptosis was significantly accelerated in p53-null Jurkat cells treated with CPFX.41
In conclusion, we confirm the findings of other groups by showing that CPFX can induce exclusive G2 cell cycle arrest and apoptosis in TK6 leukemia cells, with no significant effects in normal mitogen-stimulated Lymphs. Our data suggest that these effects are related to the production of some form of topo II-mediated DNA and/or chromatin modification, although not formation of DSBs, and subsequent activation of ATM. Induction of apoptosis by CPFX in B-cell-derived lymphoblast cells appears to be facilitated by a functional p53 pathway. CPFX may therefore have a potential use as a novel chemotherapeutic agent in the treatment of leukemia, as well as previously suggested bladder and prostate cancer.19,20 Whether or not CPFX exposure may also attenuate background levels of cellular DNA damage (produced by endogenous oxidants), thus protecting against the development of pathologies such as aging and age-related cancer is the subject of ongoing investigations.
Materials and Methods
Cell culture and treatment
TK6 (wild-type p53), WTK1 (mutant p53) and NH32 (p53-null) human lymphoblast cells,34 were cultured in RMPI 1640 medium (GIBCO, OR, USA) supplemented with L-glutamine (2 mM) and fetal bovine serum (10%). Human Lymphs were isolated from two healthy donors and stimulated by PHA to proliferate for 48 h prior to treatment and cultured as above. Cells were treated with 0-100 µg/ml CPFX (dissolved in 0.1 N HCl; Sigma Aldrich, MO, USA) or vehicle alone for 2-48 h. All treatments were carried out at least in duplicate.
Immunofluorescence
Following treatment, cells were fixed in suspension in p-formaldehyde (1%) for 15 min at 4°C and post-fixed in ice-cold ethanol (70%) for at least 1 h at −20°C. Fixed cells were incubated with BSA (1%) containing either γH2AX Ab, ATMP1981 Ab (both diluted 1:100; Millipore Corporation, MA, USA), Ser-10 phosphorylated histone H3 (H3P) or activated caspase 3 Ab (both diluted 1:100; Cell Signaling Technology, MA, USA) for 1 h at 25°C. Following washing in BSA (1%), cells were incubated with Alexa Fluor 488 secondary Ab (diluted 1:100; Molecular Probes, OR, USA) for 1 h at 25°C. Cells were counterstained with a solution of propidium iodide in PBS containing RNase (both 10 µg/ml; Sigma Aldrich) and stored at 4°C overnight. For cell nuclei images, cells were attached to slides by cytocentifugation and fixed in p-formaldehyde (1%). Nuclear DNA was stained using the DNA fluorochrome 7 -aminoactinomycin D (7-AAD; 1 µg/ml) dissolved in PBS and examined using fluorescence microscopy.
Fluorescence measurement
Cellular green (γH2AX, ATMP1981, H3P or activated caspase 3) and red (PI; nuclear DNA) fluorescence was measured using a FACScan flow cytometer (Becton Dickinson, CA, USA) with the standard emission filters for green (FL1) and red (FL3) fluorescence as described in Halicka et al.27 At least 5000 cells were counted per sample.
Identification of CPFX-induced SCC
CPFX-induced SCC were identified using the In Vivo Link Kit (TopoGEN, FL, USA) as per the manufacturer's instructions. Briefly, cells were lysed in TE buffer (10 mM Tris, pH 7.5 and 1 mM EDTA) containing sarkosyl (1%) and layered on to a cushion of cesium chloride (density of 1.5 g/ml). Following centrifugation (170000 × g for 12 h, at 25°C) in a SW50.1 rotor (Beckman Coulter, CA, USA), isolated DNA was precipitated in ethanol (100%) and resolubilized in TE buffer. DNA (5 µg) was applied to a nitrocellulose membrane (Bio-Rad, CA, USA) using a slot-blotting apparatus (Bio-Dot SF, Bio-Rad). DNA-bound topo II isoform α (topo IIα) was identified by immunoblotting with an anti-topo IIα antibody (diluted 1:1500; TopoGEN) and enhanced chemiluminescence plus reagents (GE Healthcare, NJ, USA).
Acknowledgements
H.D.H., F.T. and Z.D. are supported in part by NCI RO1 28 704.
Abbreviations
- CPFX
ciprofloxacin
- DSBs
DNA double-strand breaks
- FQs
fluoroquinolones
- GSH
reduced glutathione
- NAC
N-acetyl-L-cysteine
- Lymphs
human lymphocytes
- ATMP1981
Ser-1981 phosphorylated ATM
- PHA
phytohaemagglutinin
- ROS
reactive oxygen species
- SSBs
DNA single-strand breaks
- topo II
topoisomerase II
References
- 1.Fenton SL, Luong QT, Sarafeim A, Mustard KJ, Pound J, Desmond JC, Gordon J, Drayson MT, Bunce CM. Fibrates and medroxyprogesterone acetate induce apoptosis of primary Burkitt's lymphoma cells and cell lines: Potential for applying old drugs to a new disease. Leukemia. 2003;17:568–75. doi: 10.1038/sj.leu.2402843. [DOI] [PubMed] [Google Scholar]
- 2.Serafeim A, Holder MJ, Grafton G, Chamba A, Drayson MT, Luong QT, Bunce CM, Gregory CD, Barnes NM, Gordon J. Selective serotonin reuptake inhibitors directly signal for apoptosis in biopsy-like Burkitt lymphoma cells. Blood. 2003;101:3212–9. doi: 10.1182/blood-2002-07-2044. [DOI] [PubMed] [Google Scholar]
- 3.Meredith EJ, Holder MJ, Chamba A, Challa A, Drake-Lee A, Bunce CM, Drayson MT, Pilkington G, Blakely RD, Dyer MJ, Barnes NM, Gordon J. The serotonin transporter (SLC6A4) is present in B-cell clones of diverse malignant origin: Probing a potential anti-tumor target for psychotropics. Faseb J. 2005;19:1187–9. doi: 10.1096/fj.04-3477fje. [DOI] [PubMed] [Google Scholar]
- 4.Saunders G. Overview of drug therapy for multiple myeloma. J Oncol Pharm Pract. 2005;11:83–100. doi: 10.1191/1078155205jp160oa. [DOI] [PubMed] [Google Scholar]
- 5.Bernardi A, Jacques-Silva MC, Delgado-Canedo A, Lenz G, Battastini AM. Nonsteroidal anti-inflammatory drugs inhibit the growth of C6 and U138-MG glioma cell lines. Eur J Pharmacol. 2006;532:214–22. doi: 10.1016/j.ejphar.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Burford C, Barnes MN, Oelschlager DK, Myers RB, Talley LI, Partridge EE, Grizzle WE. Effects of nonsteroidal anti-inflammatory agents (NSAIDs) on ovarian carcinoma cell lines: Preclinical evaluation of NSAIDs as chemopreventive agents. Clin Cancer Res. 2002;8:202–9. [PubMed] [Google Scholar]
- 7.Williams GM, Iatropoulos MJ, Jeffrey AM, Shirai T. Protective effect of acetaminophen against colon cancer initiation effects of 3,2'-dimethyl-4-aminobiphenyl in rats. Eur J Cancer Prev. 2002;11:39–48. doi: 10.1097/00008469-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Bardia A, Ebbert JO, Vierkant RA, Limburg PJ, Anderson K, Wang AH, Olson JE, Vachon CM, Cerhan JR. Association of aspirin and nonaspirin nonsteroidal anti-inflammatory drugs with cancer incidence and mortality. J Natl Cancer Inst. 2007;99:881–9. doi: 10.1093/jnci/djk200. [DOI] [PubMed] [Google Scholar]
- 9.Jacobs EJ, Thun MJ, Bain EB, Rodriguez C, Henley SJ, Calle EE. A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J Natl Cancer Inst. 2007;99:608–15. doi: 10.1093/jnci/djk132. [DOI] [PubMed] [Google Scholar]
- 10.Appelbaum PC, Hunter PA. The fluoroquinolone antibacterials: Past, present and future perspectives. Int J Antimicrob Agents. 2000;16:5–15. doi: 10.1016/s0924-8579(00)00192-8. [DOI] [PubMed] [Google Scholar]
- 11.Shen LL, Kohlbrenner WE, Weigl D, Baranowski J. Mechanism of quinolone inhibition of DNA gyrase: Appearance of unique norfloxacin binding sites in enzyme-DNA complexes. J Biol Chem. 1989;264:2973–8. [PubMed] [Google Scholar]
- 12.Hooper DC, Wolfson JS. Mode of action of the quinolone antimicrobial agents. Rev Infect Dis. 1988;10(Suppl 1):S14–21. doi: 10.1093/clinids/10.supplement_1.s14. [DOI] [PubMed] [Google Scholar]
- 13.Burden DA, Osheroff N. Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochim Biophys Acta. 1998;1400:139–154. doi: 10.1016/s0167-4781(98)00132-8. [DOI] [PubMed] [Google Scholar]
- 14.Hussy P, Maass G, Tummler B, Grosse F, Schomburg U. Effect of 4-quinolones and novobiocin on calf thymus DNA polymerase alpha primase complex, topoisomerases I and II, and growth of mammalian lymphoblasts. Antimicrob Agents Chemother. 1986;29:1073–8. doi: 10.1128/aac.29.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukherjee A, Sen S, Agarwal K. Ciprofloxacin: Mammalian DNA topoisomerase type II poison in vivo. Mutat Res. 1993;301:87–92. doi: 10.1016/0165-7992(93)90029-u. [DOI] [PubMed] [Google Scholar]
- 16.Bredberg A, Brant M, Jaszyk M. Ciprofloxacin-induced inhibition of topoisomerase II in human lymphoblastoid cells. Antimicrob Agents Chemother. 1991;35:448–50. doi: 10.1128/aac.35.3.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh T, Mitsumori K, Kawaguchi S, Sasaki YF. Genotoxic potential of quinolone antimicrobials in the in vitro comet assay and micronucleus test. Mutat Res. 2006;603:135–44. doi: 10.1016/j.mrgentox.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Herbold BA, Brendler-Schwaab SY, Ahr HJ. Ciprofloxacin: In vivo genotoxicity studies. Mutat Res. 2001;498:193–205. doi: 10.1016/s1383-5718(01)00275-3. [DOI] [PubMed] [Google Scholar]
- 19.Aranha O, Grignon R, Fernandes N, McDonnell TJ, Wood DP, Jr, Sarkar FH. Suppression of human prostate cancer cell growth by ciprofloxacin is associated with cell cycle arrest and apoptosis. Int J Oncol. 2003;22:787–94. [PubMed] [Google Scholar]
- 20.Aranha O, Wood DP, Jr, Sarkar FH. Ciprofloxacin mediated cell growth inhibition, S/G2-M cell cycle arrest, and apoptosis in a human transitional cell carcinoma of the bladder cell line. Clin Cancer Res. 2000;6:891–900. [PubMed] [Google Scholar]
- 21.Miclau T, Edin ML, Lester GE, Lindsey RW, Dahners LE. Effect of ciprofloxacin on the proliferation of osteoblast-like MG-63 human osteosarcoma cells in vitro. J Orthop Res. 1998;16:509–12. doi: 10.1002/jor.1100160417. [DOI] [PubMed] [Google Scholar]
- 22.Somekh E, Douer D, Shaked N, Rubinstein E. In vitro effects of ciprofloxacin and pefloxacin on growth of normal human hematopoietic progenitor cells and on leukemic cell lines. J Pharmacol Exp Ther. 1989;248:415–8. [PubMed] [Google Scholar]
- 23.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 24.Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol Ther. 2003;2:233–5. doi: 10.4161/cbt.2.3.373. [DOI] [PubMed] [Google Scholar]
- 25.Bassing CH, Alt FW. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle. 2004;3:149–53. doi: 10.4161/cc.3.2.689. [DOI] [PubMed] [Google Scholar]
- 26.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol. 2003;79:351–8. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 27.Halicka HD, Huang X, Traganos F, King MA, Dai W, Darzynkiewicz Z. Histone H2AX phosphorylation after cell irradiation with UV-B: Relationship to cell cycle phase and induction of apoptosis. Cell Cycle. 2005;4:339–45. [PubMed] [Google Scholar]
- 28.Kurose A, Tanaka T, Huang X, Halicka HD, Traganos F, Dai W, Darzynkiewicz Z. Assessment of ATM phosphorylation on Ser-1981 induced by DNA topoisomerase I and II inhibitors in relation to Ser-139-histone H2AX phosphorylation, cell cycle phase, and apoptosis. Cytometry A. 2005;68:1–9. doi: 10.1002/cyto.a.20186. [DOI] [PubMed] [Google Scholar]
- 29.Huang X, Traganos F, Darzynkiewicz Z. DNA damage induced by DNA topoisomerase I- and topoisomerase II-inhibitors detected by histone H2AX phosphorylation in relation to the cell cycle phase and apoptosis. Cell Cycle. 2003;2:614–9. [PubMed] [Google Scholar]
- 30.Sedelnikova OA, Rogakou EP, Panyutin IG, Bonner WM. Quantitative detection of (125)IdU-induced DNA double-strand breaks with gamma-H2AX antibody. Radiat Res. 2002;158:486–92. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 31.Olive PL. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004;75:355–73. doi: 10.1016/s0091-679x(04)75014-1. [DOI] [PubMed] [Google Scholar]
- 32.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 33.Porter AG, Janicke RU. Emerging roles of caspase 3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz JL, Jordan E, Evans HH, Lenarczyk M, Liber H. The TP53 dependence of radiation-induced chromosome instability in human lymphoblastoid lines. Radiat Res. 2003;159:730–738. doi: 10.1667/rr3005. [DOI] [PubMed] [Google Scholar]
- 35.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–60. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Tanaka T, Kurose A, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation on Ser-139 in cells untreated by genotoxic agents is cell-cycle phase specific and attenuated by scavenging reactive oxygen species. Int J Oncol. 2006;29:495–501. [PubMed] [Google Scholar]
- 37.Bredberg A, Brant M, Riesbeck K, Azou Y, Forsgren A. 4-Quinolone antibiotics: Positive genotoxic screening tests despite an apparent lack of mutation induction. Mutat Res. 1989;211:171–80. doi: 10.1016/0027-5107(89)90117-6. [DOI] [PubMed] [Google Scholar]
- 38.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 39.Herold C, Ocker M, Ganslmayer M, Gerauer H, Hahn EG, Schuppan D. Ciprofloxacin induces apoptosis and inhibits proliferation of human colorectal carcinoma cells. Br J Cancer. 2002;86:443–8. doi: 10.1038/sj.bjc.6600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang X, Okafuji M, Traganos F, Luther E, Holden E, Darzynkiewicz Z. Assessment of histone H2AX phosphorylation induced by DNA topoisomerase I and II inhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin. Cytometry A. 2004;58:99–110. doi: 10.1002/cyto.a.20018. [DOI] [PubMed] [Google Scholar]
- 41.Jun YT, Kim HJ, Song MJ, Lim JH, Lee DG, Han KJ, Choi SM, Yoo JH, Shin WS, Choi JH. In vitro effects of ciprofloxacin and roxithromycin on apoptosis of Jurkat T lymphocytes. Antimicrob Agents Chemother. 2003;47:1161–4. doi: 10.1128/AAC.47.3.1161-1164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]