

Abstract
Neuronal pentraxins (NPs) define a family of proteins that are homologous to C-reactive and acute-phase proteins in the immune system and have been hypothesized to be involved in activity-dependent synaptic plasticity. To investigate the role of NPs in vivo, we generated mice that lack one, two, or all three NPs. NP1/2 knock-out mice exhibited defects in the segregation of eye-specific retinal ganglion cell (RGC) projections to the dorsal lateral geniculate nucleus, a process that involves activity-dependent synapse formation and elimination. Retinas from mice lacking NP1 and NP2 had cholinergically driven waves of activity that occurred at a frequency similar to that of wild-type mice, but several other parameters of retinal activity were altered. RGCs cultured from these mice exhibited a significant delay in functional maturation of glutamatergic synapses. Other developmental processes, such as pathfinding of RGCs at the optic chiasm and hippocampal long-term potentiation and long-term depression, appeared normal in NP-deficient mice. These data indicate that NPs are necessary for early synaptic refinements in the mammalian retina and dorsal lateral geniculate nucleus. We speculate that NPs exert their effects through mechanisms that parallel the known role of short pentraxins outside the CNS.
Keywords: retinogeniculate, neuronal pentraxins, synaptic plasticity, LTP, long-term potentiation, LTD, long-term depression, development, knock-out, retinal ganglion cell
Introduction
In the developing CNS, synapses are either strengthened and maintained or weakened and eliminated through competitive processes that require activity (Katz and Shatz, 1996; Sanes and Lichtman, 1999). Uptake of growth factors, clustering of neurotransmitter receptors, and activity-mediated gene expression have all been put forth as factors that mediate synaptic turnover and axonal refinements. Still, the molecular mechanisms underlying these cellular processes remain incompletely understood (for review, see Cohen-Corey, 2002). The neuronal pentraxins (NPs), NP1 (Schlimgen et al., 1995) and NP2 (Noland et al., 1994; Reid and Blobel, 1994; Hsu and Perin, 1995; Tsui et al., 1996), and NP receptor (NPR) (Dodds et al., 1997) were identified as synaptic proteins that bind to affinity columns of the snake venom toxin, taipoxin (that presynaptically blocks neurotransmission), and the lumenal calcium-binding protein TCBP49 (Dodds et al., 1995). NP1, NP2, and NPR are 50% identical to each other, and their C-terminal halves are 20–30% identical to classic immune system pentraxins such as the acute phase proteins, C-reactive protein (Whitehead et al., 1990), serum amyloid P protein (Dowton and McGrew, 1990), and the long pentraxin, PTX3 (Breviario et al., 1992). Neuronal pentraxins form heteromultimers (Kirkpatrick et al., 2000). Although NP1 and NP2 are secreted when expressed alone, they can be tethered to the cell membrane when expressed as heteromultimers with NPR. NP2 (also called Narp) is dramatically upregulated by neuronal activity (Tsui et al., 1996) and can induce clustering of AMPA receptors when coexpressed in heterologous cells (O'Brien et al., 1999; Xu et al., 2003). On the basis of these properties and their homology to short pentraxins in the innate immune system that mark cells for phagocytosis and degradation by opsonization, we tested the hypothesis that NPs are required for synapse refinement and elimination in the developing CNS (Dodds et al., 1995, 1997).
We generated single mutant mice that lack NP1, NP2, or NPR, double mutants, and mice that lack all three NPs. Multisite extracellular recordings revealed that spontaneous retinal activity is intact in NP knock-out (KO) mice, but levels of retinal activity were increased. Mice lacking NPs exhibited defects in patterning of eye-specific axonal projections to the dorsal lateral geniculate nucleus (dLGN). Patch-clamp recordings of retinal ganglion cells (RGCs) cultured from NP1/2 KO mice revealed an early absence of glutamatergic synaptic transmission, despite the fact that structural synapses were intact. Other cellular processes previously shown to be associated with defects in eye-specific segregation and synaptic maturation, such as pathfinding of RGCs at the optic chiasm and hippocampal long-term potentiation (LTP) and long-term depression (LTD), appeared normal in NP KO mice. These findings indicate that NPs are necessary for normal structural and physiological maturation of the retina and dLGN. We speculate that NPs mediate synaptic refinements by marking synaptic sites for turnover and degradation in response to activity.
Materials and Methods
In situ hybridization.
Hybridization was performed according to procedures described previously (Schlimgen et al., 1995). Digoxigenin-labeled cRNAs were prepared from rat and mouse NP1, NP2, and NPR cDNAs in pBluescript with T7 and T3 polymerases.
Generation of KO mice.
The generation of NP1 KO mice has been described previously (Kirkpatrick et al., 2000). The mouse genes for NP2 and NPR were identified, sequenced (GenBank accession numbers AF318618 and AF318076), and used to generate KO constructs. For NP2, this involved replacing the first exon with a neo-resistance cassette. For NPR, the second and third exons were replaced with the same cassette. Constructs were electroporated into 129/SVJII-derived embryonic stem (ES) cells, and neomycin-resistant clones were isolated and screened for the expected restriction fragment length changes by genomic Southern blot as described (Kirkpatrick et al., 2000). For NP2, ES cell genomic DNA was digested with XhoI and ApaI. For NP2 tail analysis, BamH1 digests were used with separate probes for wild-type (WT) (exon 1 probe) or recombined (neo probe) alleles. In addition, NP2 genotypes were determined by using PCR with oligos for wild-type (5′-CCGTGGGTAATTTGGAACTTTGGGGGAG, 5′-CCTATTTCTCTCACTCCTCTGCCATCACGCTG; for 390 bp) and recombinant (5′-CAACGCTTTCTCTGACCGTTTCCGAAG, 5′-TGGGGTGGGATTAGATAAATGCCTGCTC; for 451 bp) NP2 alleles. For NPR ES cell and tail analysis, genomic DNA was digested with BglII and BamH1. Correctly targeted clones were injected into blastocysts and implanted into pseudopregnant female mice. Chimeric mice were bred to obtain mice heterozygous for the modified NP2 and NPR genes, and heterozygous mice were bred to generate mice homozygous for the mutation. Homozygous NP1, NP2, and NPR mice were bred to generate double heterozygous NP1/NP2 and NP1/NPR mice. These were bred to generate double null NP1/NP2 and NP1/NPR mice. Double null mice were bred to obtain mice null for NP1 and heterozygous for NP2 and NPR. These mice were bred to obtain mice null for NP1/NP2/NPR. All mice used in these studies were the 129/SVJII, C57BL/6 mixed background of the original single KO mice.
Northern analysis.
Total RNA was electrophoresed, blotted onto nylon membranes, and hybridized with uniformly 32P-labeled DNA probes as described previously (Perin et al., 1990).
Affinity chromatography and immunoprecipitation.
Taipoxin affinity columns were generated and used as described (Schlimgen et al., 1995; Kirkpatrick et al., 2000). For chromatographies of wild-type and KO tissue, 20 mouse brains [postnatal day 21 (P21) and older] were homogenized in 0.32 m sucrose. Brain membranes were collected by 100,000 × g spin for 1 h and solubilized at 4°C in 20 mm HEPES, pH 7.4, 1% Triton X-100, and 1 mm EGTA for 1 h; insoluble material was removed with the same centrifugation. Solubilized brain membranes were adjusted to 100 mm NaCl and 1 mm free CaCl2 and run over taipoxin columns equilibrated with the same solution. The column was washed with 20 column volumes of 100 mm NaCl, 20 mm HEPES, 1 mm CaCl2, and 0.2% Triton X-100. Columns were eluted with a NaCl2 step gradient of 30 ml each of 200, 400, 600, and 800 mm and 1 m NaCl2 in 20 mm HEPES, 1 mm CaCl2, and 0.2% Triton X-100. A final elution of 1 m NaCl2, 20 mm HEPES, 10 mm EDTA, and 0.2% Triton X-100 elutes neuronal pentraxin heteromultimers. Fractions were concentrated with centriprep-30 (Millipore, Bedford, MA) concentrators, and 1/20 of each fraction was run on SDS-PAGE gels. Immunoprecipitations were performed with the same solubilized brain membranes by using an amount derived from one brain (∼10 ml solution). Twenty microliters of polyclonal sera were added to the appropriate NP, and the solution was rotated gently for 1 h at 4°C; 100 μl of Protein A/G beads (Santa Cruz Biotechnology, Santa Cruz, CA) was added for an additional 1 h. Beads were collected by centrifugation and washed with the solubilization buffer. Protein was extracted with SDS sample buffer and run on SDS-PAGE gels.
Antibodies, Western blotting, and immunofluorescence.
Four polyclonal antibodies against NP1 (raised to residues 42–57, 419–432, 292–415, and 20–432) (Schlimgen et al., 1995), a polyclonal C-terminal anti-peptide antibody to NP2 (residues 416–429) (Hsu and Perin, 1995), and three polyclonal antibodies to NPR (residues 31–57, 472–494, and 31–494) (Dodds et al., 1997) were used as described previously (Kirkpatrick et al., 2000). In addition, an antibody to recombinant NP2 that recognizes NP1 and NP2 (residues 16–429 of NP2) was used to detect and immunoprecipitate neuronal pentraxins. SDS-PAGE (12%) was used for all experiments. After separation, gels were stained with Coomassie dye or transferred electrophoretically to nitrocellulose sheets. Blots were blocked in 20% goat sera in Tris-buffered saline with Tween 20 and incubated with primary and secondary antibodies in 10% goat sera in the same buffer. Peroxidase-conjugated secondary antibodies were detected by ECL (Amersham Biosciences, Arlington Heights, IL). Antibody immunofluorescence was performed with rabbit polyclonal antibodies to NP1 (1:500), NP2 (1:3000), or NPR (1:25 or 1:100) (provided by Dr. Paul Worley). For brain tissue, animals were perfused with 4% paraformaldehyde; eye tissue was collected and immersion fixed in 4% paraformaldehyde; both eye and brain tissue were cryoprotected in 30% sucrose overnight. dLGN staining was performed on free-floating, 25-μm-thick, microtome-cut sections. Retinal staining was performed on 10-μm-thick cryosections directly on the slide. Goat anti-rabbit Alexa 488 dye (1:500) was used for secondary detection on retinal tissue. WT and KO tissues of a given age were processed in parallel.
Labeling of retinogeniculate afferents.
Mouse pups were anesthetized with isoflurane inhalant. Mice received intravitreal injections of cholera toxin-β subunit (CTβ) conjugated to Alexa 488 dye (green label) into the left eye and CTβ conjugated to Alexa 594 dye (red label) into the right eye (2–3 μl per eye; 0.5% in sterile saline; Invitrogen, Eugene, OR). Mice were transcardially perfused 24 h later with 4% paraformaldehyde (ages in given in text correspond to age at which mice were killed). Brain tissue was postfixed overnight, cryoprotected in 30% sucrose, and then sectioned coronally at 40 μm, mounted onto gelatin-coated slides, and coverslipped with Vectashield (Vector Laboratories, Burlingame, CA).
Image quantification and preparation of photomicrographs.
Images were digitally acquired with a 3900 × 3600 pixel color CCD camera (Axiocam; Zeiss, Thornwood, NY) or an RT SPOT camera. The same gains and image exposure times were used for each label in all specimens to limit variability caused by imaging procedures. Raw images of the dLGN were imported to Photoshop (Adobe) and cropped to exclude the ventral LGN and the intergeniculate leaflet. The degree of left and right eye axon overlap was quantified with the multi-threshold protocol described in Torborg et al. (2005). This technique is designed to compare overlap across a range of signal–noise values in WT versus transgenic mice. This approach best allows for direct statistical comparison of overlap between various strains of mice at different ages and has been used by others (Pak et al., 2004). After quantification, images were imported to Photoshop (Adobe) for adjustments to intensity, cropping, and alignment. In some cases, artifact was removed from outside the boundaries of the dLGN.
Multi-electrode extracellular recordings from retinal explants.
Mice were killed by decapitation on P4 or P5, and an eye was removed. The neural retina was dissected and placed with the ganglion cell side down in a recording chamber with a piece of nitrocellulose paper as support. The multi-electrode chambers (MEA-60 system; Multi Channel Systems MCS, Reutlingen, Germany) consisted of an array of 60 planar electrodes in eight rows spaced 100 μm apart for a total array size of 0.49 mm2. Retinas were perfused at room temperature for 30 min and at 34°C for another 30 min before recordings were started, and the temperature was maintained at 34°C throughout the recordings. The saline consisted of (in mm): 124 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 22.2 glucose; the pH was maintained at 7.3–7.4 by bubbling with 95%O2/5% CO2 mixed gas. Acquired voltage signals were bandpass filtered at 0.1 Hz to 3 kHz and sampled at 5 kHz (MC Rack software; Multi Channel Systems MCS). After recording, action potential waveforms from high-pass-filtered data were clustered on the basis of the first two principal components as described previously (Tian and Copenhagen, 2001) with a k-means algorithm (OfflineSorter, v.1.3; Plexon, Dallas, TX). The algorithm eliminated outlier waveforms at a threshold of 1.3 times the mean distance from the calculated cluster center. Obvious automatic sorting errors were corrected manually for each cluster.
The dataset consisted of 493 neurons recorded from 10 WT retinas and 360 neurons from 7 NP1/2 KO retinas. Timestamps for each action potential of each sorted unit were used for spike train analyses with NeuroExplorer (v.2.6; Nex Technologies, Dallas, TX), IGOR Pro (v.4.0; WaveMetrics, Lake Oswego, OR), and Excel (Microsoft, Redding, WA). Correlation indices were calculated as described in Wong et al. (1993). Different time windows for the calculation (from 10 to 200 ms) had only minor effects on the correlation index curves. Spike rates were determined based on the total number of spikes assigned to each RGC divided by the total time of the recording (5 min). Synchronous spikes between pairs of neurons (Torborg et al., 2005) were counted with a time window of 100 ms. The median value of these spike numbers for all RGC pairs at distances of ≥600 μm (i.e., at least six electrodes apart on the array) was subtracted from the median value for all RGC pairs at distances of <150 μm (i.e., one electrode apart) as a measure of the difference in correlation between near and distant RGC pairs. The median instantaneous frequencies for each RGC were calculated as the reciprocal of the interspike interval for every pair of adjacent spikes in the spike train. Interspike intervals of ≤100 ms were counted to determine the amount of spikes fired at rates of ≥10 Hz. These values were normalized by the spikes per minute value for each neuron. Autocorrelograms for the spike train of each RGC were generated with a time axis of ±10 s and a bin width of 15 ms, and a measure of the burst duration for each RGC was calculated as the full width at half height of the Gaussian fit to these autocorrelograms (Demas et al., 2003). Because RGCs can fire very short bursts of several spikes that appear different from the longer bursts that make up waves, only burst durations >0.5 s were used for determination of averages. Interwave intervals were determined from representative units for each retina. The interval was taken to be the time between peaks in the spike rate histogram. These peaks occurred during the individual bursts that make up the waves. Both the Mann–Whitney U test and Student's t test were used for statistical comparisons. Because the different parameters of spikes and waves were generated from the same dataset of recordings and compared with each other (with the null hypothesis that NP1/2 KO values did not differ from WT values), the p value accepted to indicate a significant difference was adjusted downward to 0.01 (i.e., a Bonferroni correction was applied) to protect against false positives in the tests.
Purification and culture of RGCs.
RGCs were purified as described previously (Barres et al., 1988). RGCs were purified by sequential immunopanning to >99.5% purity from P5 mice or Sprague Dawley rats (Simonsen Laboratories, Gilroy, CA). Approximately 30,000 RGCs were cultured per well in 24-well plates (Falcon, Franklin Lakes, NJ) on glass (Assistant) or Aclar 22C (AlliedSignal) coverslips coated with poly-d-lysine (10 μg/ml) followed by laminin (2 μg/ml). RGCs were cultured in 600 μl of serum-free medium, containing Neurobasal (Invitrogen, Gaithersburg, MD), bovine serum albumin, selenium, putrescine, triiodo-thyronine, transferrin, progesterone, pyruvate (1 mm), glutamine (2 mm), CNTF (10 ng/ml), BDNF (50 ng/ml), insulin (5 μg/ml), and forskolin (10 μm). Recombinant human BDNF and CNTF were generously provided by Regeneron Pharmaceuticals (Tarrytown, NY). TTX and picrotoxin from RBI. All other reagents were obtained from Sigma (St. Louis, MO).
Preparation of astrocytes.
Cortical glia were prepared as described (Ullian et al., 2001). Briefly, P1–P2 cortices were papain digested and plated in tissue culture flasks (Falcon) in a medium that does not allow neurons to survive [DMEM; fetal bovine serum (10%), penicillin (100 U/ml), streptomycin (100 μg/ml), glutamine (2 mm) and Na-pyruvate (1 mm)]. After 4 d, nonadherent cells were shaken off the monolayer, and cells were incubated for an additional 2–4 d to allow the monolayer to refill. Medium was replaced with fresh medium containing AraC (10 μm) and incubated for 48 h. Astrocytes were trypsinized and plated onto 24-well inserts (Falcon; 1.0 μm) or 10 cm tissue culture dishes.
In vitro RGC electrophysiology.
Membrane currents were recorded by whole-cell patch clamping at room temperature (18°–22°C) at a holding potential of −70 mV unless specified otherwise. Patch pipettes (3–10 MΩ) were pulled from borosilicate capillary glass (World Precision Instruments, Sarasota, FL). For recordings of synaptic or whole-cell glutamate currents, the bath solution contained (in mm): 120 NaCl, 3 CaCl2, 2 MgCl2, 5 KCl, and 10 HEPES, pH 7.3. The internal solution contained (in mm): 100 K-gluconate, 10 KCl, 10 EGTA (Ca2+ buffered to 10-6), and 10 HEPES, pH 7.3. For recordings of autaptic currents, the internal solution contained (in mm): 122.5 K-gluconate, 8 NaCl, 10 HEPES, 0.2 EGTA, 2 Mg-ATP, 0.3 Na-GTP, 20 K2-creatine phosphate, and phosphocreatine kinase (50 U/ml). Currents were recorded with pClamp software for Windows (Molecular Devices, Foster City, CA). Glutamate and CNQX (250 mm) were rapidly applied by a quartz microtube array (Superfusion System; ALA Scientific Instruments, Westbury, NY). Miniature EPSCs (mEPSCs) were analyzed with the Mini Analysis Program (SynaptoSoft, Decatur, GA) and plotted with SigmaPlot (SPSS, Chicago, IL) or Origin (Microcal, Northampton, MA).
Electrophysiology in hippocampal slices.
Transverse hippocampal slices (350 μm) were obtained from 3- to 4- week-old mice and allowed to recover in a holding chamber for at least 1 h before transfer to a recording chamber. Slices were maintained at room temperature and perfused in external solution containing (in mm): 119 NaCl, 2.5 KCl, 4 MgSO4, 4 CaCl2, 1 NaH2PO4, 26.2 NaHCO3, 10 d-glucose, and 0.1 picrotoxin (saturated with 95% O2/ 5% CO2), pH 7.4. Extracellular field EPSPs were recorded with a patch pipette filled with external solution. Schaffer collateral–commissural afferents in stratum radiatum were stimulated at 0.1 Hz with patch recording pipettes filled with extracellular solution. The field EPSP initial slope was monitored on-line with custom software. For inducing LTP, a 100 Hz/1 s tetanus was given four times with an intertrain interval of 20 s. For inducing LTD, 900 pulses were applied at 3 Hz (5 min total).
Primary hippocampal neuron culture.
Primary cultures of mouse hippocampal neurons on mixed glia were prepared from P0 mouse pups plated on poly-d-lysine/collagen-coated glass coverslips maintained according to standard protocols in Neurobasal/B27 media (Banker and Goslin, 1991). Beds of astrocytes were prepared from P4 hippocampus, grown to confluency, and then treated with AraC to inhibit cell division. Neurons were cultured for 7 d and then imaged for immunofluorescence microscopy.
Visual electrophysiology.
After overnight dark adaptation, mice were anesthetized (ketamine, 80 mg/kg; xylazine, 16 mg/kg), and the pupils were dilated (1% tropicamide, 2.5% phenylephrine HCl, 1% cyclopentolate) and anesthetized (0.5% proparacaine HCl). Electroretinograms were recorded in response to strobe flashes presented in a Ganzfeld bowl under dark- and light-adapted conditions by using protocols described in detail previously (Ren et al., 2000; Ball et al., 2002). In separate sessions, visual evoked potentials were recorded by using a previously described technique (Ren et al., 2000).
Behavioral task.
Visual learning was evaluated with a two-way active avoidance shuttle box paradigm that has been described previously (Ball et al., 2002; Gregg et al., 2003).
Retrograde labeling of RGCs.
Small (∼0.5 mm) DiI crystals or 1 μl of CTβ conjugated to a fluorescent dye, Alexa 594 (Invitrogen), were injected into the rostral and middle superior colliculus of one hemisphere in P8 NP1/2 KO mice (n = 10). To avoid leak of tracer into the opposite hemisphere, care was taken to ensure that the injections did not enter the medial superior colliculus.
Results
Developmental expression of neuronal pentraxins in the visual system
Neurons of the hippocampus, cerebellum, and cerebral cortex express NP1, NP2, and NPR (Hsu and Perin, 1995; Schlimgen et al., 1995; Tsui et al., 1996; Dodds et al., 1997), but most midbrain and thalamic structures, including the dLGN, lack NP expression by in situ hybridization. Gene chip experiments showed the presence of neuronal pentraxins in cultured purified retinal ganglion cells (data not shown). In situ hybridization for NP message on rat and mouse retinas indicated that NP1 (Fig. 1A), NP2 (Fig. 1B), and NPR (Fig. 1C) mRNAs are prominent in cells within the RGC layer and inner nuclear layer of the retina. No specific labeling was observed with sense controls (Fig. 1E). There are no gradients of NP message apparent in transverse retinal sections (Fig. 1F) or retinal whole mounts (data not shown). In situ hybridization of mouse retina showed results similar to those observed in rat, although NP1 and NPR signals were lower in mouse tissues, which was expected given the ∼10-fold lower levels of NP1 and NPR detected in mouse brain by Northern analysis.
Figure 1.
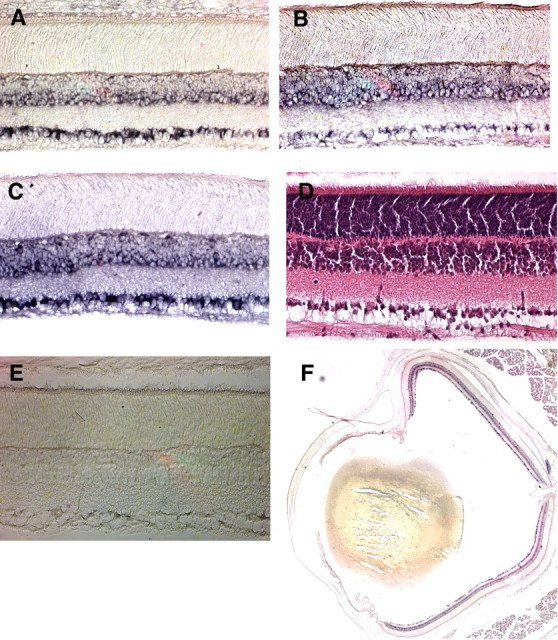
In situ hybridization for NP1, NP2, and NPR mRNAs in the retinal ganglion cell and inner nuclear layers of the retina. Transverse sections of rat eyes were hybridized with digoxigenin-labeled antisense cRNA probes to NP1 (A), NP2 (B), or NPR (C). D, Hematoxylin and eosin-stained section to reveal cell bodies and retinal layers. E, Sections were also incubated with NP1, NP2, and NPR sense probes. A representative section, with lack of specific signal, is shown here for the NP1 sense probe. F, Low-power image of NP1 mRNA localization, showing lack of a gradient for NP1 expression.
To localize NP proteins in the retina, we processed retinal sections from P5, P7, and P14 WT mice for NP1, NP2, or NPR immunohistochemistry. At each age examined, all three NPs were expressed by cells in the ganglion cell layer (Fig. 2A–I). Expression was also observed in the inner nuclear (Fig. 2G,H) and inner plexiform layers (Fig. 2B,G–I). NP2 immunoreactivity was particularly prominent, although this likely represents the higher affinity of this antibody. No gradients of NP protein expression were observed in retinal whole mounts (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Immunolocalization of NPs represents both cell-tethered NP heteromultimers and proteolytically released heteromultimers. NPs can be isolated by taipoxin chromatography from both soluble fractions (containing clipped NPR) and membrane fractions (containing transmembrane NPR) (Kirkpatrick et al., 2000). NP1, NP2, and NPR immunoreactivity was absent in transgenic mice lacking NP1, NP2, and NPR (Fig. 2J–L), indicating specificity of antibody labeling in WT mice.
Figure 2.
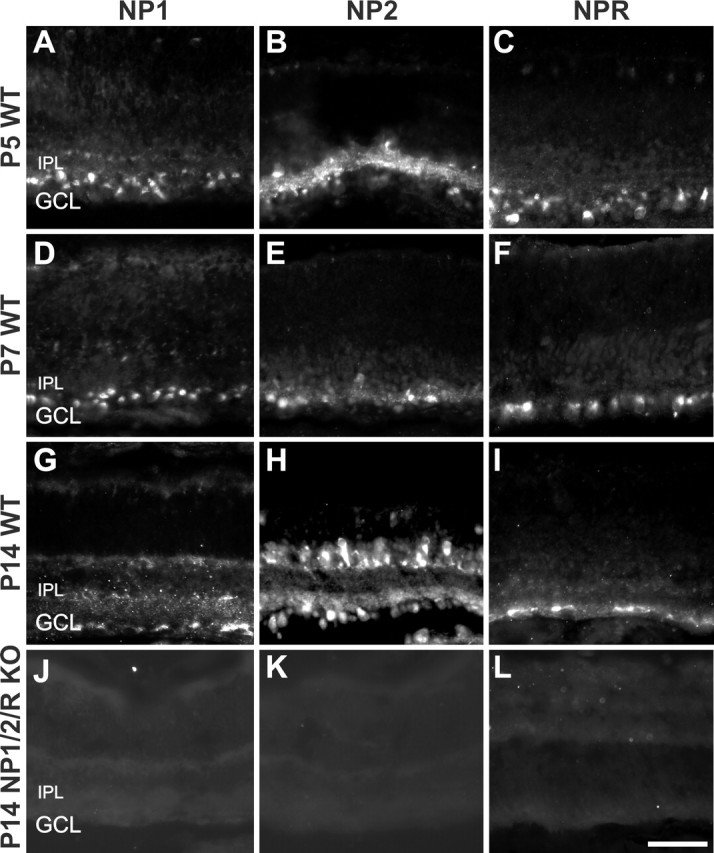
Immunofluorescence images of postnatal mouse retina stained with antibodies to NP1 (first column), NP2 (second column), or NPR (third column) at P5, P7, or P14. A–I, WT retinas. J–L, NP1/2/R KO retinas. In WT retinas (A–I), NP1-, NP2-, and NPR-immunopositive cells are seen in the ganglion cell layer at every age examined. No specific signal is seen in the retinas of NP1/2/R KO mice (J–L). IPL, Inner plexiform layer; GCL, ganglion cell layer. Scale bar, 50 μm.
We also processed WT P7 and P14 mouse brain tissue sections for NP1, NP2, and NPR immunohistochemistry (Fig. 3A–F). At P7, both NP1 and NP2 were detected in the dLGN (Fig. 3A,B). NP1 staining appeared more cellular and evenly distributed throughout the dLGN, whereas NP2 was expressed on axon fibers and was substantially lower in the innermost segment of the dLGN (Fig. 3B, asterisk). It is likely that these NP2-positive profiles in the dLGN are RGC axons. First, NP2 immunoreactivity was observed in the optic nerve (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Second, anterograde tracing of RGC axons with CTβ combined with immunolabeling for NP2 in the same tissue sections revealed a high degree of colocalization of the two labels in the dLGN (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Indeed, the intensity of the CTβ and NP2 labels varied together in the optic tract and LGN. Third, NP2 labels RGC axons in culture (data not shown). Collectively, these lines of evidence indicate that much of the NP2 immunoreactivity in the dLGN likely represents RGC axons. NPR immunoreactivity was very low from the P7 dLGN (Fig. 3C).
Figure 3.
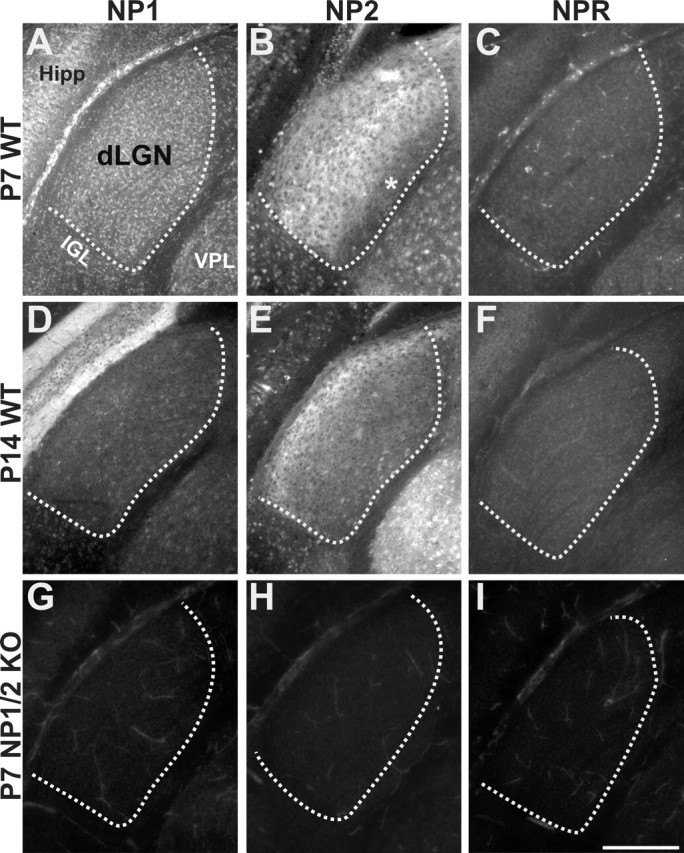
Immunofluorescence images of postnatal mouse dLGN stained with antibodies to NP1 (first column), NP2 (second column), or NPR (third column) at P7 or P14. A–F, WT retinas. G–I, NP1/2 KO retinas. A, D, NP1 is expressed in dLGN neurons at P7 and P14 but with relatively lower levels at P14. B, E, NP2 is expressed on axons projecting to the dLGN (see Results). B, At P7, axonal expression is present throughout the dLGN except for in the innermost segment (asterisk), whereas at P14 (E), NP2 expression is enriched in the outer segment of the dLGN. C, F, Low levels of NPR are present at P7 and P14. G–I, No specific label is present for NP1, NP2, or NPR in dLGNs from NP1/2 KO mice, and NPR levels are reduced in NP1/2 KO mice relative to WT mice. Hipp, Hippocampus; IGL, intergeniculate leaflet; VPL, ventral posterolateral nucleus. Coronal plane is shown; dorsal is up and medial is to the right of each panel. The boundary of the dLGN is shown with the dashed line. Scale bar, 200 μm.
At P14, levels of NP1 appeared reduced in the dLGN relative to their levels at P7 (Fig. 3, compare A, D). The levels of NP1 actually increased in the overlying hippocampus from P7 to P14, indicating that reduced levels of NP1 in the dLGN were not caused by poor staining on the older tissues. At P14, NP2 was still highly expressed on axons projecting to the dLGN; however, at P14, NP2 labeling was highest in fibers projecting to the outer shell of the dLGN, the mirror image of the pattern observed for NP2 in the P7 dLGN (Fig. 3, compare B, E). At maturity, the outer shell of the mouse dLGN receives axonal input from a subset of RGCs in the retina as well as axons from the ipsilateral superior colliculus (Grubb and Thompson, 2004a; Quina et al., 2005). Thus, NP2 immunoreactivity in the dLGN may reflect axonal input from a subset of RGCs and/or axons arising from the superior colliculus. At P14, NPR is present at slightly higher levels than at P7 (Fig. 3F).
Tissue sections from NP1/2 KO mice showed no immunoreactivity to NP1 or NP2 (Fig. 3G,H), and no increase in NPR immunoreactivity was observed in the dLGN of NP1/2 KO mice (Fig. 3I). Indeed, levels of NPR appeared reduced in NP1/2 KO mice, relative to the expression of NPR seen in WT mice (Fig. 3, compare C, F, I).
Collectively, these expression data indicate that all three NPs are expressed by mouse RGCs and that NP1 and NP2 protein are present in the early postnatal mouse dLGN. NP2 expression clearly appears axonal and shows developmental and spatial variation in the amount of NP2 that may reflect variations within subclasses of RGCs and/or superior colliculus axons. These data are supported by mRNA in situ data of mice at equivalent ages (data not shown; and http://www.stjudebgem.org).
Generation of mice that lack individual and multiple neuronal pentraxins
We previously generated mice that lack NP1 (Kirkpatrick et al., 2000) and now have generated mice that lack NP2 or NPR. The targeting constructs used to knock out NP2 and NPR by homologous recombination in ES cells are shown in Figure 4. For NP2, we targeted the first exon, coding for the translation start site and signal peptide, to block production of NP2 protein (Fig. 4A). For NPR, we targeted exons 2 and 3 (Fig. 4B). Southern blot analysis of genomic DNA from homologously recombined ES cells and chimeric, heterozygous, and homozygous mice yielded the expected mutant allele structure after recombination (Fig. 4C,D). Mice homozygous for NP2 or NPR have a substantial reduction in NP2 or NPR mRNA, and these messages are shortened as would be expected from the loss of exon 1 for NP2 or exons 2 and 3 for NPR (Fig. 4E,F). Chromatography on columns of immobilized taipoxin, which can purify NPs 1000-fold (Schlimgen et al., 1995; Kirkpatrick et al., 2000), was used to show conclusively the loss of NP2 and NPR protein in mice homozygous for each recombined allele (Fig. 4G,H). Because NPR does not bind taipoxin columns when expressed alone, loss of NPR in single, double, and triple KO mice was confirmed with immunoprecipitation to enrich NPs for detection (Fig. 4I). Single, double, and triple KO animals lacked the appropriate NPs. For taipoxin chromatographies, the amount of each NP was measured with 125I-labeled secondary antibodies (Fig. 4J). Loss of NPR causes a 50% reduction in the amount of NP1 and NP2 that can be purified, suggesting that NPR is necessary for the trafficking or membrane presentation of NP1 and NP2. All crosses generated the expected Mendelian numbers of each genotype. Mice lacking one, two, or all three NPs were viable and fertile and showed no obvious alteration in cerebral, cerebellar, or hippocampal structure and no obvious change in gait.
Figure 4.
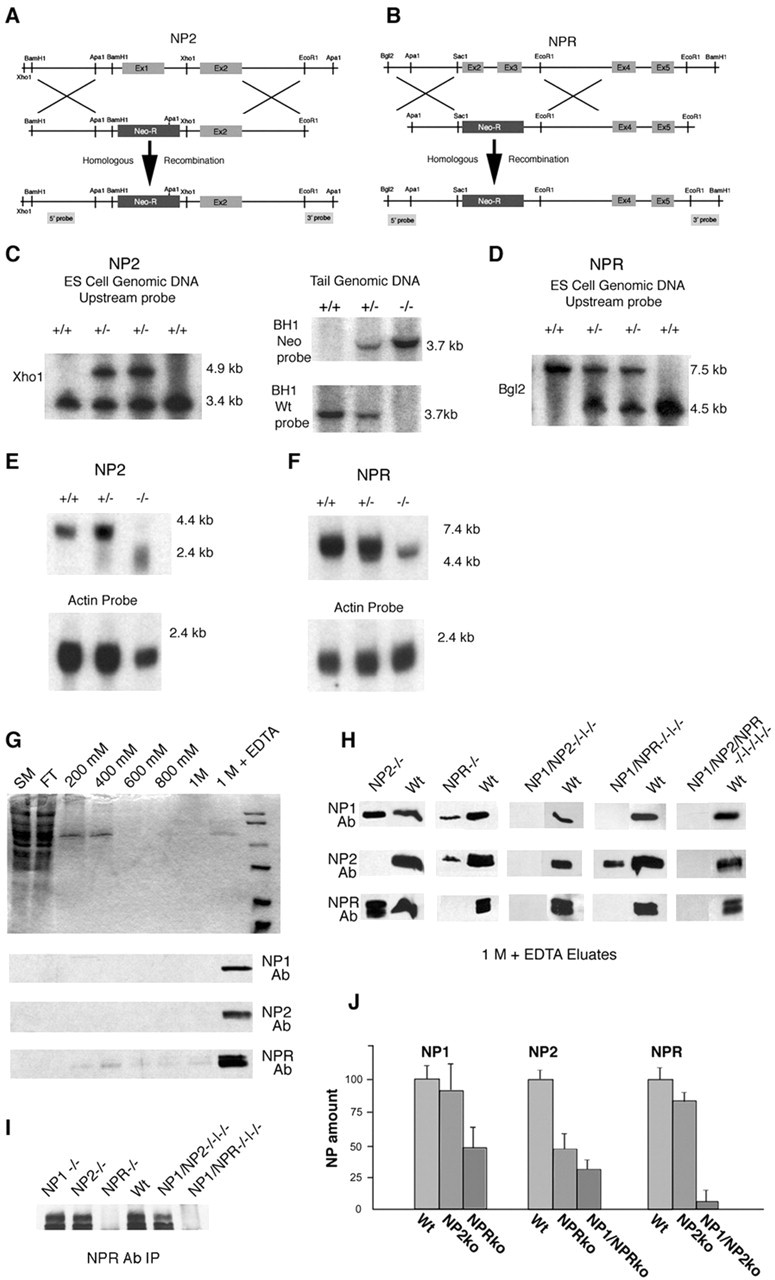
Generation of mice lacking NP2 or NPR and mice lacking multiple neuronal pentraxins. Mice lacking NP2 (A) and NPR (B) were generated through homologous recombination in ES cells with a targeting strategy. C, Alteration of genomic XhoI restriction fragment lengths in wild-type and recombined ES cells or BH1 in wild-type, heterozygous, and homozygous null NP2 mice. D, Alteration of genomic BglII restriction fragment lengths in wild-type, heterozygous, and homozygous null NPR mice. E, F, Alterations in NP2 and NPR message size and abundance in wild-type, heterozygous, and homozygous mice is shown in Northern blots. G, Chromatography of solubilized wild-type brains on taipoxin columns greatly enriches NP1, NP2, and NPR. H, Similar chromatography of solubilized brain from single, double, and triple KO mice shows the lack of the respective neuronal pentraxins. I, Loss of NPR in single, double, and triple KO mice shown by immunoprecipitation. J, Quantification of the loss of NP1, NP2, and NPR by taipoxin chromatography in select single, double, and triple KO mice. Ab, Antibody; FT, flow through; SM, solubilized membranes; BH1, BamH1; Wt, Wild-type.
Eye-specific refinement of RGC axons is perturbed in pentraxin knock-out mice
To address whether NPs play a role in the developmental refinement of synaptic connections in vivo, we examined the development of eye-specific RGC projections to the dLGN, a well established model system for exploring activity-dependent axonal refinement in the mammalian CNS (Katz and Shatz, 1996; Cohen-Cory, 2002). To visualize RGC–dLGN projections, we injected CTβ conjugated to Alexa 488 dye (green) into the left eye and CTβ conjugated to Alexa 594 dye (red) into the right eye of mice at various postnatal ages. We waited 24 h to allow for anterograde transport of the tracers and then examined the pattern of binocular label in the dLGN. In WT P4 mice, axons from the two eyes overlapped extensively; axons from the contralateral eye were distributed throughout the dLGN, where they intermingled with axons from the ipsilateral eye (Fig. 5A). In P4 NP1/2 KO mice, the pattern of RGC inputs to the dLGN at P4 appeared similar; axons from the contralateral eye were found throughout the entire dLGN, intermingled with axons from the ipsilateral eye (Fig. 5B). Multi-threshold quantitative analysis of the degree of binocular overlap in the dLGN (see Materials and Methods) revealed no significant differences between P4 WT and P4 NP1/2 KO mice (Fig. 5C).
Figure 5.
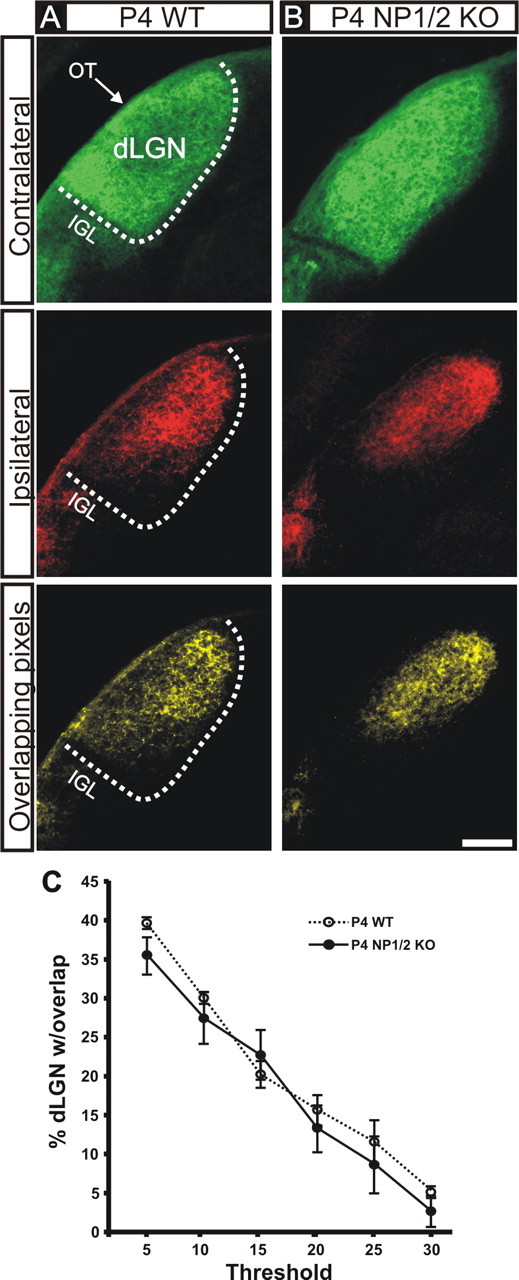
Retinogeniculate projections from the two eyes in P4 WT and P4 NP1/2 KO mice. A, B, Photomicrographs of the contralateral eye projections (top panels), ipsilateral eye projections (middle panels), and the locations in which they overlap (bottom panels) in the dLGN of WT (A) and NP1/2 KO (B) P4 mice. In both the P4 WT and P4 KO mice, axons from the two eyes are extensively intermingled in the dLGN. Coronal plane is shown; dorsal is up and medial is to the right of each panel. IGL, Intergeniculate leaflet; OT, optic tract. The boundary of the dLGN is shown with the dashed line. Scale bar, 100 μm. C, Multi-threshold analysis of the percentage of the dLGN containing overlapping axons from both eyes in the dLGN (see Materials and Methods) of WT and NP1/2 KO mice revealed no significant differences in overlap at any threshold (n = 3 mice were analyzed per genotype at P4; Student's t test; ±SEM). All images are shown at same threshold level (15% above background; see Materials and Methods).
By P10 in WT mice, axons from the two eyes were segregated; they occupied complementary, nonoverlapping regions in the dLGN (Fig. 6A). A characteristic “gap” in the dorsomedial dLGN was devoid of label from the contralateral eye, and this gap was occupied by the axons arising from the ipsilateral eye. These findings (at P4, inputs from the two eyes are overlapping; by P10, they are segregated) are in agreement with the results of previous reports that examined the development of eye-specific retino-dLGN projections in WT mice (Godement et al., 1984; Rossi et al., 2001; Muir-Robinson et al., 2002; Pfeiffenberger et al., 2005). In contrast, the dLGN of P10 NP1/2 mice lacked eye-specific segregation. Examples of retino-dLGN projections from two different P10 NP1/2 KO mice are shown in Figure 6, B and C. In both cases, the contralateral eye axons filled the entire dLGN, leading to extensive binocular overlap (Fig. 6B,C, yellow in bottom panels). The shape and extent of dLGN territory occupied by the ipsilateral eye varied somewhat from one P10 NP1/2 KO animal to the next (Fig. 6, compare B, C, middle panels), with approximately half the P10 NP1/2 KO mice showing a single ipsilateral eye projection zone similar to that shown in Figure 6B and the remaining animals showing several ipsilateral eye projection zones, as in Figure 6C; however, because in 100% of the P10 NP1/2 KO mice examined (n = 6 mice) the contralateral eye input was spread throughout the entire dLGN, eye-specific segregation was absent in every case (Fig. 6B,C, bottom panels). Quantitative analysis confirmed these observations: the degree of binocular overlap in the dLGN of P10 NP1/2 KO mice was significantly greater than in P10 WT mice, regardless of threshold (Fig. 6F).
Figure 6.

Retinogeniculate projections from the two eyes in P10 and P30 WT and NP1/2 KO mice. Except for scale, conventions are as in Figure 5. A, In P10 WT mice, inputs from the two eyes are segregated; a region in the dorsomedial dLGN is devoid of label from the contralateral eye. This region is occupied by the axons from the ipsilateral eye. Note also that axons from the ipsilateral eye are always restricted to the dorsomedial dLGN. B, C, In P10 NP1/2 KO mice, axons from the two eyes are still intermingled in the dLGN. The contralateral projection fills the entire dLGN; there is no gap present. B, In approximately half of the P10 NP1/2 mice examined, the ipsilateral eye projection comprises a single termination zone, but this zone appears larger than in WT mice. C, In the other cases, the ipsilateral eye axons are especially widespread. Despite these slight differences in patterning of the ipsilateral eye projection, in all P10 NP1/2 cases examined (n = 6 mice), the contralateral eye projection fills the entire dLGN, and thus axons from the two eyes always intermingled with those from the opposite eye, which is abnormal for mice of this age (compare bottom panels). D, Retinal inputs to the dLGN of P30 WT mice are segregated. E, Binocular inputs to the dLGN of P30 NP1/2 KO mice are also segregated (no yellow in bottom panel); however, note that compared with P10 or P30 WT mice, the ipsilateral axons are widespread and not restricted to the dorsomedial dLGN in NP1/2 KO mice (middle panels). Scale bars, 200 μm. F, Quantification of the percentage of the dLGN receiving overlapping inputs from the ipsilateral eye in WT versus NP1/2 KO mice at P10 and P30. P10 NP1/2 KO mice exhibit significantly more overlap in the dLGN than P10 WT mice, regardless of threshold. P30 KO mice have retino-dLGN inputs that are segregated to a degree similar to that of WT mice P10 and older (no significant differences observed at any threshold) (n = 4 P10 WT mice; n = 8 P10 NP1/2 KO mice; n = 2 P30 NP1/2 KO mice; Student's t test; ±SEM; ∗p < 0.0001). All images are shown at the same threshold level (15% above background; see Materials and Methods).
To test whether the retino-dLGN phenotype observed in NP1/2 KOs is modified across development, we also examined WT and NP1/2 KO mice at P30 (which is ∼2 weeks after eye opening). In contrast to the pattern seen at P10, axons from the two eyes appeared segregated in P30 NP1/2 KO mice (Fig. 6E, bottom panel). Quantitative analysis of overlap confirmed this observation (Fig. 6F); however, at P30, axons arising from the ipsilateral eye remained distributed across an abnormally large extent of the dLGN, and rather than comprising a single termination zone in the nucleus, multiple ipsilateral-eye termination zones were present (Fig. 6E, middle panel).
Alterations in the decussation patterns of RGCs at the optic chiasm can lead to defects in the patterning of eye-specific dLGN projections (Guillery and Kaas, 1971; Guillery et al., 1973). Although chiasm decussation errors are not associated with defects in ocular segregation per se, we investigated the possibility that the dLGN phenotype that we observed arises from aberrant targeting of RGC axons at the optic chiasm. Retrograde labeling studies showed that ipsilaterally projecting RGCs resided in the appropriate portion of the retina in NP1/2 KO mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Thus, the dLGN defects that we observed in NP KO mice do not arise from aberrant pathfinding at the chiasm. They arise from a failure of RGC axons to refine within the dLGN.
We also examined the pattern of retino-dLGN projections in mice lacking only NP1, NP2, or NPR, as well as in NP1/2/R triple mutant mice. To compare each genotype, we analyzed the extent of the ipsilateral eye input to the dLGN at P10, which is another established measure of the degree of synaptic refinement in the developing retino-dLGN projection (Godement et al., 1984; Huh et al., 2000; Rossi et al., 2001). The ipsilateral eye input to the dLGN of single NP1, NP2, or NPR KO mice was slightly expanded when compared with WT but was mild when compared with NP1/2 double KO mice. The defects observed in NP1/R double KO mice were also mild relative to the NP1/2 KO mice. The retino-dLGN phenotype observed in NP1/2/R triple KO mice was not more severe than that observed in the NP1/2 KO mice (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Thus, the combined loss of NP1 and NP2 appeared most relevant for retino-dLGN axonal remodeling. These data reveal that NP1 and NP2 together are required for normal retino-dLGN process refinement. This could reflect a need for NP1/2 to generate normal spontaneous retinal activity or retino-dLGN glutamatergic transmission, or both. We next explored these possibilities with electrophysiological recordings of RGCs from postnatal NP1/2 KO mice.
Spontaneous retinal activity in pentraxin knock-out retinas
Although there is debate regarding which aspects of spontaneous retinal activity are important for eye-specific segregation in the dLGN (Huberman et al., 2003; Grubb and Thompson, 2004b; Torborg et al., 2005), there is general agreement that segregation requires RGC activity: abolishing spontaneous retinal activity (Penn et al., 1998; Rossi et al., 2001; Huberman et al., 2002) prevents the segregation of eye-specific projections. To address whether retinal activity patterns are altered in NP1/2 KO mice, we performed multisite, extracellular electrophysiological recordings of spontaneous activity (Meister et al., 1991; Wong et al., 1993; Demas et al., 2003) in retinal explants from WT and NP1/2 KO P4–P5 mice. In both WT and NP1/2 KO retinas, RGCs showed robust bursts of spiking activity that were highly correlated among neighboring RGCs and spread in a wave-like manner (Fig. 7A–C). As determined previously in WT mouse retinas at these early postnatal ages (Cang et al., 2005), the waves in NP1/2 KO retinas were blocked by high concentrations of the nicotinic cholinergic receptor agonist epibatidine (10 nm; n = 2; data not shown). Eye-specific segregation of afferents in the dLGN has been hypothesized to depend on correlated activity within an eye, specifically the correlated firing of neighboring RGCs that is not correlated with activity occurring at matched retinotopic locations in the other eye (Torborg et al., 2005). In theory, the relatively long intervals that occur between retinal waves should also contribute to eye-specific segregation. We thus examined the duration between waves in WT and NP1/2 KO retinas and found that the NP1/2 KO interwave interval was not significantly different compared with the WT interval (Fig. 7D) (mean seconds ± SD: WT = 85 ± 23, n = 26 intervals from 10 retinas; NP1/2 = 72 ± 16, n = 24 intervals from 7 retinas; p > 0.05).
Figure 7.

Early postnatal NP1/2 KO mouse retinas maintain activity characteristics important for eye-specific segregation. A, Correlation indices for electrically isolated RGC spikes in a single P4 WT retina versus distance between electrodes. The correlation index represents the likelihood that a cell recorded at one electrode fired a spike within a 100 ms time window of any spike from an RGC at another electrode. Each point represents the index for one such RGC pair plotted against the inter-electrode distance; the solid line is the best-fit single exponential for the population of distances. As expected, correlations are higher for cells that are nearer one another. B, Correlation indices calculated from the spikes of all RGC pairs recorded from a single P4 NP1/2 (neuronal pentraxin double knock-out) retina are shown. As in the WT retina, correlations are high for near-neighbor cells and decrease with increasing distance between them. C, Example spike rasters from a 9 s period of recording show the responses of individual RGCs as a wave travels across a P4 NP1/2 retina (the same one quantified in B). Inset illustrates the relative position of the electrodes (1–4) on the array. Electrodes 2, 3, and 4 were 200, 500, and 700 μm from electrode 1, respectively. This wave traveled at a speed of ∼130 μm/s across the recorded retinal area. D–G, Dark gray bars show WT values, and light gray bars show NP1/2 values. D, The interwave interval (and thus wave frequency) was not significantly different in NP1/2 retinas compared with WT retinas. E, The overall spike rate of cells recorded from NP1/2 retinas was significantly higher than in WT mice. F, When the amount of firing at ≥10 Hz for each recorded cell was normalized to the spike rate of that cell, NP1/2 RGCs had slightly more of this high-frequency firing compared with WT retinas. G, As waves of activity pass across the retina, individual ganglion cells fire bursts of spikes after periods of relative silence. The mean burst duration calculated from all recorded NP1/2 RGCs was significantly longer than the mean WT burst duration. H, Information on the near-neighbor relationships of RGCs can be carried by the level of correlated activity, with the spikes of near neighbors having high correlations and those of RGCs distant from one another having lower correlations. The overall spike rate and number of synchronous spikes for RGC pairs within a given time window (100 ms here) were higher for the NP1/2 retinas compared with the WT retinas. To determine for each RGC whether a near-neighbor RGC could still be distinguished from a spatially distant RGC based on the amount of correlated activity, we calculated the difference in the amount of correlated spiking between near-neighbor pairs and distant pairs. Pairs recorded on electrodes <150 μm apart were considered near neighbors, whereas those ≥600 μm apart were considered distant from one another. The difference in the median values for each retina was plotted against the overall median spike rate. Black circles represent WT retinas, and gray triangles represent NP1/2 retinas. The solid line is a linear fit to the WT data. The proximity of the NP1/2 data points to this line indicate that the difference in correlated activity in the NP1/2 retinas is very similar to that of the WT retinas. This suggests that the correlated activity in the NP1/2 retinas still contains retinotopic information. I, Histograms of median burst durations for RGCs of WT (thick line) and NP1/2 (thin line) are shown. The distributions show that the significant difference between burst durations shown in F result from (1) slightly longer bursts in many RGCs, (2) a few RGCs with much longer bursts in the NP1/2 retinas, and (3) a large group of RGCs with shorter burst durations in the WT retinas. Analyses of retinal wave parameters were based on 5 min periods of spike activity from P4 and P5 WT retinas (n = 10) and P4 NP1/2 retinas (n = 7) recorded on a multi-electrode array. D, F, G, Bars indicate SEM; in G they represent medians and quartiles, respectively. ∗p < 0.001. n.s., Not significant.
Within each retina, the degree of spike correlation between RGCs, measured as their correlation index (Demas et al., 2003), decreased as a function of the distance between them in both WT and NP1/2 KO retinas (Fig. 7A,B). Analysis of the spike trains from individual RGCs indicated that NP1/2 KO RGCs had a significantly higher overall spike rate than RGCs from WT retinas (Fig. 7E) (median spikes per minute; 25th/75th percentiles: WT = 11.8, 5.8/23.4, n = 493 RGCs; NP1/2 = 30.6, 16.8/46.9, n = 360 RGCs; p < 0.0001) as well as a slightly higher median instantaneous firing frequency (median Hz; 25th/75th percentiles: WT = 11.2, 9.14/42.0, n = 39829 interspike intervals; NP1/2 = 18.0, 10.3/51.0, n = 72757 interspike intervals; p < 0.0001). A recent study concluded that the percentage of firing at ≥10 Hz within RGC bursts is a key parameter for eye-specific segregation of RGC axons in the dLGN (Torborg et al., 2005). NP1/2 KO RGCs fired at ≥10 Hz more than WT RGCs. To determine whether this was simply a result of the increase in overall spike rate, the amount of ≥10 Hz firing was normalized to the spike rate for each RGC. Even with normalization, the amount of ≥10 Hz firing was modestly larger in NP1/2 KO retinas (Fig. 7F) (mean normal value ± SD: WT = 2.90 ± 1.18; NP1/2 = 3.45 ± 1.08) but only by ∼20% and with a marked overlap of the populations.
Next we examined the median rate of synchronous spiking between pairs of RGCs (defined as spikes occurring in both cells within a 100 ms time window) and did this for near pairs of RGCs (located within 150 μm of each other) and distant pairs of RCGs (at least 600 μm apart). In both WT and NP1/2 KO retinas, the numbers of correlated spikes differed fivefold between near versus distant pairs (mean synchronous spikes per minute ± SD: distant WT pairs = 2.98 ± 2.56, near WT pairs = 15.8 ± 10.8; distant NP1/2 pairs = 9.22 ± 3.03, near NP1/2 pairs = 44.9 ± 5.37). The difference in these spikes was a measure of the difference in correlations between near and distant pairs. These differences are plotted against the overall spike rate for each retina in Figure 7H. The NP1/2 KO data fall near the linear fit of the WT data, indicating that the difference in correlated activity between near and distant RGC pairs in the NP1/2 KO retinas is similar to that in WT retinas. Thus, the relative difference in correlation between near versus distant RGCs is maintained in NP1/2 KO retinas. This can also be seen in the similar decreases in correlation index with distance (Fig. 7A,B). This correlation decrease was independent of the time window used, being apparent with time windows varying from 10 to 200 ms (other data not shown). It is notable that the number of synchronous spikes at near or far distances in the NP1/2 KO retinas was approximately three times greater than observed for WT retinas.
Butts and Rokhsar (2001) examined wave data from mammalian retinas and found that the bursts of action potentials carry most of the retinotopic information. The precise timing of spikes within those bursts does not add significantly more information about RGC retinotopy. RGCs in NP1/2 KO mice exhibited, on average, longer burst durations compared with WT RGCs (Fig. 7G) (mean seconds ± SD: WT = 2.04 ± 0.70, n = 256; NP1/2 = 2.35 ± 0.69, n = 209; p < 0.0001). The NP1/2 KO burst population, although significantly longer on average than WT, had a similar structure to the WT burst population (Fig. 7I). What it lacked were the shorter bursts below ∼1.5 s in duration that were more numerous in a subset of RGCs in the WT retina. These shorter bursts carry less retinotopic information than the longer bursts (Butts and Rokhsar, 2001). Thus, the NP1/2 KO retinas have bursts that should carry retinotopic information similar to the WT bursts.
Together these data indicate that loss of NPs impacts cholinergically driven, spontaneous activity in the early postnatal retina. Correlated firing of RGCs was intact in KO retinas, but the levels of activity were elevated. Below (see Glutamatergic synaptic maturation is perturbed in RCGs from pentraxin KO mice) we discuss which aspects of retinal wiring and synaptic organization might lead to these perturbed retinal activity patterns and consider whether increased levels of retinal activity could impact eye-specific segregation of RGC axons in the dLGN. Of course, a critical requirement for eye-specific segregation is that RGC activity be transmitted faithfully to the postsynaptic neurons. To begin to address whether glutamatergic synaptic transmission is altered in pentraxin KO mice, we recorded synaptic activity from WT and NP1/2 KO RGCs in culture with patch clamping.
Glutamatergic synaptic maturation is perturbed in RGCs from pentraxin KO mice
RGCs purified from WT and NP1/2 KO mice were cultured in the presence or absence of WT astrocyte feeding layers (Ullian et al., 2001). After 2 weeks, endogenous mEPSCs were recorded by patch clamping RGCs (Ullian et al., 2001; Christopherson et al., 2005). WT RGCs cultured in the presence of glia showed high rates of spontaneous mEPSCs of various amplitudes (Fig. 8A). In contrast, RGCs from NP1/2 double KO mice cultured in the presence of glia showed few synaptic currents with only small amplitudes (Fig. 8B). The number of synaptic current events recorded per minute in both the presence and absence of glia is graphed in Figure 8C. As shown previously (Ullian et al., 2001), culturing purified WT RGCs with glia increases the number of synaptic events approximately sevenfold. This increase in the number of synaptic events with glial coculture does not occur with NP1/2 KO RGCs, even after 2 weeks in culture. In addition to the difference in the frequency of mEPSCs, the amplitude distributions of the mEPSCs recorded in the presence of glia for WT and NP1/2 KO RGCs are markedly different. The amplitudes of mEPSCs for WT versus NP1/2 KO RGCs are graphed in Figure 8D as a frequency count of mEPSCs versus amplitude size. NP1/2 KO RGCs had small mEPSCs and completely lacked moderate and large mEPSCs at 2 weeks. Only when NP1/2 RGCs were left in culture with WT glia for extended periods of time (4 weeks) did the number and size of mEPSCs recover to normal levels (Fig. 8G,H). Indeed, after 4 weeks in culture, NP1/2 RGC recordings had more moderately sized events than in the WT control of the same age (Fig. 8H). This may reflect variability in mEPSC size or indicate a shift in the event sizes in older KO RGCs.
Figure 8.
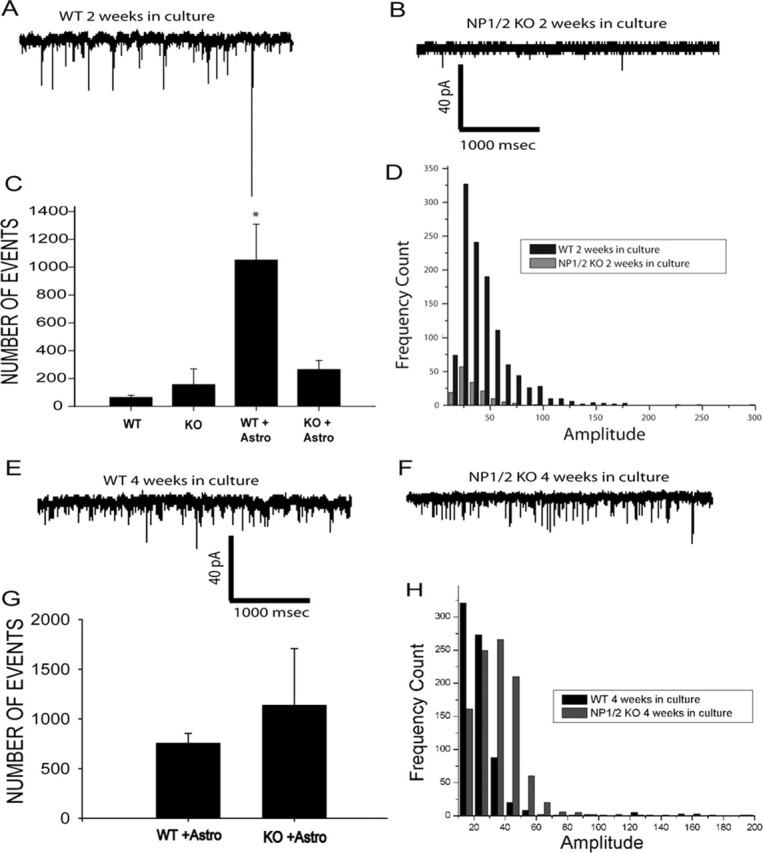
Retinal ganglion cells from NP1/2 KO animals show a delay in the formation of functional synapses in vitro. A, Patch-clamp recording of an RGC cultured with a feeding layer of astrocytes for 2 weeks. Numerous spontaneous mEPSCs are seen (downward deflections). B, Example of recordings of synaptic activity in NP1/2 KO RGC cultures after 2 weeks with astrocyte feeding layers. Fewer synaptic events are seen. Three sets of recordings from KO and WT cultures with between five and six neurons per condition were recorded for a total of 15 neurons per genotype. C, Quantification of the rate of spontaneous synaptic activity seen in RGC cultures from WT or KO mice. In both WT and KO, low rates of activity are seen in purified RGC cultures without astrocyte feeding layers. Astrocyte feeding layers significantly increase the frequency of synaptic activity in WT cultures (WT + Astro), but there is not a corresponding increase in KO cultures (KO + Astro) (WT + Astro, 1105 ± 347 events per minute vs KO + Astro, 293 ± 60 events per minute; p = 0.013; Kruskal–Wallis ANOVA on ranks). D, Frequency histogram of spontaneous synaptic events recorded in WT and KO RGC cultures after 2 weeks. The KO events (open bars) are much smaller amplitude compared with WT events (black bars; KO + Astro, 2 weeks in culture, 26 ± 0.2 pA vs WT + Astro, 2 weeks in culture, 44 ± 0.8 pA; Mann–Whitney rank sum test). The largest events likely represent multiquantal release (Paulsen and Heggelund, 1996). E, Example of synaptic activity measured in WT cultures with feeding layers of astrocytes after 4 weeks. High rates of spontaneous synaptic activity are still seen. F, Example of spontaneous synaptic activity seen in KO RGCs with feeding layers of astrocytes at 4 weeks in culture. High-frequency events are now seen in contrast to the cultures after 2 weeks. G, Quantification of the rate of synaptic activity seen in WT and KO RGC cultures with astrocyte feeding layers. No statistically significant differences in the rates of activity are seen between WT and KO cultures (WT + Astro, 1135 ± 570 events per minute vs KO + Astro, 756 ± 98 events per minute; p > 0.5; Kruskal–Wallis ANOVA on ranks). H, Frequency histograms of spontaneous synaptic activity measured in WT or KO cultures after 4 weeks. Astro, Astrocyte.
We also explored the number of synaptic contacts between RGCs as measured by colocalization of presynaptic protein synaptotagmin 1 and postsynaptic density-95 protein (PSD-95) (Fig. 9) (Ullian et al., 2001; Christopherson et al., 2005). At both 2 and 4 weeks in culture, NP1/2 KO RGCs cultured with WT astrocyte feeding layers had similar numbers of synaptic structures as RGCs cultured from WT mice (Fig. 9E,F). Collectively, these data indicate that the development of glutamatergic synaptic function, but not formation of presynaptic and postsynaptic contacts, is significantly delayed in the absence of NP1 and NP2.
Figure 9.
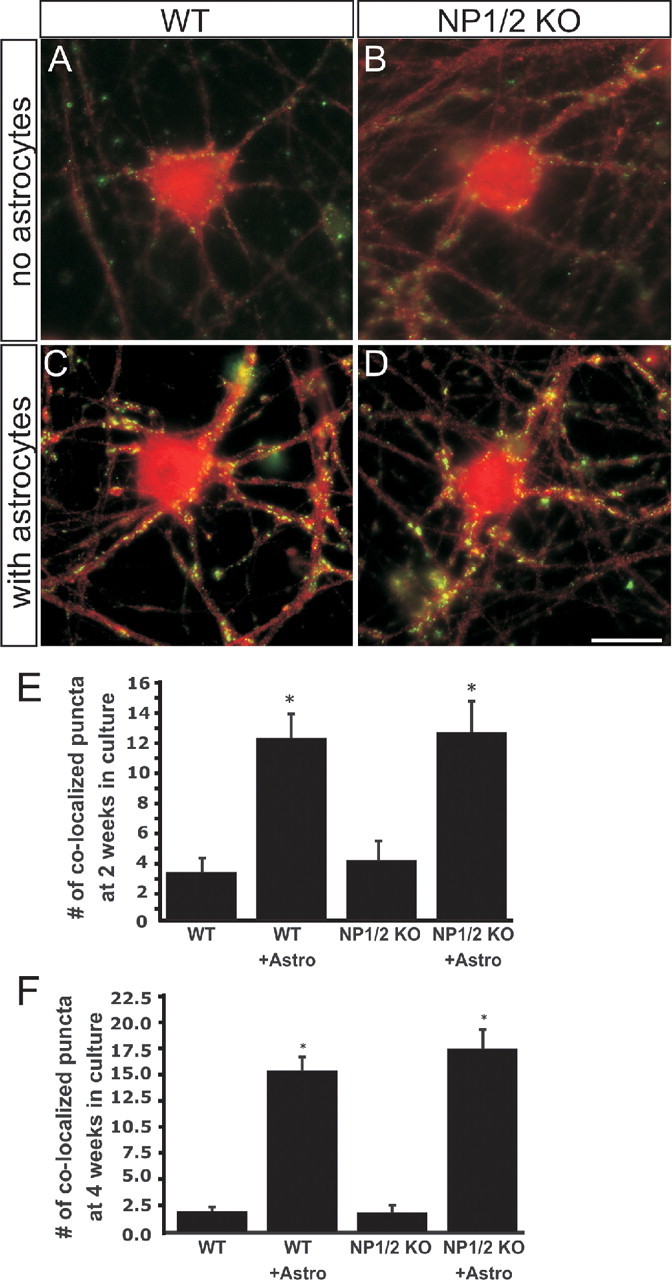
The number of synapses formed by RGCs cultured from NP1/2 KO mice is normal. A, B, RGC cultured without astrocytes and stained for synaptotagmin 1 (red) and PSD-95 (green) from WT (A) or NP1/2 KO (B) mice. C, D, RGC cultured with astrocytes and stained for synaptotagmin (red) and PSD-95 (green) from WT (C) or NP1/2 KO (D) mice. Scale bar, 20 μm. E, F, Number of synapses per field identified by colocalization of PSD-95 and synaptotagmin 1 at 2 weeks in culture (E) or 4 weeks in culture (F). Twenty cells were quantified per condition. These values are typical of RGCs in culture (Ullian et al., 2001; Christopherson et al., 2005). All values are mean ± SEM.
Hippocampal plasticity and visual function are intact in NP knock-out mice
Mice lacking components of major histocompatibility complex I signaling exhibit defects in retino-dLGN refinement, despite the presence of normal retinal waves (Huh et al., 2000). Major histocompatibility complex I KO mice also exhibit enhanced LTP and a lack of LTD in the late postnatal (P20–P40) hippocampus, suggesting that synaptic changes similar to those that mediate LTP and LTD in the hippocampus can play a role in retino-dLGN refinement (Huh et al., 2000; Boulanger et al., 2001). We applied the same paradigm as Huh et al. (2000) and evaluated hippocampal LTP and LTD in the late postnatal hippocampus of NP KO mice. We did not observe any defects in basal synaptic strength, paired-pulse facilitation, LTP, or LTD in the hippocampus of P20–P40 NP1/2 KO mice (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Thus we conclude that the defects observed in the LGN of NP KO mice are associated with aspects of synaptic plasticity that are separate or of a different time scale than those of hippocampal LTP and LTD.
We also tested whether there is a persistent requirement for NPs in visual system synaptic transmission. Visual transmission and visually mediated behavior were normal in adult NP1/2 KO mice (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), which is at variance with the idea that the dLGN defects that we observed are caused by a permanent loss in transmission of retinal activity to postsynaptic targets.
It may be that NPs are required for activity-dependent synaptic modifications only in the early postnatal brain; however, we did not find differences in AMPA receptor clusters on 7-day-old hippocampal neurons cultured from WT, NP1/2 double KO, or NP1/2/R triple KO mice (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Thus the requirement of NPs for synaptic and axonal refinements does not appear to be global. We conclude that NPs regulate the functional maturation of RGC synapses, spontaneous retinal activity (and perhaps retinal wiring), and eye-specific retino-dLGN axonal refinements.
Discussion
NPs are expressed in the developing visual system, and NP1/2 KO mice exhibit defective eye-specific retino-dLGN axonal refinement. NP KO mice show a lack of binocular segregation at P10. By P30, retino-LGN projections from the two eyes are segregated, but the spatial arrangement of eye-specific territories in the LGN is aberrant. The quality and age-dependence of these phenotypes are remarkably similar to the phenotypes of animals that had retinal activity blocked or altered from P1 to P10 but then were allowed a period of recovery, during which retinal activity returned to normal (Huberman et al., 2002; Muir-Robinson et al., 2002). This observation led us to investigate the patterns of spontaneous retinal activity in early postnatal NP KO mice. Those experiments revealed that several parameters of retinal activity were significantly altered by the deletion of NP1 and NP2; in particular, the overall levels of RGC spiking activity were markedly increased. Does this elevated RGC activity cause the retino-dLGN refinement defects observed in NP KO mice? Torborg et al. (2005) concluded that bursts of spikes at ≥10 Hz and high amounts of spike correlation between RGC pairs situated close together are critical requirements for eye-specific segregation. Both parameters were as great (and in some cases, greater) in NP1/2 KOs as compared with WT mice. It is possible that the overall increase in spontaneous retinal activity levels observed in NP1/2 KO mice may have exceeded an upper limit for eye-specific segregation; however, overall levels of RGC activity do not impact eye-specific segregation as long as correlation differences between near versus distant RGCs are maintained (Stellwagen and Shatz, 2002; Torborg et al., 2005). Indeed, Torborg et al. (2005) found that adenylyl cyclase activation within the retina causes a several-fold increase in correlated RGC spiking, but this treatment does not disrupt the segregation of eye-specific projections (Stellwagen and Shatz, 2002). Thus the model for activity-driven eye-specific segregation put forth by Torborg et al. (2005) cannot alone explain the phenotypes that we observed in NP KO mice.
It is worth noting that activity levels can impact the outgrowth and branching of RGCs (Goldberg et al., 2002), so the increased spiking observed in NP KO retinas may have altered retino-dLGN refinement indirectly via effects on RGC axon growth. In any case, the data presented here reveal a previously unrecognized requirement for NPs in regulating levels of cholinergically driven spontaneous retinal activity. In future experiments, it will be interesting to address which aspects of intra-retinal wiring are altered in NP KO mice to give rise to increased RGC spiking. Loss of NPs could increase retinal activity levels by enhancing the intrinsic excitability of RGCs or the amacrine cells that drive spontaneous activity. Because cholinergic transmission from amacrine cells mediates early retinal waves through the activation of nicotinic acetylcholine receptors present on RGCs (Penn et al., 1998; Feller et al., 1996), one hypothesis is that NPs regulate RGC excitability by mediating levels of nicotinic acetylcholine receptors.
NPs may also affect retino-dLGN refinement by directly affecting the structural and functional development of retino-dLGN synapses. Unlike P0–P10 spontaneous retinal waves, which depend on cholinergic transmission (Feller et al., 1996; Cang et al., 2005), the retino-dLGN synapse is glutamatergic. Here we began to address the role of NPs in maturation of glutamatergic synapses by using cultured RGCs, which form only glutamatergic synapses. Purified RGCs cultured from NP1/2 KO mice formed normal numbers of presynaptic and postsynaptic glutamatergic synapses, but there was a conspicuous lack of spontaneous synaptic transmission in these cultures until 4 weeks in vitro. Although RGC–RGC synapses in vitro may not completely resemble RGC–dLGN neuron synapses in vivo, our present results suggest that lack of NPs does not prevent the formation of glutamatergic synaptic contacts between RGC axons and geniculate cells. Nonetheless, NPs may be necessary for the early development of glutamatergic sensitivity of dLGN neurons. If dLGN cells are unresponsive to RGC glutamatergic input from P0 to P10, this could explain the lack of eye-specific segregation seen in NP KO mice at P10. A non-mutually exclusive hypothesis is that NPs are required for normal synaptic transmitter release from RGCs in the early postnatal period. Both possibilities are compatible with our observation that NP1/2 KO mice lack eye-specific segregation at P10, but that segregation is rescued by P30. The fact that ipsilateral eye projections were segregated but abnormally fragmented in P30 NP KO mice can be explained by the fact that eye-specific segregation can occur at ages older than P10, provided that retino-dLGN transmission is restored (Huberman et al., 2002; Muir-Robinson et al., 2002), whereas the patterning of RGC axons into single, stereotyped eye-specific termination zones can only occur in the early postnatal period (P0–P10), when guidance cues such as ephrin-As are present in the dLGN (Huberman et al., 2005).
Finally, in trying to determine how NPs influence neuronal excitability and axonal refinement, it is worth considering the homology of NPs to acute-phase and C-reactive proteins in the immune system that mark cells for phagocytosis and degradation (Garlanda et al., 2005) and the known regulation of NP2 by activity (Tsui et al., 1996; Xu et al., 2003). NPs may regulate structural refinements of axons in response to activity by marking specific synaptic sites for degradation and turnover. An excessive number of synapses in the retina of NP1/2 KO mice would explain the increased RGC spiking during early wave activity that we described here. As discussed above, because increased retinal activity levels cannot easily explain the lack of eye-specific refinement that we observed (Stellwagen and Shatz, 2002; Torborg et al., 2005), NPs may be required downstream of activity to induce synapse elimination and eye-specific pruning. Indeed, lack of synapse elimination in the retina and dLGN can parsimoniously explain both the increased retinal activity and the lack of eye-specific pruning in the early postnatal dLGN described here. Future studies will attempt to resolve these questions definitively. We suspect that the results shown here represent just one example of a more general role for NPs in regulating synaptic refinement and plasticity in the early postnatal CNS, and they underscore previous findings (Huh et al., 2000) that first identified a role for immune-related proteins in brain development and plasticity.
Footnotes
*L.B., A.D.H., and E.M.U. contributed equally to this work.
This work was supported by National Institutes of Health (NIH) Grant NS039283 (M.P.), National Eye Institute Grants EY11369 (B.C.) and R01 11310 (B.A.B.), National Institute on Drug Abuse Grant R01 DA15043 (B.A.B.), Sandler Family Supporting Foundation (E.M.U.), NIH Grants HD042191 and HD039886 (M.N.), NIH Grants MH63394 (R.C.M.) and MH065752 (D.S.), That Man May See and Research to Prevent Blindness (D.C.), Medical Research Service, Department of Veteran Affairs (S.B., N.P.), and The Stanford University Regenerative Medicine Interdisciplinary Training Fellowship 1-T90 DK070103 (A.D.H.).
References
- Ball SL, Powers PA, Shin H-S, Morgans CW, Peachey NS, Gregg RG (2002). Role of the β2 subunit of voltage dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci 43:1595–1603. [PubMed] [Google Scholar]
- Banker G, Goslin K (1991). In: Culturing nerve cells Cambridge, MA: MIT.
- Barres BA, Silverstein BE, Corey DP, Chun LL (1988). Immunological, morphological and electrophysiological variation among retinal ganglion cells purified by panning. Neuron 1:791–803. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Huh GS, Shatz CJ (2001). Neuronal plasticity and cellular immunity: shared molecular mechanisms. Curr Opin Neurobiol 11:568–578. [DOI] [PubMed] [Google Scholar]
- Breviario F, d'Aniello EM, Golay J, Peri G, Bottazzi B, Bairoch A, Saccone S, Marzella R, Predazzi V, Rocchi M, Della Valle G, Dejana E, Mantovani A, Introna M (1992). Interleukin-1-inducible genes in endothelial cells: cloning of a new gene related to C-reactive protein and serum amyloid P component. J Biol Chem 267:22190–22197. [PubMed] [Google Scholar]
- Butts DA, Rokhsar DS (2001). The information content of spontaneous retinal waves. J Neurosci 21:961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang J, Renteria RC, Kaneko M, Liu X, Copenhagen DR, Stryker MP (2005). Development of precise maps in visual cortex requires patterned spontaneous activity in the retina. Neuron 48:797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005). Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S (2002). The developing synapse: construction and modulation of synaptic structures and circuits. Science 298:770–776. [DOI] [PubMed] [Google Scholar]
- Demas J, Eglen SJ, Wong RO (2003). Developmental loss of synchronous activity in the mouse retina is independent of visual experience. J Neurosci 23:2851–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds D, Schlimgen AK, Lu S-Y, Perin MS (1995). Novel reticular calcium binding protein is purified on taipoxin columns. J Neurochem 64:2339–2344. [DOI] [PubMed] [Google Scholar]
- Dodds D, Omeis I, Cushman SJ, Helms JA, Perin MS (1997). Neuronal pentraxin receptor, a novel integral membrane pentraxin that interacts with neuronal pentraxin 1 and 2 and taipoxin-associated calcium binding protein 49. J Biol Chem 272:21488–21494. [DOI] [PubMed] [Google Scholar]
- Dowton SB, McGrew SD (1990). Rat serum amyloid P component. Biochem J 270:553–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller MB, Wellis DP, Stellwagen D, Werblin FS, Shatz CJ (1996). Requirement for cholinergic transmission in the propagation of spontaneous retinal waves. Science 272:1182–1187. [DOI] [PubMed] [Google Scholar]
- Garlanda C, Bottazzi B, Bastone A, Mantovani A (2005). Pentraxins at the crossroads between innate immunity, inflammation, matrix deposition, and female fertility. Annu Rev Immunol 23:337–366. [DOI] [PubMed] [Google Scholar]
- Godement P, Salaun J, Imbert M (1984). Prenatal and postnatal development of retinogeniculate and retinocollicular projections in the mouse. J Comp Neurol 230:552–575. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GT, Barres BA (2002). Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron 33:689–702. [DOI] [PubMed] [Google Scholar]
- Gregg RG, Mukhopadhyay S, Candille SI, Ball SL, Pardue MT, McCall MA, Peachey NS (2003). Identification of the gene and the mutation responsible for the nob (no b-wave) phenotype. Invest Ophthalmol Vis Sci 44:378–384. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Thompson ID (2004a). Biochemical and anatomical subdivision of the dorsal lateral geniculate nucleus in normal mice and in mice lacking the beta2 subunit of the nicotinic acetylcholine receptor. Vision Res 44:3365–3376. [DOI] [PubMed] [Google Scholar]
- Grubb MS, Thompson ID (2004b). The influence of early experience on the development of sensory systems. Curr Opin Neurobiol 14:503–512. [DOI] [PubMed] [Google Scholar]
- Guillery RW, Kaas JH (1971). A study of normal and congenitally abnormal retinogeniculate projections in cats. J Comp Neurol 143:73–100. [DOI] [PubMed] [Google Scholar]
- Guillery RW, Scott GL, Cattanach BM, Deol MS (1973). Genetic mechanisms determining the central visual pathways of mice. Science 179:1014–1016. [DOI] [PubMed] [Google Scholar]
- Hsu Y-C, Perin MS (1995). Human neuronal pentraxin II (NPTX2): conservation, genomic structure and chromosomal localization. Genomics 28:220–227. [DOI] [PubMed] [Google Scholar]
- Huberman AD, Stellwagen D, Chapman B (2002). Decoupling eye-specific segregation from lamination in the lateral geniculate nucleus. J Neurosci 22:9419–9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman AD, Wang G-Y, Liets LC, Collins OA, Chapman B, Chalupa LM (2003). Eye-specific retinogeniculate segregation independent of normal neuronal activity. Science 300:994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman AD, Murray KD, Warland DK, Feldheim DA, Chapman B (2005). Ephrin-As mediate targeting of eye-specific projections to the lateral geniculate nucleus. Nat Neurosci 8:1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riqueime PA, Brotz TM, Shatz CJ (2000). Functional requirement for Class 1 MHC in CNS development and plasticity. Science 290:2155–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996). Synaptic activity and the construction of cortical circuits. Science 274:1133–1138. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, Matzuk MM, Dodds DC, Perin MS (2000). Biochemical interactions of the neuronal pentraxins. J Biol Chem 276:17786–17792. [DOI] [PubMed] [Google Scholar]
- Meister M, Wong RO, Baylor DA, Shatz CJ (1991). Synchronous bursts of action potentials in ganglion cells of the developing mammalian retina. Science 252:939–943. [DOI] [PubMed] [Google Scholar]
- Muir-Robinson G, Hwang BJ, Feller MB (2002). Retinogeniculate axons undergo eye-specific segregation in the absence of eye-specific layers. J Neurosci 22:5259–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland TD, Friday BB, Maulit MT, Gerton GL (1994). The sperm acrosomal matrix contains a novel member of the pentaxin family of calcium-dependent binding proteins. J Biol Chem 269:32607–32614. [PubMed] [Google Scholar]
- O'Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P (1999). Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron 23:1–20. [DOI] [PubMed] [Google Scholar]
- Pak W, Hindges R, Lim YS, Pfaff SL, O'Leary DD (2004). Magnitude of binocular vision controlled by islet-2 repression of a genetic program that specifies laterality of retinal axon pathfinding. Cell 119:567–578. [DOI] [PubMed] [Google Scholar]
- Paulsen O, Heggelund P (1996). Quantal properties of spontaneous EPSCs in neurones of the guinea-pig dorsal lateral geniculate nucleus. J Physiol 496:759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn AA, Riquelme PA, Feller MB, Shatz CJ (1998). Competition in retinogeniculate patterning driven by spontaneous activity. Science 279:2108–2112. [DOI] [PubMed] [Google Scholar]
- Perin MS, Fried VA, Mignery GA, Südhof TC, Jahn R (1990). Phospholipid binding by a synaptic vesicle protein homologous to the regulatory domain of protein kinase C. Nature 345:260–263. [DOI] [PubMed] [Google Scholar]
- Pfeiffenberger C, Cutforth T, Woods G, Yamada J, Renteria RC, Copenhagen DR, Flanagan JG, Feldheim DA (2005). Ephrin-As and neural activity are required for eye-specific patterning during retinogeniculate mapping. Nat Neurosci 8:1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quina LA, Pak W, Lanier J, Banwait P, Gratwick K, Liu Y, Velasquez T, O' Leary DD, Turner EE (2005). Brn3a-expressing retinal ganglion cells project specifically to thalamocortical and collicular visual pathways. J Neurosci 25:11595–11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MS, Blobel CP (1994). Apexin, an acrosomal pentaxin. J Biol Chem 269:32615–32620. [PubMed] [Google Scholar]
- Ren J-C, LaVail MM, Peachey NS (2000). Retinal degeneration in the nervous mutant mouse. III. Electrophysiological studies of the visual pathway. Exp Eye Res 70:467–473. [DOI] [PubMed] [Google Scholar]
- Rossi FM, Pizzorusso T, Porciatti V, Marubio LM, Maffei L, Changeux J-P (2001). Requirement of the nicotinic acetylcholine receptor b2 subunit for the anatomical and functional development of the visual system. Proc Natl Acad USA 98:6453–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999). Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22:389–442. [DOI] [PubMed] [Google Scholar]
- Schlimgen AK, Helms JA, Vogel H, Perin MS (1995). Neuronal pentraxin, a secreted protein with homology to acute phase proteins of the immune system. Neuron 14:519–526. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Shatz CJ (2002). An instructive role for retinal waves in the development of retinogeniculate connectivity. Neuron 33:357–367. [DOI] [PubMed] [Google Scholar]
- Tian N, Copenhagen DR (2001). Visual deprivation alters development of synaptic function in inner retina after eye opening. Neuron 32:439–449. [DOI] [PubMed] [Google Scholar]
- Torborg CL, Hansen KA, Feller MB (2005). High frequency, synchronized bursting drives eye-specific segregation of retinogeniculate projections. Nat Neurosci 8:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui CC, Copeland NG, Gilbert DJ, Jenkins NA, Barnes C, Worley PF (1996). Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci 16:2463–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA (2001). Control of synapse number by glia. Science 291:657–660. [DOI] [PubMed] [Google Scholar]
- Whitehead AS, Zahedi K, Rits M, Mortensen RF, Lelias JM (1990). Generation of cDNA clones, structural analysis, and induction of mRNA during inflammation. Biochem J 266:283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RO, Meister M, Shatz CJ (1993). Transient period of correlated bursting activity during development of the mammalian retina. Neuron 11:923–938. [DOI] [PubMed] [Google Scholar]
- Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O'Brien RJ, Worley P (2003). Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron 39:513–528. [DOI] [PubMed] [Google Scholar]