

Abstract
IRAK-1 is a critical modulator regulating innate immunity signaling processes. However, the physiological substrates for IRAK-1 remain poorly defined. In this report, we have demonstrated that IRAK-1 is a kinase responsible for the constitutive phosphorylation and inactivation of the Nuclear Factor of Activated T-cell (NFAT). Expression of IRAK-1 suppressed NFAT reporter activity. Correspondingly, the levels of both nuclear NFATc1 and NFATc4 were constitutively elevated in IRAK-1-/- cells. Furthermore, the phosphorylation of NFATc4 at the S168PS170P site was significantly diminished in IRAK-1-/- cells. Mechanistically, we observed that IRAK-1 interacted with NFATc4 via the C-terminus of IRAK-1 and the N-terminal NHR region of NFATc4. IRAK-1 mutants that ablated either its kinase activity or its interaction with NFATc4 failed to suppress NFAT reporter activity. The expression level of COX-2, which is under the control of NFAT, was elevated in IRAK-1-/- cells. Functionally, ApoE-/-/IRAK-1-/- mice were protected from high-fat-diet induced hypertension and atherosclerosis. Taken together, our findings reveal NFAT molecules as novel physiological targets for IRAK-1.
Keywords: Inflammation, signaling, NFAT, IRAK-1, cardiovascular disease
INTRODUCTION
IRAK-1 was initially discovered as a kinase forming a close association with the intracellular domain of interleukin-1 receptor (Cao et al., 1996). Since IL-1 is one of the critical inflammatory cytokines, IRAK-1 has drawn great attention in the inflammation field. The significance of IRAK-1 was further appreciated when it was later found to be shared by the Toll-Like-Receptor (TLR) mediated innate immunity signaling pathways (Deng et al., 2003; Gan and Li, 2006). Recent studies have shown that other pathways such as GPCR mediated pathway (Wang et al., 2006a), CD26 signaling (Ohnuma et al., 2005), and insulin signaling pathway (Kim et al., 2005) may all share IRAK-1 as one of their key signaling components.
Despite its significant and broad involvement in signaling processes, the substrates of IRAK-1 have not been well defined. Sequence alignment and structural prediction indicate that the kinase domain of IRAK-related molecules may adopt a tertiary fold resembling cyclin-dependent-kinases (CDKs) (Kuglstatter et al., 2007; Wang et al., 2006b), which suggests that IRAK-1 may prefer to phosphorylate a serine/threonine residue immediately flanked by a proline residue, sequence known as the TP or SP motif. Supporting this argument, biochemical analyses indicate that IRAK-1 can auto-phosphorylate itself at the Proline-Serine-rich region (Kollewe et al., 2004). In addition, a few previously defined IRAK-1 downstream target molecules such as STAT3, IRF5 and IRF7 all contain Serine-Proline motifs (Huang et al., 2004; Schoenemeyer et al., 2005; Uematsu et al., 2005).
Nuclear Factor of Activated T-cell (NFAT) is a family of widely expressed transcription factors regulating diverse cellular functions such as adipocyte differentiation, cardiac hypertrophy and cardiovascular functions, neuronal development and angiogenesis (Graef et al., 2001; Ho et al., 1998; Molkentin et al., 1998; Zaichuk et al., 2004). So far five isoforms of NFAT have been identified sharing significant sequence similarity, namely NFATc1(NFAT2), NFATc2(NFAT1), NFATc3 (NFAT 4) and NFATc4 (NFAT3) (Crabtree and Olson, 2002; Im and Rao, 2004). Their activation is primarily achieved by calcenurin-mediated dephosphorylation. Once dephosphorylated and activated, NFAT can be subsequently re-phosphorylated by multiple export kinases including GSK-3β and cyclin dependent kinases (CDK) which help to export NFAT out of the nucleus. In untreated resting cells, NFAT molecules are constantly kept in the constitutively phosphorylated and inactive states by various maintenance kinases including casein kinase 1 and other yet-to-be-defined kinases (Macian, 2005). Despite extensive studies on NFAT, the maintenance kinases responsible for the constitutive NFAT phosphorylation have not been fully defined (Macian, 2005). Within the N-terminal NFAT-Homology-Region (NHR), there are multiple Ser-Pro rich motifs (Macian, 2005), which may potentially serve as phosphorylation sites for IRAK-1.
We therefore tested the hypothesis that IRAK-1 may contribute to the constitutive phosphorylation and inactivation of NFAT. Using murine cells harvested from wild type and IRAK-1-/- mice, we studied the status of NFAT phosphorylation and nuclear distribution, as well as downstream gene expression. Furthermore, we examined the molecular domains required for interaction between IRAK-1 and NFAT molecules. We conclude that IRAK-1 plays a novel role in mediating NFAT phosphorylation and function. Our findings bear significant physiological relevance since NFAT molecules are involved in the pathogenesis and regression of various inflammatory diseases including atherosclerosis and cardiovascular complications.
MATERIALS AND METHODS
Plasmids and antibodies
The wild type pFlag-IRAK-1 plasmid, pFlag-IRAK-1B and the C-terminal deletion pFlag-IRAK-1ΔC plasmids were as described (Hu et al., 2002). The pFlag-IRAK-1-D340N and the pFlag-IRAK-1-C203S plasmids were generated using the GeneEditor site-directed mutagenesis kit (Promega, WI). The pNFAT-SEAP plasmid and the pRL-null were purchased from Clontech and Promega respectively. pEGFP-NFATc4 was from Addgene (Addgene plasmid 10961) (Ichida and Finkel, 2001), and was used to generate the pEGFP-NHR and pEGFP-ΔNHR plasmids, which encode the NFATc4 N-terminal NHR region and the NFATc4 with a truncation of the NHR region respectively (figure 4). All constructs were confirmed by automated sequencing. The antibodies specific for NFATc1, NFATc4, p-NFATc4-Ser168ProSer170Pro, GAPDH, GFP, and Lamin B were purchased from Santa Cruz. The anti-IRAK1 polyclonal antibody was from Upstate. The anti-FLAG monoclonal antibody was from Sigma.
Figure 4.

IRAK-1 forms a complex with NFATc4 via the C-terminus of IRAK-1 and the N-terminus of NFATc4. A) The endogenous IRAK-1 and NFATc4 interact with each other. Equal amounts of cell lysates from human THP-1 cells were immunoprecipitated with anti-IRAK-1 antibody. The immunoprecipitates were separated on SDS-PAGE, and blotted with anti-NFATc1, anti-NFATc4, or IRAK-1 antibody. B). The NFATc4 N-terminal NHR region is required for its interaction with IRAK-1. MAT-2 cells were transfected with pEGFP-NFATc4, pEGFP-NHR, or pEGFP-ΔNHR plasmid. Twenty four hours after the transfection, equal amounts of total cellular lysates were prepared and used to perform immunoprecipitation with anti-GFP antibody. Immunoprecipitated proteins were separated on SDS-PAGE, and blotted with anti-IRAK-1 or anti-GFP antibody. C). The C-terminus of IRAK-1 is necessary for its interaction with NFATc4. MAT-2 cells were transfected with either pFlag-IRAK-1 or pFlag-IRAK-1ΔC plasmid. Twenty four hours after the transfection, equal amounts of total cellular lysates were prepared and used to perform immunoprecipitation with anti-Flag antibody. Immunoprecipitated proteins were separated on SDS-PAGE, and blotted with anti-Flag or anti-GFP antibody.
Cell culture, isolation and culture of mouse embryonic fibroblast (MEF) cells and bone marrow derived macrophages (BMDM)
MEF cells were elicited from E13.5 C57BL/6 wild type or IRAK-1-/- mice (Thomas et al., 1999) according to standard protocol (Rui et al., 2001). The isolation and culture of murine BMDM, maintenance of THP-1 and MAT-2 cells (Hela cells stably transfected with TLR2) were as described (Su et al., 2007).
Cell transfection and luciferase reporter assay
MAT2 cells were maintained as described (Su et al., 2007). 125 ng of either IRAK expression plasmids or the empty pCMV-Flag plasmid was co-transfected with 375 ng/well of the pNFAT-SEAP reporter plasmid as well as 25 ng pRL-null renillar control plasmid using Lipofectamine (Invitrogen). pRL-null renillar plasmid was used to normalize the transfection efficiency. Twenty-four hours post-transfection, the secreted SEAP activities in the culture medium and the renillar luciferase activities within cellular lysates were assayed per the manufacturer’s specifications (Clontech and Promega, respectively) and measured using a luminometer (Molecular Devices).
Preparation of Nuclear Extracts, co-immunoprecipitation and western blot
Isolation of total cellular and nuclear extracts, immunoprecipitation, and western blot were as described previously (Huang et al., 2004).
Real-time PCR analyses
Total cellular RNAs were harvested using the Trizol method as previously described (Huang et al., 2004). Reverse transcription and subsequent real-time PCR analyses were performed using the cyber green method on a BIO-RAD IQ5 thermocycler. The relative levels of COX2 messages were calculated based on the ΔΔCt value using GAPDH as the control. The relative level of COX2 in untreated resting wild type cells was adjusted to 1 and served as the basal reference value.
High-fat-diet feeding and visualization of atherosclerosis plaques
ApoE-/- and APoE-/-/IRAK-1-/- mice were fed with high-fat-diet (Harlan Teklad 94059) for 3 month. Blood pressures were measured by the tail cuff method using the Hatteras MC-4000 Blood Pressure Analysis System (Cary, NC). At the end of the feeding regimen, mice were euthanized with isoflurane and perfused with PBS. The entire mouse aorta was dissected from the proximal ascending aorta to the bifurcation of the iliac artery under a dissecting microscope. Adventitial fat was removed, and the aorta was opened longitudinally, pinned flat onto black dissecting wax, stained with Sudan IV, and photographed at a fixed magnification. The photographs were digitized, and total aortic areas and lesion areas were calculated by using Adobe Photoshop version 7.0 and NIH Scion Image software. The results were reported as a percentage of the total aortic area that contained lesions. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Tech.
Statistical analyses
Statistical significance was determined using the unpaired 2-tailed Student’s t-test or 1-way ANOVA corrected for multiple comparisons where appropriate. P values less than 0.05 were considered statistically significant.
RESULTS
IRAK-1 suppresses NFAT transcriptional activity
In order to determine whether IRAK-1 may regulate NFAT function, we performed the luciferase reporter assay. As shown in Figure 1, co-transfection of wild type IRAK-1 plasmid with the NFAT-responsive reporter pNFAT-SEAP plasmid led to a dramatic and significant suppression of the NFAT-SEAP reporter activity. In contrast, co-transfection of IRAK-M plasmid with the NFAT-SEAP reporter plasmid had no inhibitory effect on NFAT reporter activity. This finding suggests that IRAK-1 can negatively modulate NFAT function.
Figure 1.
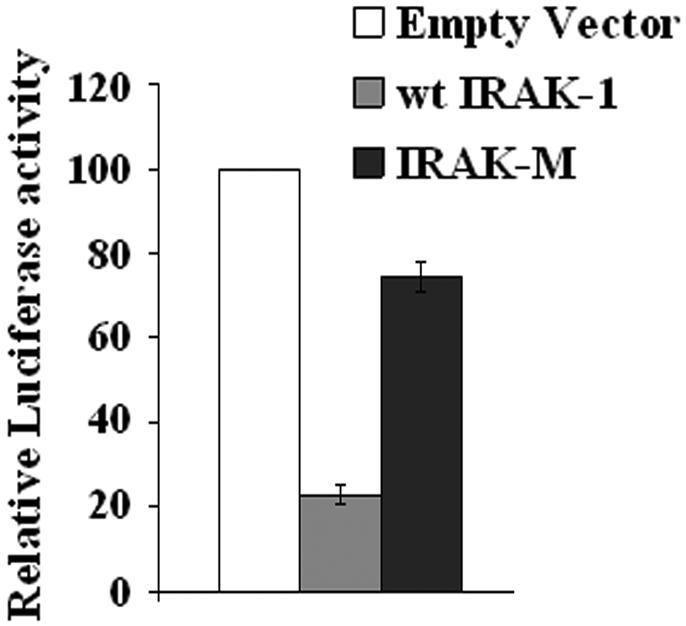
IRAK-1 specifically inhibits NFAT transcriptional activity. MAT2 cells were transfected with the NFAT-SEAP reporter plasmid, the pRL-null renilla luciferase control plasmid, and 125 ng of the empty pFlag vector, pFlag-IRAK-1, or pFlag-IRAK-M plasmid as described in the Materials and Methods. 24 hrs after the transfection, the secreted SEAP luciferase activities in the culture medium were measured and normalized with the renillar luciferase activities within the cell lysates. The relative NFAT-SEAP reporter activity in cells transfected with the empty pFlag vector was set at an arbitrary value of 100. The plot represents the average of three independent experiments. *p<0.01.
IRAK-1 deletion leads to elevated NFAT nuclear distribution
We subsequently examined the nuclear levels of NFAT in wild type and IRAK-1-/- cells. As shown in Figure 2, there was no NFATc1 or NFATc4 in untreated resting wild type nuclear lysates. Following Pam3CSK4 challenge, there was a slight increase of nuclear NFATc4 in the wild type MEFs, potentially due to the activation of calcium signaling and calcenurin. In contrast, IRAK-1-/- MEFs have constitutively noticeable levels of both NFATc1 and NFATc4 at the resting state, as well as after stimulation. This result suggests that IRAK-1 is responsible for constitutively maintaining NFATc4 at a resting state by phosphorylating NFATc4. The total as well as the cytoplasmic levels of NFATc4 remained similar (data not shown).
Figure 2.

NFATc1 and NFATc4 nuclear levels are elevated in IRAK-1-/- cells. Wild type or IRAK-1-/- MEFs were treated with 100ng/ml Pam3CSK4 for the indicated time periods. Equal amounts of prepared nuclear extracts were analyzed by SDS-PAGE and Western blot for NFATc1 and NFATc4. The levels of lamin B were probed using the anti-lamin B antibody and served as equal loading controls.
IRAK-1 contributes to NFATc4 phosphorylation at the S168PS170P site
Upon examining the primary amino acid sequences of NFAT molecules, we noticed the presence of multiple Serine-Proline motifs within the N-terminal NFAT-Homology-Region (NHR) on all NFAT members. Since IRAK-1 can auto-phosphorylate itself at its Serine-Proline rich region, we tested whether IRAK-1 may be involved in phosphorylating NFAT molecules at their Serine-Proline motifs. There is a commercial antibody available that can specifically detect phosphorylated NFATc4 at its Ser-Pro site (S168PS170P). We therefore probed for the levels of phosphorylated NFATc4 at the S168PS170P site in wild type and IRAK-1-/- MEFs. Total cellular lysates were harvested from wild type and IRAK-1-/- MEFs. The levels of phosphorylated NFATc4 at S168PS170P position were detected by Western blot. As shown in Figure 3, NFATc4 was constitutively phosphorylated at the S168SP170P site in untreated resting wild type cells, but not in IRAK-1-/- MEFs. Following Pam3CSK4 challenge, there was a slight decrease in NFATc4 phosphorylation in wild type cells, potentially due to the activation of calcium signaling and calcenurin by TLR2 independent of IRAK-1. In contrast, NFATc4 remained unphosphorylated at the S168SP170P site in IRAK-1-/- MEFs. This finding is consistent with our above data showing persistent nuclear localization of NFATc4 in untreated resting IRAK-1-/- MEFs.
Figure 3.

NFATc4 S168PS170P phosphorylation is controlled by IRAK-1. Wild type and IRAK-1-/- MEFs were treated with 100ng/ml Pam3CSK4 for the indicated time periods. Equal amounts of harvested total cell lysates were analyzed by SDS-PAGE and Western blot using anti-NFATc4-phosphor- S168PS170P antibody. The levels of total NFATc4 were probed using anti-NFATc4 antibody. The relative levels of phosphorylated NFATc4 vs the total NFATc4 were quantified through densitometry, and plotted at the bottom panel. The plot represents the average values of three experiments.
IRAK-1 physically associates with NFATc4
Next we asked whether IRAK-1 may form a close complex with NFATc4, and consequently contribute to NFATc4 phosphorylation. The co-immunoprecipitation experiment was performed using anti-IRAK-1 antibody with human monocytic THP-1 cells. Co-immunoprecipitated cell lysates were separated on SDS-PAGE, and the presence of NFATc1 and NFATc4 were detected by Western blot. As shown in Figure 4A, we can detect both NFATc4 and NFATc1 in the IRAK-1 immuno-complex in untreated resting cells.
We further studied the domains involved in their interaction. The pEGFP-NFATc4, pEGFP-NHR, and pEGFP-ΔNHR plasmids were generated as described in the Materials and Methods, which encode respectively the EGFP fusion protein with the wild type full-length NFATc4, the NHR region only, or the truncated NFATc4 with the deletion of the N-terminal NHR region (Figure 4B). These plasmids were transiently transfected into MAT-2 cells as described (Li et al., 2004). NFAT fusion proteins were immunoprecipitated from cell lysates using the anti-GFP antibody, and separated on SDS-PAGE. The presence of IRAK-1 in the immunoprecipitates was detected through western blot. As shown in Figure 4B, we can detect IRAK-1 in the immuno-complex of the wild type EGFP-NFATc4 as well as the EGFP-NHR. However, we can not detect IRAK-1 within the EGFP-ΔNHR complex. Our result indicates that the N-terminal NHR region of NFATc4 is required for its interaction with IRAK-1. Subsequently, in order to determine the IRAK-1 domain involved in the interaction, we co-transfected MAT-2 cells with the pEGFP-NFATc4 plasmid and either the pFlag-IRAK-1 or the pFlag-IRAK-1ΔC plasmid. Following immunoprecipitation using anti-GFP antibody, we separated the immunoprecipitates on SDS-PAGE and probed the presence of expressed IRAK-1 proteins using anti-Flag antibody. As shown in Figure 4C, deletion of the C-terminal region of IRAK-1 ablated its interaction with NFATc4. Taken together, we conclude that the N-terminal NHR region of NFATc4 and the C-terminal region of IRAK-1 are required for their interaction.
Functional IRAK-1 is required for suppressing NFATc4 activity
Although IRAK-1 is an active kinase, the physiological relevance for its kinase activity has not been clearly defined. We therefore studied whether an intact and functional IRAK-1 is required for functionally suppressing NFAT activity. Using the luciferase reporter assay as described above, we co-transfected the cells with NFAT-SEAP reporter plasmid and either the wild type IRAK-1, the kinase-dead IRAK-1(D340N), IRAK-1B which is also kinase-dead, or IRAK-1ΔC which fails to associate with NFATc4. The normalized reporter SEAP activities were plotted in Figure 5. We observed that only the wild type IRAK-1 can suppress the NFATc4 reporter activity. In contrast, the kinase-dead IRAK-DN, IRAK-1B, or IRAK-1ΔC mutant all failed to suppress NFAT reporter activity.
Figure 5.
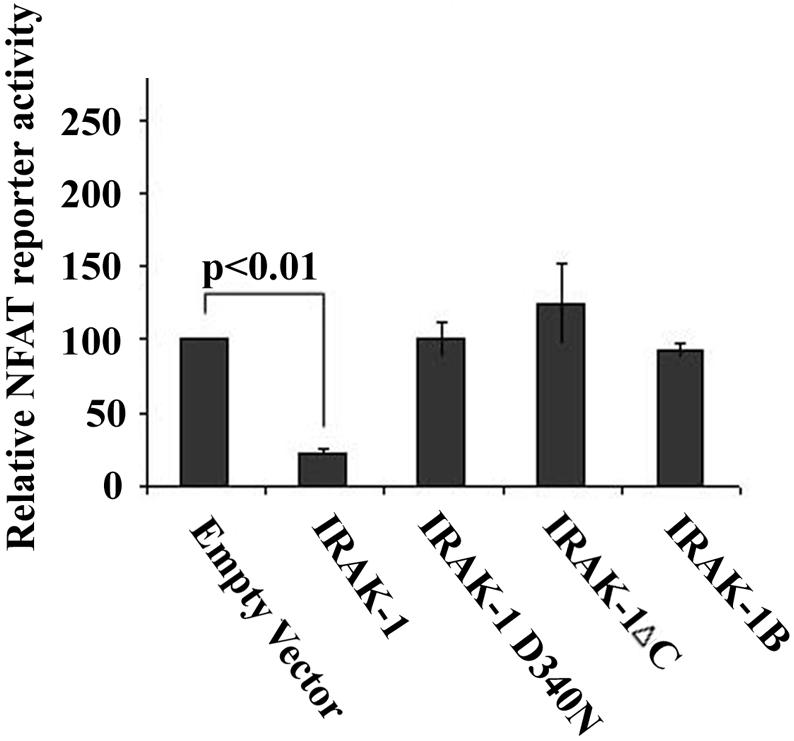
IRAK-1 variants fail to inhibit NFAT transcriptional activity. MAT2 cells were transfected with the NFAT-SEAP reporter plasmid, the pRL-null renilla luciferase control plasmid, and either the empty pFlag vector, pFlag-IRAK-1, pFlag-IRAK-1D340N, pFlag-IRAK-1ΔC, or pFlag-IRAK-1B plasmid as described in the Materials and Methods. 24 hrs after the transfection, the secreted SEAP luciferase activities in the culture medium were measured and normalized with the renillar luciferase activities within the cell lysates. The relative NFAT-SEAP reporter activity in cells transfected with the empty pFlag vector was set at an arbitrary value of 100. The plot represents the average of five independent experiments.
The IRAK-1 deletion leads to increased COX2 expression
NFAT has been linked with the expression of COX2 gene (Yiu and Toker, 2006). We therefore tested whether IRAK-1 may contribute to the modulation of COX2 gene expression. The wild type or IRAK-1-/- BMDMs were stimulated with Pam3CSK4 for six hours, and the cellular RNAs were harvested from the untreated and treated cells. The levels of COX2 messages were determined by real-time PCR analyses. As shown in Figure 6, both the untreated resting and stimulated IRAK-1-/- cells expressed significantly higher levels of COX2 message compared with the wild type cells.
Figure 6.
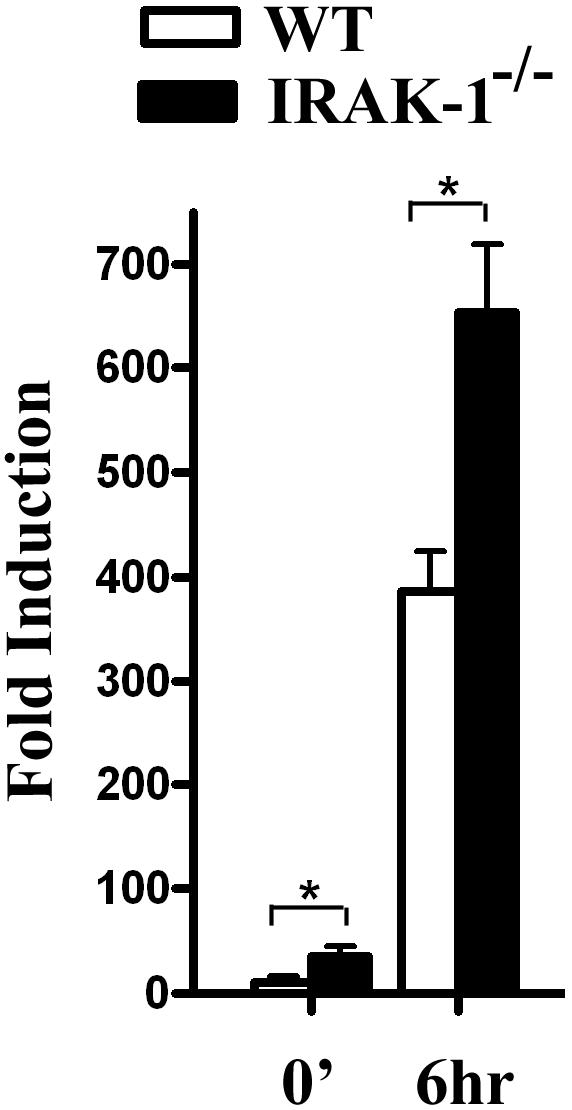
IRAK-1 deletion leads to increased COX2 expression. BMDMs from wild type and IRAK-1-/- mice were treated with Pam3CSK4 for six hours. Equal amounts of harvested total RNAs were used to perform real-time PCR analyses of COX2 expression. The relative fold induction of COX2 message was normalized with GAPDH. The plot represents three independent experiments. * p<0.01.
IRAK-1-/- mice were protected from high-fat-diet (HFD)-induced atherosclerosis and hypertension
COX2 is an important enzyme responsible for generating various prostogladins such as PGE2 which are cardio-protective by preventing thrombosis. We therefore tested whether IRAK-1-/- mice may be protected from high-fat-diet-induced atherosclerosis and hypertension. ApoE-/- and ApoE-/-/IRAK-1-/- mice were fed with the Western diet for a four-month period. The blood pressure was measured by the tail-cuff method. The deposition of lipid plaques on the artery vessel wall was visualized by Sudan-IV staining. As shown in Figure 7, ApoE-/-/IRAK-1-/- mice had significantly lower blood pressure and lipid plaque deposition compared with ApoE-/- mice.
Figure 7.

IRAK-1 deletion alleviates high-fat-diet-induced cardiovascular complications. Ten 8-weeks-old male ApoE-/- and ApoE-/-/IRAK-1-/- mice were fed with high-fat-diet as described in the Materials and Methods for three month. A) Blood pressures were measured by the tail-cuff method. *p<0.02. B) Dissected mice aortae were stained with Sudan IV. The percentage of lesion areas vs the total surface aorta areas are presented.
DISCUSSION
We have collected several lines of evidence indicating that NFATc4 is a physiological target of IRAK-1. First, IRAK-1 inhibits NFAT reporter activity. Second, IRAK-1 forms a complex with NFATc4 via the N-terminal NHR region of NFATc4 and the C-terminus of IRAK-1. Third, IRAK-1 contributes to the constitutive NFATc4 phosphorylation at the S168PS170P site. Consequently, the deletion of IRAK-1 leads to constitutive NFATc4 nuclear localization and elevated downstream gene expression such as COX2. IRAK-1-/- mice were also protected from high-fat-diet-induced cardiovascular complication.
Our data indicate that IRAK-1 may serve as a constitutively active kinase phosphorylating NFATc4 and maintaining NFATc4 in an inactive state. There are two classes of kinases that contribute to the regulation of NFAT, the maintenance kinases and the export kinases (Macian, 2005). The maintenance kinases primarily help to keep NFAT in the phosphorylated and inactive state. In contrast, the export kinases phosphorylate the once-active NFAT family proteins and return them back to the inactive state (Macian, 2005). Multiple phosphorylation motifs within the N-termini of NFAT molecules are subjected to phosphorylation by either the maintenance kinases or the export kinases. For example, Casein kinase 1 (CK1) serves as a maintenance kinase phosphorylating the serine-rich regions (SRRs) of NFAT (Macian, 2005). On the other hand, glycogen synthase kinase 3-beta (GSK3β) was shown to be the export kinase phosphorylating the Serine-Proline motifs of NFAT (Macian, 2005). However, the maintenance kinases responsible for constitutively phosphorylating the Ser-Pro motifs of NFAT have not been clearly identified. Intriguingly, a recent study primarily employing the inhibitor approach reveals that ERK5 and mTOR may be responsible for phosphorylating NFATc4 S168PS170P site (Yang et al., 2008). In vitro assays demonstrated that ERK5 or mTOR can phosphorylate NFATc4. Furthermore, ERK5-/- cells had elevated levels of nuclear NFATc4 following cyclosporine A treatment (Yang et al., 2008). However, the patterns of NFATc4 phosphorylation and nuclear distribution were similar among naïve untreated resting wild type and ERK5-/- cells, suggesting that ERK5 may not be fully responsible for the constitutive phosphorylation of NFATc4 in the resting cells. It is therefore likely that multiple kinases are involved in NFATc4 phosphorylation under distinct cellular context and stimulation, in order to ensure a stringent control of NFATc4 activation. We herein demonstrate that NFATc4 is constitutively phosphorylated at the S168PS170P site in the untreated resting wild type, but not the IRAK-1-/- cells. Consequently, untreated resting IRAK-1-/- cells have constitutively higher levels of nuclear NFATc4. Our data indicate that IRAK-1 is a maintenance kinase responsible for phosphorylating NFATc4 in untreated resting cells. This conclusion is further corroborated by our finding that IRAK-1 forms a constitutive complex with NFATc4. As a consequence, IRAK-1-/- cells have constitutively higher levels of nuclear NFATc4 at the resting state, leading to elevated expression of COX2. However, it remains likely that IRAK-1 and ERK may also be closely related during the phosphorylation of NFATc4. We have previously reported that IRAK-1 deficiency selectively attenuates ERK activation (Su et al., 2007). Therefore, IRAK-1 may contribute to the regulation of NFATc4 phosphorylation both directly and indirectly, which explains the striking difference we observed in NFATc4 phosphorylation and nuclear distribution among the untreated naïve wild type and IRAK-1-/- cells.
Phenotypically, we found that HFD-fed ApoE-/-/IRAK-1-/- mice have reduced blood pressure and atherosclerotic plaque deposition in the arteries. Excessive NFAT activities have been linked with the risk of hypertrophic growth and hypertension (Molkentin et al., 1998). However, cardiovascular complications can be triggered by multiple factors and involve multiple cells and tissues. Consequently, distinct and seemingly opposite physiological outcomes may occur due to the differential activation of NFAT-mediated signaling pathways following various challenges in distinct cells or tissues. It is likely that the contribution of NFAT to hypertension may be more prominent when angiotensin or other hypertrophy-inducing factors are highly elevated (Olson, 2004). On the other hand, when the major cardiovascular risk factor is high-fat-diet, the hypertrophic proliferation of smooth muscle cells may not be a predominant issue. Instead, the deposition of lipid plaques, altered regulation of endothelia, fibroblasts and macrophages, as well as elevated thrombosis may be the primary contributors for hypertension (Mullick et al., 2008). Under this scenario, the elevation of NFAT activity and increased downstream gene expression such as COX-2 may actually help to prevent lipid deposition and thrombosis, therefore exerting a beneficial effect. This reminds of major cardiovascular complications people encountered when the COX2 inhibitor was initially developed and administered (Salzberg and Weir, 2007). Clearly, further detailed analyses regarding the relevance of IRAK-1 mediated NFATc4 regulation in various cells such as endothelial cells, smooth muscle cells, mononuclear cells, and T cells are warranted.
Our study also reveals that IRAK-1 may be involved in the regulation of multiple NFAT molecules such as NFATc1 and NFATc4. Although our current study has focused on defining the molecular details regarding the interaction of IRAK-1 and NFATc4, our unpublished studies using T cells indicate that NFATc2 is also subjected to the regulation of IRAK-1 following T Cell Receptor (TCR) ligation. It is therefore tantamount and necessary to study the differential contribution of IRAK-1 to distinct NFAT molecules and relevant downstream gene expression in primary macrophages, endothelial cells, smooth muscle cells, and T cells. Furthermore, it will be important to define whether different ligands (such as angiotensin, TCR ligands, etc) may elicit distinct IRAK-1 mediated NFAT regulation within individual cell type.
Acknowledgments
This work is supported in part by grants from the National Institute of Health. We thank technical assistance from Lin Zhang during the course of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996;271:1128–31. doi: 10.1126/science.271.5252.1128. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Deng C, Radu C, Diab A, Tsen MF, Hussain R, Cowdery JS, Racke MK, Thomas JA. IL-1 receptor-associated kinase 1 regulates susceptibility to organ-specific autoimmunity. J Immunol. 2003;170:2833–42. doi: 10.4049/jimmunol.170.6.2833. [DOI] [PubMed] [Google Scholar]
- Gan L, Li L. Regulations and roles of the interleukin-1 receptor associated kinases (IRAKs) in innate and adaptive immunity. Immunol Res. 2006;35:295–302. doi: 10.1385/IR:35:3:295. [DOI] [PubMed] [Google Scholar]
- Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev. 2001;11:505–12. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]
- Ho IC, Kim JH, Rooney JW, Spiegelman BM, Glimcher LH. A potential role for the nuclear factor of activated T cells family of transcriptional regulatory proteins in adipogenesis. Proc Natl Acad Sci U S A. 1998;95:15537–41. doi: 10.1073/pnas.95.26.15537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Jacinto R, McCall C, Li L. Regulation of IL-1 receptor-associated kinases by lipopolysaccharide. J Immunol. 2002;168:3910–4. doi: 10.4049/jimmunol.168.8.3910. [DOI] [PubMed] [Google Scholar]
- Huang Y, Li T, Sane DC, Li L. IRAK1 serves as a novel regulator essential for lipopolysaccharide-induced interleukin-10 gene expression. J Biol Chem. 2004;279:51697–703. doi: 10.1074/jbc.M410369200. [DOI] [PubMed] [Google Scholar]
- Ichida M, Finkel T. Ras regulates NFAT3 activity in cardiac myocytes. J Biol Chem. 2001;276:3524–30. doi: 10.1074/jbc.M004275200. [DOI] [PubMed] [Google Scholar]
- Im SH, Rao A. Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol Cells. 2004;18:1–9. [PubMed] [Google Scholar]
- Kim JA, Yeh DC, Ver M, Li Y, Carranza A, Conrads TP, Veenstra TD, Harrington MA, Quon MJ. Phosphorylation of Ser24 in the pleckstrin homology domain of insulin receptor substrate-1 by Mouse Pelle-like kinase/interleukin-1 receptor-associated kinase: cross-talk between inflammatory signaling and insulin signaling that may contribute to insulin resistance. J Biol Chem. 2005;280:23173–83. doi: 10.1074/jbc.M501439200. [DOI] [PubMed] [Google Scholar]
- Kollewe C, Mackensen AC, Neumann D, Knop J, Cao P, Li S, Wesche H, Martin MU. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J Biol Chem. 2004;279:5227–36. doi: 10.1074/jbc.M309251200. [DOI] [PubMed] [Google Scholar]
- Kuglstatter A, Villasenor AG, Shaw D, Lee SW, Tsing S, Niu L, Song KW, Barnett JW, Browner MF. Cutting Edge: IL-1 receptor-associated kinase 4 structures reveal novel features and multiple conformations. J Immunol. 2007;178:2641–5. doi: 10.4049/jimmunol.178.5.2641. [DOI] [PubMed] [Google Scholar]
- Li T, Hu J, Li L. Characterization of Tollip protein upon Lipopolysaccharide challenge. Mol Immunol. 2004;41:85–92. doi: 10.1016/j.molimm.2004.03.009. [DOI] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–28. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullick AE, Soldau K, Kiosses WB, Bell TA, 3rd, Tobias PS, Curtiss LK. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J Exp Med. 2008;205:373–83. doi: 10.1084/jem.20071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnuma K, Yamochi T, Uchiyama M, Nishibashi K, Iwata S, Hosono O, Kawasaki H, Tanaka H, Dang NH, Morimoto C. CD26 mediates dissociation of Tollip and IRAK-1 from caveolin-1 and induces upregulation of CD86 on antigen-presenting cells. Mol Cell Biol. 2005;25:7743–57. doi: 10.1128/MCB.25.17.7743-7757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EN. A decade of discoveries in cardiac biology. Nat Med. 2004;10:467–74. doi: 10.1038/nm0504-467. [DOI] [PubMed] [Google Scholar]
- Rui L, Fisher TL, Thomas J, White MF. Regulation of insulin/insulin-like growth factor-1 signaling by proteasome-mediated degradation of insulin receptor substrate-2. J Biol Chem. 2001;276:40362–7. doi: 10.1074/jbc.M105332200. [DOI] [PubMed] [Google Scholar]
- Salzberg DJ, Weir MR. COX-2 inhibitors and cardiovascular risk. Subcell Biochem. 2007;42:159–74. doi: 10.1007/1-4020-5688-5_7. [DOI] [PubMed] [Google Scholar]
- Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA, Golenbock DT. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem. 2005;280:17005–12. doi: 10.1074/jbc.M412584200. [DOI] [PubMed] [Google Scholar]
- Su J, Xie Q, Wilson I, Li L. Differential regulation and role of interleukin-1 receptor associated kinase-M in innate immunity signaling. Cell Signal. 2007;19:1596–601. doi: 10.1016/j.cellsig.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, Cao Z, Wasserman SA. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol. 1999;163:978–84. [PubMed] [Google Scholar]
- Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–23. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G. Association of beta-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2006a;7:139–47. doi: 10.1038/ni1294. [DOI] [PubMed] [Google Scholar]
- Wang Z, Liu J, Sudom A, Ayres M, Li S, Wesche H, Powers JP, Walker NP. Crystal structures of IRAK-4 kinase in complex with inhibitors: a serine/threonine kinase with tyrosine as a gatekeeper. Structure. 2006b;14:1835–44. doi: 10.1016/j.str.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Yang TT, Yu RY, Agadir A, Gao GJ, Campos-Gonzalez R, Tournier C, Chow CW. Integration of protein kinases mTOR and extracellular signal-regulated kinase 5 in regulating nucleocytoplasmic localization of NFATc4. Mol Cell Biol. 2008;28:3489–501. doi: 10.1128/MCB.01847-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu GK, Toker A. NFAT induces breast cancer cell invasion by promoting the induction of cyclooxygenase-2. J Biol Chem. 2006;281:12210–7. doi: 10.1074/jbc.M600184200. [DOI] [PubMed] [Google Scholar]
- Zaichuk TA, Shroff EH, Emmanuel R, Filleur S, Nelius T, Volpert OV. Nuclear factor of activated T cells balances angiogenesis activation and inhibition. J Exp Med. 2004;199:1513–22. doi: 10.1084/jem.20040474. [DOI] [PMC free article] [PubMed] [Google Scholar]