

Abstract
Lipids play important roles in cellular dysfunction leading to disease. Although a major role for phospholipids is in defining the membrane permeability barrier, phospholipids play a central role in a diverse range of cellular processes and therefore are important factors in cellular dysfunction and disease. This review is focused on the role of phospholipids in normal assembly and organization of the membrane proteins, multimeric protein complexes, and higher order supercomplexes. Since lipids have no catalytic activity, it is difficult to determine their function at the molecular level. Lipid function has generally been defined by affects on protein function or cellular processes. Molecular details derived from genetic, biochemical, and structural approaches are presented for involvement of phosphatidylethanolamine and cardiolipin in protein organization. Experimental evidence is presented that changes in phosphatidylethanolamine levels results in misfolding and topological misorientation of membrane proteins leading to dysfunctional proteins. Examples are presented for diseases in which proper protein folding or topological organization is not attained due to either demonstrated or proposed involvement of a lipid. Similar changes in cardiolipin levels affects the structure and function of individual components of the mitochondrial electron transport chain and their organization into supercomplexes resulting in reduced mitochondrial oxidative phosphorylation efficiency and apoptosis. Diseases in which mitochondrial dysfunction has been linked to reduced cardiolipin levels are described. Therefore, understanding the principles governing lipid-dependent assembly and organization of membrane proteins and protein complexes will be useful in developing novel therapeutic approaches for disorders in which lipids play an important role.
Keywords: Membrane protein folding, phosphatidylethanolamine, cardiolipin, mitochondria, oxidative phosphorylation
8.1 Introduction
Deciphering the role of lipids in normal and dysfunctional cell function is a more complex and challenging task than defining similar roles for specific proteins. Roles for individual lipids and lipid classes are diverse and widespread throughout a cell where individual proteins have narrow and well-defined functions that usually are localized to discrete cellular locations. Lipids define the essential membrane permeability barrier of cells and internal organelles. The membrane lipid bilayer is a dynamic non-covalent supermolecular organization of individual lipid molecules whose combined physical and chemical properties define the matrix within which membrane proteins are organized. Lipids govern the folding, organization, and final structure of all membrane proteins. Lipids directly influence and modulate the function of membrane proteins and a large number of amphitropic proteins that reversibly interact with the membrane surface. They act as metabolic signaling molecules and are the substrates for posttranslation modification of proteins.
Lipids exert their influence on cellular processes through their diverse chemical spectrum and the cooperative physical properties of lipid mixtures of variable composition (Dowhan, 1997; Dowhan et al., 2008). The complexity of the lipidome is often not fully appreciated. Eukaryotic cells contain phospholipids, sphingolipids, and glycolipids with a wide variation in the hydrophilic headgroups as well as diverse fatty acid compositions. Add to this complexity other lipids such as steroids and fatty acid derived cellular products, and the complexity of the lipidome equals or exceeds that of the proteome. While the basic characteristics and structure of the proteome among all organisms are largely conserved, lipid complexity increases significantly when all forms of life are considered. Through the efforts of LIPID MAPS in the United States (Schmelzer et al., 2007) and the European Lipodomics Initiative (van Meer et al., 2007), the composition of the lipidome is rapidly approaching the level of detail available for the proteome. This type of structural information will be essential for complete understanding of the molecular basis for normal lipid function and lipid-related cellular dysfunction in disease.
Lipids are not covalently bound in membranes but rather interact dynamically to form transient arrangements with asymmetry both perpendicular and parallel to the plane of the lipid bilayer. The fluidity, supermolecular-phase propensity, lateral pressure and surface charge of the bilayer matrix is largely determined by the collective properties of the complex mixture of individual lipid species, some of which are shown in Fig. 8.1. Lipids also interact with and bind to proteins in stiochiometric amounts affecting protein structure and function. The broad range of lipid properties coupled with the dynamic organization of lipids in membranes multiplies their functional diversity in modulating the environment and therefore the function of membrane proteins.
Fig. 8.1.

Structure of glycerol-based lipids. Stick drawing represents carbon backbone of headgroup (bold) and fatty acid chains. “R” represents hydrocarbon chains ranging from 10 to 22 in length with and without double bonds. Headgroups are attached to the sn-3 position of glycerol and the nature chirality is noted for the sn-2 position
The pleiotropic function of even a single lipid and its widespread distribution within a given cell poses difficult challenges to defining roles for a lipid in cellular processes. Unlike proteins lipids display no inherent catalytic activity and are not encoded by genes so that many of the initial clues to lipid function outside of forming membrane bilayers have been derived from in vitro studies. Lipid function is many times defined by an effect on a biological process or enzyme reaction elicited by addition of a lipid. Often not considered in such experiments is the effect of the physical properties of a lipid since within any give set of lipids with the same hydrophilic domains exists a large array of possible hydrophobic domains (such as the diverse fatty acid composition of phospholipids). Although there is a large body of information describing the physical properties (phase properties, supermolecular organization, fluidity, etc.) of individual lipids and lipid mixtures, it is still not clear how to translate this information into a biological function or mechanism. Is the affect of a lipid on a process studied in vitro due to its physical properties, its chemical properties or an artifact due to adding a lipid with the wrong properties? Therefore, studying lipid-protein interaction only in vitro has serious limitations. However, studies in vivo pose additional obstacles. Genetic approaches are indirect since mutations must be made in genes that encode for biosynthetic enzymes along a pathway leading to a final lipid product. Such mutations result in changes in multiple lipid intermediates many of which may have important functions. In many cases such mutations are lethal causing loss of membrane integrity before more specific functions are affected and identified. The cumulative effect of loss of diverse functions often results in cell death and in complex phenotypes, especially in eukaryotic cells containing multiple organelles. However, in many cases viable lipid mutants in culture and even in whole animals have been generated. Defining lipid function requires biochemical dissection of biological processes as influenced by lipids and must be based on full knowledge of the physical and chemical properties of lipids. The in vitro properties must be verified by and be consistent with the properties of cells whose lipid metabolism and composition have been subjected to molecular genetic manipulation. Molecular genetic manipulation of cellular lipid metabolism has been most widely applied to bacteria and S. cerevisiae (Dowhan et al., 2004), but such approaches are increasingly successful in mammalian cell culture and whole animals.
Membrane proteins represent at least 30% of the all currently sequenced genomes and represent 60 percent of drug targets. In addition at least an equal number of proteins transiently interact with the membrane surface. More than half the drug targets pursued by pharmaceutical companies are related to membranes or membrane bound proteins (Drews, 2006). Effective drug design is dependent on understanding membrane protein structure and the rules that govern the folding and assembly of native and mutant membrane proteins as well as the principles governing interaction of “soluble” proteins with the membrane. In the past decade major advances have been made towards understanding the mechanisms by which polytopic membrane proteins fold and assemble in cellular membranes. However, the role that lipids play in the folding and assembly of membrane proteins, in the higher order organization of molecular machines, and in stabilizing final functional organization of proteins has only recently received attention. Through genetic manipulation of cellular lipid composition, it is now clear that membrane lipid composition is a determinant in the folding and topological organization of membrane proteins. Recent advances in detailed structural analysis of membrane proteins coupled with genetic manipulation of lipid composition has demonstrated that lipids play a specific role as integral components of multisubunit membrane protein complexes and higher order organization of complexes into molecular machines. Therefore, how lipids influence folding, assembly and function of proteins will be useful in developing novel therapeutic approaches for disorders involving proteins that associate with the membrane.
This review will focus on a combination of molecular genetic and biochemical studies on the role of primarily phosphatidylethanolamine (PE) and cardiolipin (CL) in the folding and organization of individual membrane proteins and multicomponent supercomplexes. The results of such studies will be related to the known and possible involvement of lipids in diseases resulting from lack of proper organization of membrane proteins. Rather than being an inclusive review of protein-lipid interactions, the aim is to select specific well-documented examples of lipid-protein interactions to illustrate the broader role of lipids in determining cellular function.
8.2 Lipid-Assisted Protein Folding
8.2.1 Experimental Evidence for Lipid-Assisted Folding of Proteins
Molecular chaperones, traditionally proteins, facilitate the folding of proteins by interacting non-covalently with non-native folding intermediates and not with either the native or totally unfolded protein. When folding is complete, molecular chaperones are not required to maintain proper conformation. However, molecular chaperone function is not restricted to proteins (Bogdanov et al., 1996; Ellis, 1997). Specific lipids are able to interact with partially folded proteins in a transient manner in either de novo protein folding or protein renaturation in vitro similar to that of protein molecular chaperones.
The most compelling evidence for a specific role of phospholipids in membrane protein folding is the requirement for PE in the folding of the integral membrane protein lactose permease (LacY) of Escherichia coli (Bogdanov and Dowhan, 1999). LacY is organized within the inner cytoplasmic membrane as twelve transmembrane domains (TMs) connected by alternating solvent exposed cytoplasmic and periplasmic domains with both the N- and C-terminus oriented inward (Fig. 8.2). Normal assembly of LacY occurs into E. coli membranes containing an abundance of PE (70% with the remainder being about 20% phosphatidylglycerol (PG) and 5% CL) so that a separation between phospholipid-assisted and unassisted folding pathways cannot be distinguished in vivo. Studies of the lipid-assisted folding of periplasmic domain P7 connecting segments TMVII and TMVIII of LacY was made possible by a conformation specific monoclonal antibody directed to domain P7, viablemutants of E. coli lacking PE, and the development of a modification of the Western blotting procedure (Eastern-Western blot) that allowed renaturation of proteins in the presence of lipids on a solid support (Bogdanov et al., 1996, 1999). The loss of uphill energy-dependent transport function of LacY in mutant cells lacking PE was initially correlated with misfolding of domain P7 that is crucial for uphill energy dependent transport of substrate (Sun et al., 1996). Sodium dodecyl sulfate polyacrylamide gel electrophoresis (which only partially denatures LacY) followed by Western blotting analysis (using the conformation specific monoclonal antibody) of LacY from PE-containing cells demonstrated that the protein could be separated from PE (as determined by absence of radiolabeled phospholipid) and still retain its native structure with respect to domain P7. However, LacY from cells lacking PE was not detected by the conformation specific monoclonal antibody but was detected by a polyclonal antibody. Thus LacY is “denatured” in vivo with respect to the P7 domain by assembly in a non-native lipid environment. Therefore, once information was imparted during folding of LacY in vivo in the presence of PE, this lipid was no longer required for maintenance of proper conformation of the protein.
Fig. 8.2.

Topological models for LacY as a function of lipid composition. Topology of LacY in PE-containing (+PE) and PE-lacking (-PE) E. coli cells is illustrated in the upper left and right diagrams, respectively. The topology of LacY after initial assembly in cells lacking PE followed by post-assembly synthesis of PE is shown in the bottom diagram. The sequences within the rectangular shaded areas (TMs) define the amino acids that lie within the 27 Å hydrocarbon core of the bilayer excluding the lipid head groups. The darkly shaded TMs are those that undergo rearrangements as a function of membrane lipid composition (see text). TMs (Roman numerals), extramembrane domains (P in black for periplasmic and C in gray for cytoplasmic domains as in PE-containing cells), N-terminus (NT) and C-terminus (CT) are shown. The minus signs in TMVII represent the negative amino acids that salt bridge in PE-containing cells with positive residues in TMX and TMXI
The misfolding of LacY due to assembly in PE-lacking cells was corrected by employing the Eastern-Western blotting technique in which LacY was exposed to hydrated phospholipids during renaturation from sodium dodecyl sulfate on a solid support followed by probing with a conformation specific monoclonal antibody (Bogdanov and Dowhan, 1999; Bogdanov et al., 1996, 1999). LacY regained the native conformation of domain P7, which was absent during in vivo assembly, after renaturation in the presence of specifically PE. Anionic lipids PG and CL and “foreign” lipids such as phosphatidylcholine (PC) did not support proper refolding. Proper refolding did not occur with protein extensively denatured by sodium dodecyl sulfate-urea treatment. In addition monoclonal antibody recognition of LacY assembled either in vitro (Bogdanov and Dowhan, 1998) or in vivo (Bogdanov et al., 2002) in the absence of PE could be restored after its insertion and assembly by the initiation of PE synthesis in the absence of new synthesis of LacY.
The interaction between lipids and partially folded LacY during refolding was found to be structurally specific. Both the chemical properties of the individual lipid molecules and the collective properties of phospholipid mixtures were determinants supporting proper protein folding (Bogdanov et al., 1999). Minimal requirements for refolding were lipid mixtures in the bilayer state containing diacyl phospholipids with an ionizable primary amine (i.e., PE, phosphatidylserine (PS), and mono- and dimethyl-PE, but not PC). Non-bilayer prone forms of PE were only effective if mixed with an excess of bilayer forming lipids. Stereoisomers of amino-containing lipids with either unnatural backbone or head group configuration did not support proper refolding. Therefore, there is a specific requirement for an ionizable amine-containing phospholipid of natural chirality and preference for bilayer organization to facilitate proper folding of LacY into its native conformation. Thus PE appears to facilitate in vitro refolding and in vivo folding into a fully native conformation by interacting with LacY via a transient non-covalent interaction with a folding intermediate and fulfills the minimum requirements of a molecular chaperone.
Is lipid-assisted folding a widespread phenomenon and possibly applicable to soluble proteins? The erythrocyte membrane contains about 20-mole % of PE that is almost exclusively localized in the inner leaflet and is in contact with highly concentrated heme-containing proteins. The refolding of the denatured soluble and heme-containing enzyme horseradish peroxidase (HRP) was followed in the presence and absence of liposomes made up of different phospholipids (Debnath et al., 2003). Remarkably, dimyristoyl-PE (a bilayer-forming PE) was able to drastically increase the yield of renatured enzyme relative to refolding in the absence of liposomes. However, dioleoyl-PE, which does not favor bilayer organization, did not support proper refolding. PCs containing a wide range of fatty acids were either non-supportive of refolding or inhibited refolding relative to folding in the absence of liposomes. Moreover Trp117 quenching through energy transfer with the heme moiety indicated that the denatured protein after dimyristoyl PE-assisted folding assumed an overall conformation similar to that of the native protein with the heme moiety in a native-like conformation. Therefore, LacY and the peroxidase share common requirements for proper refolding dependent on lipids.
Based on all these results lipids can function as non-protein molecular chaperones or lipochaperones that specifically mediate the folding of proteins thereby extending the definition of chaperones to other biomolecules in addition to proteins (Bogdanov and Dowhan, 1999).
8.2.2 Lipochaperones and Protein Folding Disorders
8.2.2.1 Alzheimer’s and Scapies Diseases
Even under healthy conditions the energetic balance between folding and misfolding pathways of membrane proteins is very delicate and fragile (Sanders and Myers, 2004). Therefore, membrane protein folding can be re-routed in the pathophysiological direction by both mutations and changes in protein environment including the membrane lipid composition. Errors in insertion (Milenkovic et al., 2007), folding (Lin and Liu, 2006), localization and intracellular trafficking (Aridor and Hannan, 2000), processing (Fadiel et al., 2007) or degradation (Gelman and Kopito, 2003), and turnover (Sambamurti et al., 2006) of integral transmembrane proteins are responsible for numerous diseases including neurodegenerative cerebral amyloidoses, cystic fibrosis and others (Harrison et al., 2007). Misfolded proteins fail to be degraded and become prone to formation of toxic aggregates. Diverse disorders such as Alzheimer’s disease and prion/scrapie disease (Monaco et al., 2006), cystic fibrosis (Mendoza and Thomas, 2007) and even cataract formation (Crabbe, 1998) and type 2 diabetes (Hayden et al., 2005) arise from protein misfolding and are grouped together under the category of conformational diseases.
Prion and scrapie diseases are linked with the conformational transition of normally monomeric α-helical cellular prion protein, PrPc, to a ß-sheet-rich pathogenic form, PrPSC, which is prone to aggregation. A similar conformational transition of the normal cellular form of α-helical amyloid peptide (αAP (1-40)) into the disease-specific largely ß-sheet form of amyloid peptide (ßAP (1-40)) occurs in Alzheimer’s disease, which results in amyloid deposits (Monaco et al., 2006). So far more than 19 different mutations in the human PrP gene have been linked with inherited prion diseases (Monaco et al., 2006). However, the molecular event triggering the spontaneous conversion of wild-type andmutant PrPC forms to the infectious PrPSC isoform is still unknown. Since αAP is normally produced as a soluble peptide, the question arises as to what conditions induce conversion to the ßAP form and aggregation of the peptide? Thus the search for molecules interacting with cellular forms of PrP or Alzheimer amyloid peptide is a major effort in the study of transmissible amyloidoses.
An abundance of evidence exists suggesting that specific membrane lipids might serve as templates or nucleation sites, which play an important role in these two diseases by promoting the pathological folding of these proteins in vivo. Syrian hamster prion protein PrP has a high affinity for negatively charged phospholipid membranes, but it does not bind to membranes made of only zwitterionic PC (Sanghera and Pinheiro, 2002). The association of human (Morillas et al., 1999) or Syrian hamster PrP (Sanghera and Pinheiro, 2002) to negatively charged membranes is accompanied by an increase in ß-sheet structure, which results mostly from electrostatic lipid-protein interactions while binding of PrP to zwitterionic membranes composed of saturated PC mixed with cholesterol and sphingomyelin in a raft-mimicking ratio leads to a stabilization of α-helical structure through predominant hydrophobic lipid-protein interactions. Therefore electrostatic lipid-PrP interactions appear to promote ß-sheet formation, while hydrophobic lipid-protein interactions seem to preserve α-helix formation (Sanghera and Pinheiro, 2002).
A growing number of observations indicate that at least some pathological effects of the ßAP (1-40), the major component of Alzheimer plaques, could be mediated by peptide-lipid interactions. This peptide binds (Yanagisawa et al., 1995) to ganglioside GM1 containing membranes and upon binding undergoes a rapid conformational transition from random coil to an ordered conformation rich in ß-sheet structure which was not detected upon binding to ganglio-side-free liposomes composed of zwitterionic phospholipids (PC), acidic phospholipids (PG and PS) or the isolated oligosaccharide moiety of the ganglioside (Choo-Smith and Surewicz, 1997). Furthermore, GM1 tends to accumulate in certain regions of neurons forming ganglioside-rich domains in neural membranes. Since GM1-bound ßAP (1-40) is associated with early diffuse plaques, it was suggested that GM1-bound peptide may act as a template that facilitates self-aggregation of ßAP peptides as a precursor to formation of a mature amyloid plaques (Yanagisawa, 2005). Therefore, ganglioside appears to act as an anti-chaperone by inducing a misfolding event. Taken together, these data suggest that structural propensities of AP and PrP peptides are determined by the nuances of specific lipid environments and major conformational changes in prion and Alzheimer proteins may involve lipids as auxiliary molecules in the pathogenesis of these diseases.
8.2.2.2 Lipid Basis for Cystic Fibrosis
Lack of Phe508 in the first nucleotide binding domain (NBD-1) of the cystic fibrosis transmembrane conductance regulator (CFTR) is the molecular basis for the most common form of cystic fibrosis (Mendoza and Thomas, 2007). Isolated NBD-1 lacking Phe508 displays a kinetic defect in folding as evidenced by a dramatic reduction in the refolding efficiency starting from an unfolded state (Qu et al., 1997). This mutant form of CFTR fails to reach the plasma membrane and is retained in the endoplasmic reticulum (ER) membrane due to either illicit interactions between the mutated domain and molecular chaperones or other cellular factors in the ER (Cheng et al., 1990). The ability of acrylamide to quench the fluorescence of Trp496 located within the wild-type NBD-1 was drastically suppressed in the presence of PS while PC failed to protect this residue from acrylamide quenching (Eidelman et al., 2002). However, the changes in secondary structure due to a Phe508 mutation resulted in protection of Trp496 in the presence of both PC and PS. Thus the wild-type NBD-1 interacts selectively with PS while NBD-1 carrying the Phe508 mutation loses the ability to discriminate between these two phospholipids (Eidelman et al., 2002). This observation demonstrates that specific phospholipid-protein interactions are critical in maintaining the defined structure of the NBD-1 domain and that mutations that change the dynamics of this interaction may be the molecular basis for misfolding of the protein. The authors suggested that this lipid-specific effect on the conformation of the Phe508 mutant form of NBD-1 may have pathophysiological significance for cystic fibrosis and postulated that the trafficking defect for mutant CFTR might be based on aberrant interactions with PC since changes in cellular content of PC appear to have an drastic effect on mutant CFTR maturation and trafficking in vivo (Eidelman et al., 2002). The quantitative replacement of a large fraction of the PC with phospholipid analogues whose headgroups correspond to an analogue supplement (either 2-aminobutanol, methylethanolamine or 3-aminopropanol) in the media resulted not only in an increase in the total amount of mutant CFTR, but also in an effect on maturation leading to an increase in higher molecular weight forms of CFTR in a dose-dependent manner. In contrast cells expressing wild-type CFTR responded to the various supplements with either little change or reductions in CFTR level, indicating that choline headgroup replacement differentially affects the maturation and stability of wild-type and mutant CFTR.
8.2.2.3 Lipid Involvement in Biogenesis of CD1 Molecules
Almost a decade ago, immunology faced a paradigm shift when it became apparent that specialized CD1 - restricted T lymphocytes recognize not only peptides but also lipid antigens (Gumperz, 2006; Joyce, 2001). Antigenic lipid - CD1 complexes can present foreign glycolipid antigens to a special subpopulation of T cells, the CD1-restricted invariant natural killer T (NKT) lymphocytes (Brutkiewicz et al., 2003; Parekh et al., 2005). Human CD1d molecules can present several types of foreign lipid and glycolipid components of the cell wall of pathogenic bacteria, mycobacteria and protozoa lipids. Exogenous lipids of foreign origin with large oligosaccharide headgroups or long alkyl chains require internalization via the endocytic system to the lysosomes prior to binding to CD1d molecules (Porcelli, 2001; Roberts et al., 2002).
In vitro CD1d molecules appear to bind a wide variety of lipids that vary in the chemical properties (Joyce, 2001). The crystallographic structure of CD1d protein shows a narrow hydrophobic ligand-binding cleft specifically designed to bind lipids rather than peptides (Koch et al., 2005). The lipid ligand of CD1d as initially isolated from in vivo sources is highly restricted to phosphatidylinositol (PI) and the glycosaminylated PI (GPI) of GPI-linked proteins (De Silva et al., 2002; Joyce et al., 1998). However, repeated attempts to activate NKT cells by the addition of purified PI or GPI have failed (De Silva et al., 2002; Molano et al., 2000). The fact that NKT cells do not recognize the CD1d-PI/GPI complex implies that they are not the ligand recognized by NKT cells (De Silva et al., 2002). The apparent exclusive association of PI with CD1d in vivo occurs during assembly in the ER membrane (De Silva et al., 2002). This paradox can be explained as follows. Loading of the lipid-binding site exclusively with PI/GPI occurs in the ER and is maintained through the normal secretory pathway to the plasma membrane. Upon endocytosis and passage through the lysosomes, CD1d protein exchanges lipids loaded in the ER for lipids of foreign origin that have also been internalized via the endocytic system. CD1d protein with its newly acquired lipid ligand is then recycled to the plasma membrane where the antigenic lipid-CD1d complex is recognized by NKT cells. This temporal association of ER lipids with CD1 protein during biogenesis can satisfy all structural and physiological requirements of a molecular chaperone by: first, protecting and preserving the integrity of the large hydrophobic lipid antigen-binding groove from collapse during trafficking; second, occupying the site to prevent premature lipid antigen loading; third, occupying the binding site with easily dissociated ligand that later might be exchanged for a lipid antigen in the lysosomes (De Silva et al., 2002; Joyce, 2001; Park et al., 2004). Thus, assembly of CD1 with cellular lipids in the ER is an evolutionarily conserved feature of a chaperone-like role for lipids involved in biogenesis of CD1.
8.2.2.4 Lipid Involvement in Diabetes
Because of its importance to human health, insulin has been one of the most studied biological molecules. However, important details of the folding and assembly, storage and release of insulin remain unresolved. Insulin is synthesized as a single stranded immature precursor (preproinsulin) that contains an N-terminal signal sequence. After processing of the signal sequence, disulfide bonds between the A and B chains and within the A chain are formed followed by folding into mature monomeric reduced proinsulin form. Proinsulin exits from the Golgi network as a zinc associated hexamer and is stored in secretory granules where it is converted by proteolysis to the less soluble hexameric insulin crystalline form (Dodson and Steiner, 1998). The hexameric complex gradually dissolves to deliver the monomeric, bioactive form of insulin into the bloodstream stimulated by elevated plasma glucose levels. The crystalline forms of insulin can be either amorphic (irreversible misfolded form of insulin) or native (which can be monomerized in a bioactive form). Several lines of evidence suggest that sulfatides promote the productive folding of reduced proinsulin into its native crystalline form, indicating that a sulfatide (3′-sulfo-galactosylceramide) possesses molecular chaperone-like activity. Sulfatides are acidic glycosphingolipids that present primarily within the islets of Langerhans or ß cells of the pancreas where insulin is produced and in the nervous system.
Insulin-dependent diabetes mellitus is an autoimmune disease and high titers of auto-antibodies against both insulin and sulfatide were found in patients with insulin-dependent diabetes (Andersson et al., 2002; Buschard et al., 2005). Sulfatide and insulin are present in the same cellular compartments and share the same intracellular trafficking pathways (Buschard et al., 2005; Fredman et al., 2000). The inhibition of sulfatide synthesis with chloroquine and fumonisine B1 leads to inhibition of insulin granule formation in vivo (Fredman et al., 2000). Sulfatide binds directly to insulin, and sulfatide, but not its galactosylceramide precursor, was able to compete with monoclonal antibodies directed against a subdomain of the insulin molecule (Osterbye et al., 2001). Sulfatide specifically promotes in vitro refolding of proinsulin into its zinc-dependent hexameric form while without a sulfatide or in the presence of galactosylceramide, proinsulin dimers and hexamers were nearly absent (Osterbye et al., 2001). Sulfatide and not galactosylceramide specifically mediates the conversion of insulin hexamers to the biological active monomers at neutral pH, the pH at the ß-cell surface. The monomerization process is a dynamic equilibrium between insulin hexamers, dimers, and monomers. It appears that sulfatide is responsible for pushing the equilibrium toward the insulin monomer. Thus sulfatide has a dual role in supporting productive folding of the proinsulin form and promoting insulin monomerization. This is the first description of a lipid molecule acting as a molecular chaperone on the unfolded precursor of a protein (proinsulin) and demonstrating a functional interaction with mature protein (insulin) later in storage and release of the bioactive form.
Since sulfatide is the only glycolipid that so far has been associated with insulin-dependent diabetes mellitus, the therapeutic effect of sulfatide on the development of diabetes in the non-obese diabetic mouse was tested (Buschard et al., 2001). Diabetes was prevented in these mice by administration of sulfatide or its precursor, galactosylceramide. The mice were treated with either sulfatide, galactosylceramide (which is converted to sulfatide in the Golgi) or GM1 (a negatively charged glycosphingolipid lacking sulfate) or phosphate buffered saline control. Among all the control-treated mice 93% became diabetic. However sulfatide or galactosylceramide reduced the diabetes incidence by 50%. In contrast 86% of GM1-treated mice became diabetic. Therefore either administered or newly synthesized sulfatide is able to prevent diabetes in non-obese diabetic mice.
The composition and organization of membrane lipid species are altered in several human diseases (Alemany et al., 2007; Vigh et al., 2005). For example, in insulin-dependent diabetes mellitus massive changes occur in lipid composition of the rat myocardium causing a 46% increase in PI levels and a 22% decrease in PE content, which was prevented by insulin treatment after induction of the diabetic state (Han et al., 2000). The pathology of diabetes is not easily explained simply by a defect in glucose uptake by cells. Such drastic changes in membrane lipid composition could result in many unrecognized effects on the assembly, organization and function of membrane proteins.
Rearrangement of membrane lipid microdomains was recently recognized as crucial for proper compartmentalization and localization of insulin signaling. Insulin resistance due to the inhibition of insulin signaling can be caused by elimination of insulin receptors from caveolae microdomains induced by an accumulation of ganglioside GM3 (Kabayama et al., 2007), which triggers dissociation of the insulin receptor and caveolin-1 complex. The pharmacological inhibition of GM3 biosynthesis by a specific glucosylceramide synthase inhibitor resulted in almost complete recovery of insulin signaling (Kabayama et al., 2005), which may provide a new approach to treat insulin resistance. Such interference with glycosphingolipid biosynthesis not only enhanced insulin sensitivity (Aerts et al., 2007) but also improved glucose tolerance (Zhao et al., 2007).
8.3 Lipid-Dependent Membrane Protein Topogenesis
8.3.1 Membrane Protein Topological Organization
A fundamental architectural principle of the structure of polytopic membrane proteins is membrane topology, i.e. the number of TM segments and their orientation relative to the membrane bilayer. Numerous algorithms exist that predict TM segments of membrane proteins based on the length of hydrophobic domains along the amino acid sequence (Elofsson and von Heijne, 2007). Such in silico approaches are about 85% accurate in predicting the alternating orientation of TMs connected by extramembrane domains. However, absolute orientation with respect to the membrane bilayer is less predictable unless the sidedness of the N-terminal and/or C-terminal extramembrane domains is known. Because these algorithms rely on cumulative short-range interactions and cannot incorporate the effects of long-range interactions such as stabilizing a charged hydrophilic domain within the membrane bilayer by salt bridging to another membrane domain, the organization of a significant number of membrane proteins cannot be accurately predicted. There has been extensive investigation aimed at understanding the features of the amino acid sequence that determine the insertion and orientation of membrane protein, but the role of the membrane lipid composition as a putative topological determinant has been largely ignored. However, membrane lipids manifest a diverse array of hydrophilic and hydrophobic surfaces and positive and negative charges that can influence the folding and orientation of integral membrane proteins. Therefore, in silico predictions of membrane protein topology is only a starting point providing a framework within which topology can be experimentally determined. Since protein-lipid interactions have now been established as a determinant of membrane protein topogenesis, the molecular basis for some diseases resulting from mutations in membrane proteins may be a result of topological misorientation of proteins.
8.3.2 Properties of Lipids That Determine Protein Topogenesis
Although topogenic signals encoded in the protein sequence of membrane proteins are primary determinants of final protein organization in the membrane, the topological organization of several twelve TM spanning secondary transporters of E. coli is dramatically influenced by the membrane lipid composition thus making the properties of the lipid bilayer a factor in determining protein topology. Development of viable E. coli strains with altered phospholipid composition (Dowhan et al., 2004) and advanced methods for determining protein topology, in particular the substituted cysteine accessibility method as applied to TM determination (Bogdanov et al., 2005), revealed that membrane lipid composition is a critical determinant of topology. The topologies of the N-terminal six-TM helical bundle of LacY (Bogdanov et al., 2002) (Fig. 8.2) and the N-terminal two-TM hairpin of phenylalanine permease (Zhang et al., 2003) and γ-aminobutyrate permease (Zhang et al., 2005b) are inverted with respect to the membrane bilayer and the remainder of each protein when assembled in membranes lacking PE, the major phospholipid of this organism, and containing only the anionic phospholipids PG and CL.
What specific structural, chemical or phase-forming features of lipid molecules are important for proper TM topogenesis? Structural features, individual chemical properties, and the collective physical properties of lipids in association with each other must be considered when assessing protein-lipid interactions. For instance PE is a zwitterionic glycerophosphate-based diacyl lipid with no net charge (Fig. 8.1) that favors the formation of a typical membrane bilayer structure when both fatty acids are saturated, but favors non-bilayer structures with increasing unsaturation of its fatty acids and with increasing temperature. However, the charge properties of PE are somewhat dampened by the formation of an internal charge paired ring between the phosphate and amine moieties, which cannot be formed by the zwitterionic phospholipid PC due to the trimethylated amine. PC on the other hand is nearly always bilayer forming and like PE capable of countering the high negative charge density contributed to thebilayer surface by net negatively charged phospholipids such as PS, CL, phosphatidic acid, PG or PI. The first three can form non-bilayer structures in the presence of divalent cations while all contribute to a net negative membrane surface. Finally, monoglucosyldia-cylglycerol (MGlcDG) has no charge character (Fig. 8.1) and can be either bilayer or non-bilayer forming depending on temperature and fatty acid content. Diglucosyldiacylglycerol (DGlcDG) is similar to MGlcDG but only forms bilayer structures. The hydrophilic headgroup of these glycolipids are vastly different in their structure from that of phospholipids and although not charged can still participate in hydrogen bonding and dilution of overall membrane surface charge as does PE.
Strikingly, the replacement of PE in vivo by the foreign lipids MGlcDG (Xie et al., 2006) or DGlcDG (Bogdanov, Xie and Dowhan, unpublished) by introducing the Acholeplasma laidlawii MGlcDG and DGlcDG synthases, respectively, into an E. coli mutant lacking PE restored wild type TM topology of the N-terminal helical bundle of LacY. While replacement of PE by MGlcDAG restored the kinetic parameters of uphill transport of LacY, substitution of PE by DGlcDG did not. Similar to the effects of DGlcDG, LacY reconstituted into proteoliposomes containing PC and PG appears to display native topology but failed to show uphill transport function (Wang et al., 2002) while PG alone mimicked the aberrant orientation of LacY in PE-lacking cells. Therefore, TM topology is sensitive to the charge density on the membrane surface i.e. neutral MGlcDG and DGlcDG and zwitterionic PE and PC all dilute the negative charge of the membrane surface created by PG and CL and at the same time support native LacY topology. Moreover, the fact that charged but neutral PC and PE and the uncharged glycolipids support native topology strongly suggests that the net charge character of the membrane surface is a more important topological determinant than the structures of the headgroups. Finally, the phase-forming physical properties of the lipids appear not to be a factor in determining topology since MGlcDG and PE tend to be non-bilayer forming while DGlcDG and PC tend to be bilayer forming. Lack of structural specificity further suggests that binding of lipids to specific protein sites is less likely a factor in determining orientation than the properties of the bilayer matrix within which LacY is assembled. Since the predetermined molar ratio of charged/uncharged lipids is the most important lipid determinant of the TM topology, it is tempting to speculate that during the course of evolution both proteins and lipids co-evolved together in the context of the lipid environment of membrane systems in which both are mutually dependent on each other. Although it is surprising that E. coli is tolerant to such major changes in membrane lipid composition, these results emphasize the importance of similar charge properties of lipids in determining membrane protein topology rather than a strict structural requirement, as observed for proteins.
Phenylalanine permease and γ-aminobutyrate permease topology is also lipid dependent and secondary transporters for proline, melibiose, tryptophan and lysine are also defective in uphill transport in E. coli cells lacking PE (Bogdanov and Dowhan, unpublished) suggesting that function and possibly topology of a broad spectrum of transporters is dependent on membrane lipid composition. The orientation of OEP7, an outer envelope protein of spinach chloroplasts, was inverted with respect to its native orientation when reconstituted in liposomes made of a total lipid extract of chloroplasts containing mainly the phospholipids PC and PG (Schleiff et al., 2001). However when the ratio of these two lipids was adjusted to mimic the high PC content of the chloroplast outer membrane, then native topology was achieved. These results strongly support a general dependence of protein topology on lipid composition across species.
8.3.3 Lipid-Triggered TM Molecular Switch
The utilization of strains with tightly regulated inducible promoters controlling the expression of phospholipid biosynthetic enzymes not only allows for controlling steady state membrane lipid composition but dynamic switching of lipid composition. Use of a strain in which PE content can be controlled in a temporal manner revealed surprising topological dynamics of proteins after stable membrane insertion. PE is required to maintain correct relative orientations (Bogdanov et al., 2002) of the N- and C-terminal halves of LacY each independently folded (Nagamori et al., 2003) into a compact bundle of two six TM α-helices connected by the long hydrophilic cytoplasmic domain C6. Reintroduction of PE after assembly of LacY in vivo after membrane insertion and folding of LacY triggers a conformational change (see Fig. 8.2) resulting in a lipid-dependent restoration of uphill transport function and a near complete restoration of the wild type topological orientation (Bogdanov et al., 2002). Five of the six TMs regain native topological organization (Bogdanov et al., 2008). TMII adopts a “U”-shaped mini-loop configuration partially inserted into the cytoplasmic side of the membrane and allows the domains flanking TMII (i.e. P1 and C2) to remain on the same cytoplasmic side of the membrane with TMIII and the adjacent P3 domain adopting a proper topology in the cells with restored PE levels. TMVII, which is exposed to the periplasm in PE-lacking cells, re-inserts into the membrane after introduction of PE into cells. This result clearly demonstrates that changing the lipid composition of the membrane can induce large topology inversions of TMs in complex polytopic proteins after stable assembly.
Why can some proteins or protein domains undergo large TM movement but others cannot? What structural elements enable topological transitions dependent on lipid composition? In order for the N- and C-terminal six TM helical bundles to respond to the lipid environment independent of each other, either during initial assembly or during a change in lipid environment, there must exist flexible hinge regions between the independently folding domains. TMVII flanked by domains C6 and P7 (Fig. 8.2) was found to behave as a required molecular hinge by exiting the membrane to the periplasm in PE-lacking cells to allow sufficient flexibility so that the two halves of LacY could respond differentially to membrane lipid composition. TMVII displays low hydrophobicity due to two Asp residues that are normally salt bridged to neighboring TMs in the crystal structure. The low hydrophobicity of TMVII allows thermodynamically stable solvent exposure in PE-lacking cells. Increasing the hydrophobicity of TMVII by electrostatic neutralization of the Asp240 to an Ile within this hinge prevented TMVII from being released into the periplasm in PE-deficient cells and simultaneously blocked the inversion of the N-terminal bundle of the protein (Bogdanov et al., 2008). Finally, TMVII inserts back into the membrane upon reorganization of LacY after synthesis of PE (Fig. 8.2). Retention of aberrant orientation of TMI after introduction of PE was possible by a secondary hinge region where TMII assumed a mini-loop organization, which does not span the membrane bilayer, most likely facilitated by its high Gly content and a “kinked” structure (Abramson et al., 2004). Therefore, TM switching appears to rely on the intrinsic structural flexibility provided by TMVII as a mobile molecular hinge, which is necessary and sufficient for TM rearrangement in response to changes in lipid environment.
The inverted topology of the N-terminal two-TM hairpin of phenylalanine permease of E. coli once established in PE-lacking cells can also be changed in a reversible manner in response to alterations in PE levels (Zhang et al., 2003). An abnormally long TMIII of phenylalanine permease, which forms a “U”-shaped mini-loop in PE lacking cells, appears to provide the molecular hinge in this case. Therefore, TMs on either side of a flexible hinge region can organize independently of each other in response to lipid environment, whereas those proteins without such a hinge region (LacY with a mutation in TMVII) either cannot assume different topologies or cannot fold and are degraded. These results clearly demonstrated that the lipid composition is a determinant of TM orientation and challenges the dogma that once TM orientation is established during assembly it is static and not subject to change.
The results also lead to an interesting general conclusion about the dynamic structure of polytopic membrane proteins. Several other membrane proteins have highly flexible domains containing apparent hinge regions, which allow TM movement associated either with their biogenesis or function (Kanki et al., 2002; Lu et al., 2000; Moss et al., 1998; Zhang, 2001). The human P-glycoprotein is localized to mammalian cytoplasmic membranes and is an ATP-binding cassette transporter responsible for multidrug resistance. Like LacY (Abramson et al., 2004; Guan and Kaback, 2006) it is a highly flexible protein that undergoes large conformational changes during its catalytic cycle (Zhang, 2001) or biogenesis (Moss et al., 1998). In its native host, the protein exhibits twelve TMs with both the N- and C-terminus exposed to the cytoplasm. When expressed in E. coli, the N-terminal half of the protein assumes the same topology as in the native host. However, TM7 no longer spans the membrane and TMs 8-12 assume an inverted orientation. Therefore the whole C-terminal half, which includes the nucleotide-binding domain, is misoriented within bacterial membranes (Linton and Higgins, 2002).
8.3.4 Lipid-Protein Interactions that Determine Topology
What features of the lipid bilayer and what features of the amino sequence of integral membrane proteins determine orientation in the membrane? The initial topological decision appears to be made by the translocon which provides the permissive environment required for concurrent membrane insertion of TMs and orientation of flanking regions to the extramembrane space according to the positive-inside and/or charge difference rule (von Heijne, 1989). The fate of TMs after clearance of the translocon must follow thermodynamically driven routes involving direct interaction of the TMs and associated extramembrane domains with the surrounding lipid bilayer (Hessa et al., 2005; White and von Heijne, 2005). TMs passively partition into the bilayer based on their affinity for the hydrophobic lipid core of the membrane while flanking aromatic and charged residues position themselves near and within the aqueous-membrane interface, respectively (McKenzie et al., 2006; White and von Heijne, 2005). Therefore, early on TM interactions and folding events are impacted by the properties of the surrounding lipids, which further decode the topogenic signals within the nascent chain sequences to influence TM orientation and final folding events.
The final TM topology of polytopic membrane proteins is dictated primarily by the encoded amino acid sequence. Charged residues flanking the hydrophobic TMs are major determinants of the gross topology of polytopic membrane proteins and can in most cases be described by the statistically derived and experimentally confirmed positive inside rule (von Heijne, 1986; von Heijne, 1989), which states that loops retained on the cytoplasmic side of the membrane are enriched in positively charged residues compared to loops translocated across the membrane. However, it is not clear how positively charged residues exert their effect on topology, why they are retained in the cytosol, and what cellular factors govern their topological disposition. Although the positive inside rule discounts the importance of negatively charged residues, negatively charged residues can be topologically active if they are present in high numbers (Nilsson and von Heijne, 1990), flank a marginally hydrophobic TM (Delgado-Partin and Dalbey, 1998) or lie within a window of six residues from the end of a highly hydrophobic TM (Rutz et al., 1999). Several negative residues are required to translocate a cytoplasmic domain with even a single positive residue (Nilsson and von Heijne, 1990). Since the orientation of a membrane protein can be reversed either by the addition or removal of a single positively charged residue (Gafvelin and von Heijne, 1994) or by introduction of negatively charged residues near the ends of TMs (Rutz et al., 1999), the relative topological power of the charged residues needs further clarification. Recent studies on lipid-dependent topological organization of LacY strongly indicate that lipid-protein charge interactions modulate the topological signal potential of charged residues in extramembrane domains of proteins.
A distinguishing feature for the three permeases thus far established to be topologically responsive to membrane lipid composition is the presence of both conserved positively and negatively residues within cytoplasmic domains that are sensitive to lipid composition suggesting that protein-lipid interactions may be a determinant of final topology. The conservation of negatively charged residues within the N-terminal TM helical bundle of LacY across the sugar permease family and the low level of acidic residues within the cytoplasmic loops of the C-terminal five-TM helical bundle of LacY was postulated to be critical for these negative residues in lipid-dependent topogenesis (Bogdanov et al., 2008). The cytoplasmic extramembrane domains of LacY strictly follow the positive inside rule. It was shown previously that extramembrane domains containing only positively charged residues were more stabilized facing the cytoplasm as the anionic phospholipid content (PG and CL) was increase in vivo (van Klompenburg et al., 1997), which raised questions of how the presence of acidic residues in the cytoplasmic domains of the N-terminal bundle of LacY would be responsible for topological inversion in cells lacking PE and containing only anionic phospholipids. Remarkably, conversion of any one of the six acidic residues distributed among domains C2, C4, and C6 (see Fig. 8.2) to a neutral amino acid (increase of net charge by plus one) prevented topological inversion of the whole N-terminal bundle in PE-lacking cells (Bogdanov et al., 2008). However, in order to induce topological inversion in normal PE-containing cells, net charge had to be changed from plus two to minus two in all three cytoplasmic domains of the N-terminal bundle, i.e. a change from net plus six to net minus six for the extramembrane surface of this large domain. In all cases increasing the hydrophobicity of TMVII prevented topological inversion.
These results provide several new insights into how final topology of a polytopic membrane protein is determined and the factors that make protein domains sensitive to membrane lipid composition. Protein-lipid charge interactions must be considered as an important topological determinant particularly for extramembrane domains containing a mixture of negatively and positively charged residues. For such domains the net positive charge of the protein domains or the negative charge density of the membrane surface effects TM topological orientation in a complementary manner. Final topology is determined by cooperative short-range and long-range protein-lipid and protein-protein interactions that occur well after the nascent polypeptide exits the translocon. The N-terminal bundle behaves as a single TM unit in response to topogenic signals within the protein and to its environment. Increasing the positive charge within either the C4 or C6 domain or the hydrophobicity of TMVII affects the topology of all upstream sequences with the former changes being dependent on protein-lipid interactions. These interactions occur during late folding events most likely independent of interactions between the protein and the translocon. Therefore, candidate proteins that may be subject to lipid dependent topological changes would be those containing TM bundles with cytoplasmic domains containing a mixture of negatively and positively charged amino acids connected by a flexible hinge region (an abnormally hydrophilic TM or an unusually long TM) to TM domains attached to primarily positively charged cytoplasmic domains.
The fact that the topology of fully assembled LacY can be changed by a change in lipid environment establishes that protein topology remains dynamic both during and after assembly. Although such lipid induced topological changes are unlikely in E. coli, this proof of principle observation has important implication for membrane proteins in eukaryotic cells. The lipid composition is significantly different between the plasma membrane and internal organelles. Thus during intracellular protein trafficking a membrane protein is exposed to different lipid environments that could affect topological organization post membrane insertion to either activate a latent activity or inactivate a protein. Similarly, local changes in lipid composition could result in large topological changes affecting function and stability. As shown in the LacY model system, single amino acid changes can result in vastly different topological responses to the lipid environment, which could be the molecular basis for some membrane protein related pathologies that may be due to mutations resulting in changes in lipid-dependent topogenic signals.
8.3.5 Lipids and Topological Disorders
Experimental findings in prokaryotes and eukaryotes suggest a conserved mechanism for establishing TM orientation within membrane proteins. TM segments of polytopic membrane proteins once membrane-inserted are generally considered stably oriented due to the assumed large free energy barrier to topological reorientation of adjacent extramembrane domains. Therefore, a topological “mistake” such as a segment being trapped on the “wrong” side of the membrane might be difficult to correct (Sanders and Myers, 2004). Moreover, TM domain insertion into the ER membrane proceeds simultaneously with glycosylation of extramembrane domains, which provides accuracy of topogenesis and contributes to topological stability by trapping a domain on one side of the membrane. Furthermore, in many cases topological mistakes made in the ER result in misfolded proteins that would be subject to rapid degradation by the proteosome (Sanders and Myers, 2004). Therefore, the general assumption is that TM topology is not easily perturbed by single point mutations and topological errors should be rare in disease-related misfolding (Sanders and Myers, 2004).
However, proteins in mammalian cells manage to escape from quality control in the ER to adopt alternative or dual topology in different intracellular membrane compartments. There are an increasing number of examples of proteins that are expressed in different topological forms with different functions. For example, ductin was found in two different orientations in cellular membranes, one of which serves as the subunit of the vacuolar H+-ATPase and the other serves as a component of the microsomal connexin channel of gap junctions (Dunlop et al., 1995). Several members of the cytochrome P450 superfamily and NADPH cytochrome P450 reductase are also expressed on both the cell surface and in the ER membrane in different topological orientations (Levy, 1996; Zhu et al., 1999). Several other membrane proteins are expressed in more than one topological form within one membrane type, such as the prion protein (Hegde et al., 1998) that can result in neurodenegeration and P-glycoprotein (Moss et al., 1998) that can result in failure of cells to pump drugs out. Several viral envelope proteins adopt at least two different topological forms with respect to the membrane with only one isoform involved in cellhost fusion (Lambert and Prange, 2001; McGinnes et al., 2003).
The plasma membrane ATP-dependent P-glycoprotein is a broad spectrum multidrug antiporter of the plasma membrane, and its expression in many cancer cell lines causes multidrug resistance, which may be responsible for the failure of cancer chemotherapy. Discrepancy exists between two experimentally determined orientations of the P-glycoprotein depending on the mammalian system used to express and probe orientation; as noted earlier, the orientation of the protein expressed in E. coli is also different than in its native host. Different topologies were observed in cell-free protein synthesis system supplemented with ER membranes from dog pancreas (Skach et al., 1993; Zhang and Ling, 1991), Xenopus oocytes (Skach et al., 1993), Chinese hamster ovary cells (Zhang, 1996), and in E. coli (Beja and Bibi, 1995; Linton and Higgins, 2002) and human cell line (Loo and Clarke, 1995) expression systems. The major difference between these topologies is the membrane sidedness loop of C8 (between TM8 and TM9). TM8 in the C-terminal half does not contain an efficient stop-transfer signal (Zhang, 1996) and loop C8 linking TM 8 and TM9 contains a hidden glycosylation site (Zhang and Ling, 1991), which if glycosylated would stabilize topology in this region of the protein. The cytoplasmic location of the loop C8 was supported by both epitope mapping and cysteine scanning assays using human cell lines (Georges et al., 1993; Loo and Clarke, 1995) while extracellular location was demonstrated in dog pancreatic ER microsomes (Sahin-Toth et al., 1996; Skach et al., 1993) and further confirmed by limited proteolysis in Chinese hamster ovary cells (Zhang, 1996). The protein adopts a mixed topology with respect to this domain in Xenopus oocytes (Moss et al., 1998) also determined by a protease susceptibility assay. The glycosylation site within domain C8 is normally either sterically hidden by the membrane associated translation machinery (Zhang and Ling, 1991) or temporarily shielded by cytoplasmic protein factors (Zhang et al., 1995). Failure to glycosylate the protein in the ER could make the topology of this region of the protein unstable and sensitive to differences in membrane environment along the normal secretory pathway. Therefore, mutations that eliminate a functional glycosylation sites or introduce a new topogenic signals could result in an alternate topology for a protein at its final location in the cell.
In contrast to conformational disorders, experimental evidence of lipid effects on topology of protein membranes is lacking. However the existence of variations in lipid composition between different intracellular compartments may have relevance to membrane protein misorientation. Is topological fixed during co-translationally membrane insertion in the ER or does further remodeling of topology occur with changes in lipid composition as proteins move through different organelles to their final destination? The topogenesis of polytopic membrane proteins specifically routed to membrane domains having distinctive lipid compositions, such as lipid rafts, may be also evolutionarily “tailored” to the lipid composition of these domains (Sanders and Myers, 2004). Mis-targeting of proteins to such domains or lack of proper topogenic signals for raft proteins could be pathologically significant. Therefore, changes in membrane lipid composition either locally or during intracellular movement of proteins along the organelle-based secretory pathway can be potentially added to ligand binding (Ikeda et al., 2005), substrate binding (Gouffi et al., 2004) and membrane depolarization (Jakes et al., 1998) as modes for inducing such changes.
It is also generally assumed that alteration of protein topology requires extensive sequence changes (Sanders and Myers, 2004). Indeed most mutations result in subtle local conformational changes (Milenkovic et al., 2007). However, data are accumulating that even a single amino acid substitution within a membrane-flanking domain can profoundly alter TM topology that in turn would affect proper trafficking and processing due to aberrant localization of the targeting motif of the precursor protein. While a single mutation of either one of the N-terminal Lys or Arg residues to Ala in the lung-specific surfactant protein C precursor produced mixed orientation, double mutation resulted in complete reversal of orientation, thereby directing the targeting motif to the lumen of the ER instead of the cytosol (Mulugeta and Beers, 2003). Amazingly while the double mutant was retained in the ER, single mutants produced a mixed pattern of both ER (double mutant-like) and vesicular (wild type-like) expression, demonstrating that proper trafficking and processing of lung-specific surfactant protein C requires cytosolic localization of the targeting motif of the precursor protein. This study provides a likely precedent for a mechanism in disorders associated with misorientations of integral membrane proteins. For the growing number of lung associated pathological disorders associated with protein mutations in the membrane-flanking region, topological alteration should be considered as one significant contributing factor.
Hereditary spherocytosis is a common human inherited hemolytic anemia caused by mutations in erythrocyte anion exchanger type 1 known also as Band 3 protein. The substitution of the highly conserved Met663 to positively charged Lys located in the extracellular boundary of TM8 was recently described in a patient with the disease (Lima et al., 2005). This novel Band 3 Tambau mutant is retained in a pre-medial Golgi compartment likely due misorientation.
The vitelliform muscular dystrophy type 2 (VMD2) gene mutated in Best muscular dystrophy encodes a four TM protein termed bestrophin-1. The vast majority of known disease-associated alterations cluster near or within predicted TMs. Three out of eighteen extramembrane-associated mutations resulted in severe effects in ER membrane insertion. Four out of twelve TM-associated mutations showed an altered glycosylation pattern demonstrating that substitution of hydrophobic residues by positively charged amino acids exerts severe effects on TM properties and therefore membrane protein topology. These facts suggest that defective membrane integration or misorientation of bestrophin-1 may represent a potential disease mechanism for a subset of Best muscular dystrophy-related mutations (Milenkovic et al., 2007).
The three N-terminal TM helices of the Glu/Asp transporter are encoded by exons 2, 3 and 4, respectively. The loss of exon 3 results in the three instead of two TMs and inverts the whole topology of the protein (Huggett et al., 2000). Moreover, this splice variant encodes a functional transporter with inverted orientation within the plasma membrane. In some neurological disorders the release of glutamate due to anoxia was found to be largely due to an inverse operation of the transporter.
Pro-apoptotic proteins Bax (recruited from cytoplasm upon induction of apoptosis) and Bak have been reported to form a supermolecular pore in the outer mitochondrial membrane, which is large enough to release of cytochrome c and other proteins to the cytoplasm to initiate the apoptotic cascade (Kim et al., 2004). Upon induction of apoptosis Bax translocates and inserts into the outer mitochondrial membrane such that α-helices 5, 6 and 9 insert into the bilayer (Annis et al., 2005). The molecular mechanism by which anti-apoptotic Bcl-2 antagonizes the action of the proapoptotic proteins is still not completely understood. However unlike Bax, Bcl-2 constitutively resides in membranes as a monotopic protein anchored to the membrane via helix 9. This form of Bcl-2 is most likely inactive in preventing Bax oligomerization. However, during apoptosis preexisting, membrane-bound Bcl-2 appears to change membrane topology from a tail-anchored to multispanning form (Kim et al., 2004) in which cytoplasmic helices 5 and 6 become TMs. Bcl-2 most likely prevents productive oligomerization of membrane-bound Bax only when they are both multispanning transmembrane proteins (Dlugosz et al., 2006). Topologically changed Bcl-2 continues to inhibit apoptosis until the concentration of membrane-embedded Bax exceeds that of Bcl-2 (Dlugosz et al., 2006; Leber et al., 2007) allowing excess Bax to form a pore. Therefore, Bcl-2 membrane topology is not fixed during or immediately after biosynthesis and upon induction of apoptosis undergoes a major TM rearrangement.
8.4 Lipids in Organization of Protein Complexes
8.4.1 Lipids as Integral Components of Protein Complexes
In addition to providing the amphipathic bilayer matrix within which membrane proteins reside, phospholipids are also specifically integrated between and within the subunits of oligomeric protein complexes. Phospholipids are structurally and functionally important components in the energy-transducing multimeric complexes of the bacterial cytoplasmic membrane and the inner mitochondrial membrane. For instances CL (Fig. 8.1) is essential for optimum activity of inner mitochondrial membrane proteins including NADH dehydrogenase, the cytochrome bc1 complex, ATP synthase, cytochrome c oxidase, and the ATP/ADP translocase (for reviews and references see (Mileykovskaya et al., 2005; Schlame et al., 2000)). CL is also specifically integrated into the structure of E. coli succinate dehydrogenase and formate dehydrogenase-N (Jormakka et al., 2002; Yankovskaya et al., 2003).
The Saccharomyces cerevisiae ubiquinol:cytochrome c oxidoreductase or cytochrome bc1 complex (Complex III), which is highly homologous to the mammalian complex, is a component of the inner mitochondrial membrane. The b subunit, which is encoded by mtDNA, and the c1 and Rieske iron-sulfur protein subunits, which are encoded by nuclear DNA, make up the catalytic core of the complex. An additional 7 non-identical and non-catalytic nuclear encoded subunits, three heme groups, and two quinones make up the remainder of the complex (see (Hunte et al., 2008) for review). Complex III exists as a dimer in which fourteen phospholipid molecules have been identified in the 2.3-Å-crystal structure (Fig. 8.3A) (Hunte, 2005). These specifically localized phospholipid molecules are four CL, two PI, six PE, and two PC molecules. Six of these phospholipids localize within the oligomeric structure of the complex. Two PE molecules (one per monomer as seen in the front center of Fig. 8.3A) are at the interface between the two monomers making contact with the b subunits of both monomers. The two PE molecules are near two of the CL molecules (one per monomer as seen in the front center of Fig. 8.3A). Each PI molecule is intercalated between the three catalytic subunits of each monomer. PI acyl chains engulf the transmembrane helix of the Rieske subunit near the point of movement of its extrinsic domain and may dissipate torsion forces during the catalytic cycle of the complex. The remaining phospholipids lie at the surface of the complex as immobilized annular lipids in direct contact with the TMs of Complex III. CL and PE on the right front side and left rear side of Fig. 8.3A are annular lipids. These surfaces containing CL and PE form a cavity as shown in Fig. 8.3B that was proposed to interface with Complex IV in the formation of a supercomplex as described later. Proton uptake sites associated with quinone reduction lie near this CL binding site. Several mutations in this site either reduce electron transfer activity or reduce stability of associated subunits (Hunte, 2005; Lange et al., 2001; Palsdottir and Hunte, 2004).
Fig. 8.3.
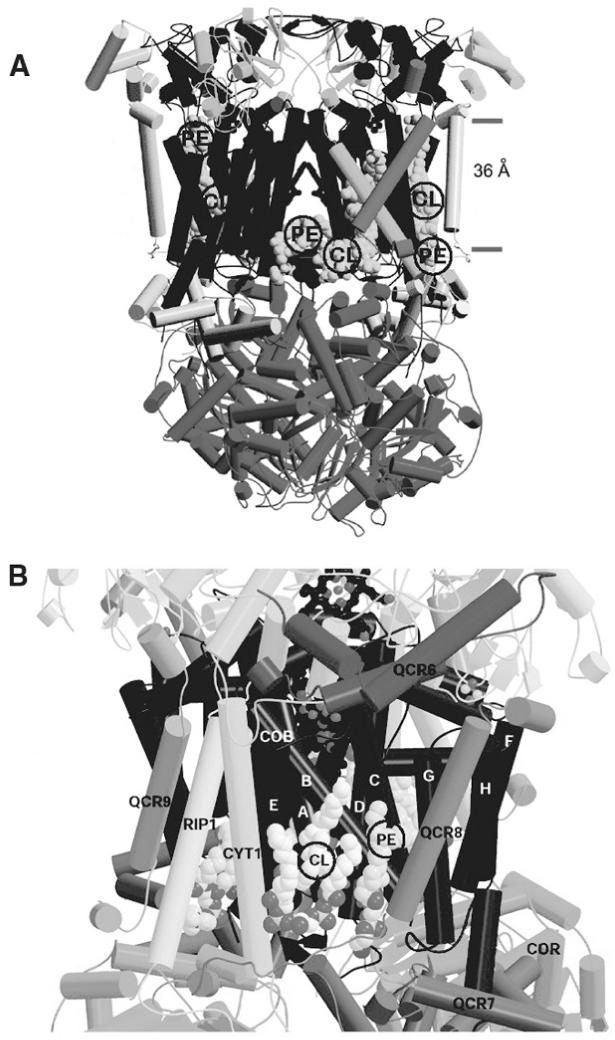
Crystal structure of Complex III dimer and model of putative Complex III surface that interacts with Complex IV from S. cerevisiae. (A) Dimer of Complex III based on the crystal structure (figure adapted from (Hunte, 2005)) with the interface between monomers in the center. α-Helices of different subunits within the dimer are shown as rods of different shading connected by non-helical domains. Note the positions of CL and PE (all circled) in the center front (also on back center but not shown) between the dimers and on the front (right) and back (left) sides of the diagram. The two bars on the right show the 36-Å width of the membrane. (B) The putative interface of Complex III monomer (figure adapted from (Pfeiffer et al., 2003)) with Complex IV with the various subunit domains labeled. This view, with a cavity containing PE and CL (both circled), corresponds to the right and left sides of the view shown in (A) and is positioned within the membrane bilayer. Bottom of both diagrams faces the mitochondrial matrix
The crystal structure of bovine cytochrome c oxidase (Complex IV) homo-dimer has been determined to a resolution of 1.8 Å(Shinzawa-Itoh et al., 2007). This integral membrane protein complex composed of thirteen different subunits per monomer is responsible for the reduction of molecular oxygen to water during aerobic respiration, with concomitant proton pumping across the mitochondrial inner membrane. A combination of high resolution X-ray structure analysis of the integral lipids in bovine Complex IV with mass spectroscopy analysis of their chain lengths and the positions of the unsaturated bonds of the hydrophobic tails provides understanding of structural and functional roles played by these lipids in the Complex IV (Shinzawa-Itoh et al., 2007). Thirteen lipids, including two CLs, one PC, three PEs, four PGs and three triacylglycerols were resolved in Complex IV. One CL, two PEs and one PG participate in the stabilization of the Complex IV dimer. However, the contacts made by PE and PG molecules between monomers are much weaker than those of CL, which interacts with subunits III and VIa within one monomer and bridges to the other monomer at subunits I and II. The four acyl chains of CL interact through van der Waals contacts with hydrophobic amino acid residues belonging to both monomers, and the two phosphate groups interact with both monomers via hydrogen bonds. Thus the dimer state of the bovine Complex IV is primarily stabilized by CL and subunit VIa. The bovine heart and liver Complex IV isozymes differ in that the N-terminal domains of their respective subunits VIa are not the same. The phosphorylated Thr11 residue of bovine heart Complex IV, which is proposed to stabilize the conformation of the N-terminal domain, is missing in liver isozyme. Therefore, the stability of the dimeric form of Complex IV may be different between the two isozymes and in vivo dimerization might respond differently to metabolic signals (Schmidt et al., 1997; Taanman and Capaldi, 1993). A third CL (not resolved in crystal structure of bovine heart cytochrome c oxidase) was found by photolabeling experiments with arylazido-containing CL analogues (Sedlak et al., 2006). This CL is located between subunits VIIa and VIIc near the entrance to the putative proton pumping channel, which contains a conserved aspartate. The authors suggested that this CL molecule could potentially function as a proton antenna to facilitate proton entry into the channel thus explaining the CL requirement for full enzymatic activity.
The high content of PG in Complex IV is remarkable in light of the low level (less than 1%) of this phospholipid in the inner membrane of mitochondria and its apparent absence from other mitochondrial complexes. Analysis of the X-ray structure of Complex IV revealed palmitate as one of the acyl chains of the two PG molecules of each dimer. These acyl chains lie near the putative O2 transfer pathway in subunit III of each monomer of the Complex IV dimer. The other acyl chain of these PG molecules is vaccenate (cis-Δ11-octadecenoate), which appears to play a critical role in the specific binding of PG to this site. Only vaccenate-containing PG is found in bovine Complex IV in spite of the abundance of oleate (cis-Δ9-octadecenoate) in mitochondrial phospholipids. The X-ray structure demonstrates that only cis-vaccenate derivatives of PG will fit into the hydrophobic grooves of subunit III near the O2 transfer pathway. All these data together suggest a unique role for PG bound to subunit III in the O2 transfer process (Shinzawa-Itoh et al., 2007).
Cytochrome c oxidase is a highly conserved enzyme; the three core subunits (I, II and III) encoded by the mitochondrial genome in eukaryotes has high amino acid sequence homology with prokaryotic and eukaryotic species. Detailed analysis of evolutionary conservation of lipid-binding sites in cytochrome c oxidase (Qin et al., 2007) shows that lipid binding sites are specific and selective for both headgroups and alkyl tails. Strikingly, the overlay of bovine and bacterial crystal structures shows identical positions containing fatty acid chains of phospholipid or triacylglycerol in both enzymes. The headgroups of lipids interact with positively charged (Arg, Lys or His), aromatic (Trp or Tyr), or other polar residues (Thr, Ser, Gln or Asn) of cytochrome c oxidase at the membrane interface. The hydrophobic tails of these lipids are inserted into shallow grooves on the protein surface and are stabilized by precise fits through van der Waals contacts with hydrophobic residues (Qin et al., 2007). A similar analysis of the Complex III dimer crystal structure shows very similar protein lipid interactions (Palsdottir and Hunte, 2004). Therefore, these protein-lipid associations are not the result of random or weak interactions but are extensive over whole lipid molecules and represent an integral part of the overall structure of these complexes.
8.4.2 Pathological Effects of Reduced CL Levels
Therefore, any pathological state in which the above lipids, and in particular CL and PG, are significantly reduced (ischemia, hypothyroidism, aging, and heart failure; see (Chicco and Sparagna, 2007) for detailed review) would be expected to affect electron transfer efficiency and mitochondrial energy production. Mitochondrial dysfunction and diseases associated with inhibition of catalytic activity due to the loss of CL have been described for both Complexes III and IV. Decrease in Complex III activity coupled with a decrease in the content of CL was demonstrated for mitochondria isolated from rat heart subjected to ischemia and reperfusion presumably due to the oxidative damage of specifically heart mitochondrial CL. Remarkably, Complex III activity of mitochondria was restored to pre-ischemia levels after fusion of the mitochondria with CL-containing liposomes, while PC-, PE-, and oxidized CL-containing liposomes failed to restore the activity. It was suggested that the loss of Complex III activity results from oxidation of the high content of tetralinoleate-containing CL in heart mitochondria by oxygen free radicals produced upon reperfusion after ischemia (Petrosillo et al., 2003, 2005).
A similar connection between a decrease in CL levels and the age-linked decline of rat heart mitochondrial cytochrome c oxidase activity was demonstrated. Treatment of heart mitochondria from aged rats with CL-liposomes restored their lower cytochrome c oxidase activity to the level of young control rats (Paradies et al., 1997b). Again, no restoration of activity was found after treatment with other phospholipids or with peroxidized CL. CL level in heart mitochondria was shown to be regulated by thyroid hormone (Mutter et al., 2000). A decrease in cytochrome c oxidase activity in heart mitochondria isolated from hypothyroid rats can be also completely restored to the level of control rats by exogenously added CL but not by other phospholipids (Paradies et al., 1997a)
Decreased Complex I activity in fatty liver mitochondria isolated from rats fed with a choline-deficient diet to model in animals nonalcoholic fatty liver disease could also be completely restored to the level of control livers by exogenously added CL (Petrosillo et al., 2007). Under conditions of a choline-deficient diet the mitochondrial content of CL decreased due to reactive oxygen species-induced CL oxidation. Although no high-resolution crystal structure of the entire Complex I is available, these findings strongly suggest the presence of functionally important CL molecules in the complex.
8.4.3 Lipid Involvement in Supercomplex Formation
Kinetic and structural analysis of the mammalian mitochondrial respiratory chain suggests that the individual Complexes I-IV (NADH:ubiquinone oxidoreductase, succinate:ubiquinone oxidoreductase, bc1 complex (ubiqunol:cytochrome c oxidoreductase), and cytochrome c oxidase, respectively) exist in mitochondria under physiological conditions in equilibrium with supercomplexes or “respirasomes” composed of all of the above individual complexes (see (Mileykovskaya et al., 2005) for references). Depending on metabolic conditions electron transfer in the respiratory chain would occur (1) via substrate channeling of the small carriers, (ubiquinone, between Complexes I (or II) and III or cytochrome c between Complexes III and IV) due to supercomplex formation or (2) via random diffusion of these small carriers between individual complexes independently imbedded in the lipid bilayer (Genova et al., 2005). In S. cerevisiae mitochondria this equilibrium appears to be shifted to supercomplex organization of Complexes III and IV (Mileykovskaya et al., 2005), which may also contain Complex II as well as two peripheral NADH dehydrogenases (Boumans et al., 1998); S. cerevisiae lack Complex I and utilize the peripheral NADH dehydrogenases. F1F0-ATP synthase (Complex V) uses the electrochemical proton gradient generated in respiration to produce ATP.
The 3D structure for a respiratory supercomplex I1III2IV1 (Fig. 8.4) from bovine heart mitochondria (Schafer et al., 2007) and a 2D model of S. cerevisiae supercomplex IV1III2IV1 (Heinemeyer et al., 2007) have been recently proposed based on projection maps generated from single particle analysis of negatively stained samples visualized by electron microscopy. In both structures Complex III exists as a dimer, and Complex IV exists as a monomer. This is in agreement with numerous results demonstrating that the Complex III dimer represents one structural and functional unit (Hunte et al., 2008; Xia et al., 2007). In S. cerevisiae two Complex IV monomers are bound to opposite ends of the dimer of Complex III. In the bovine supercomplex the Complex III dimer interacts with Complex I and Complex IV monomers. The Complex IV face interacting with Complex III in the bovine supercomplex is the face forming the dimer with itself in the X-ray structure (Schafer et al., 2007). In contrast, in the 2D model of the S. cerevisiae supercomplex, an analogous side of the Complex IV was proposed to be oriented away from Complex III (Heinemeyer et al., 2007). It was suggested that in mammals the dimerization of Complex IV under different metabolic conditions might play a functional role, competing with supercomplex formation using the same contact interface (Schafer et al., 2007). In both structures the putative binding site for a mobile carrier (for ubiquinone or cytochrome c in bovine or only cytochrome c in S. cerevisiae) of each individual complex is in close proximity with the corresponding binding site of the neighboring complex (Fig. 8.4) supporting a substrate channeling mechanism in the supercomplex.
Fig. 8.4.
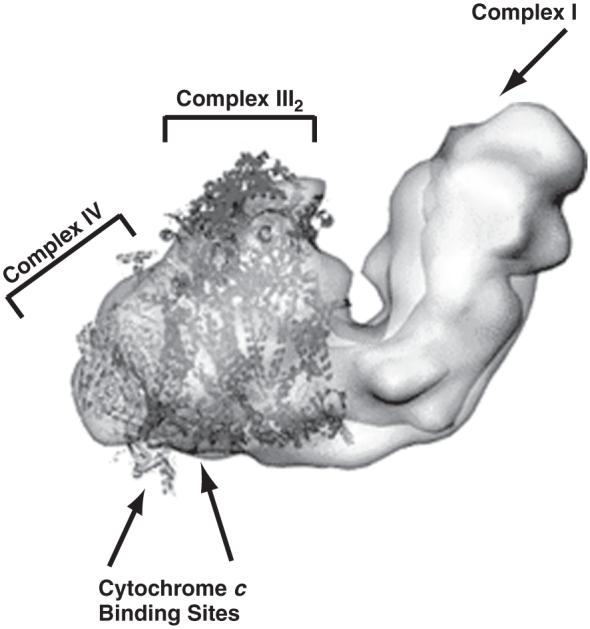
3D structure of bovine heart supercomplex I 1III2IV1. The globular 3D structure of the supercomplex is overlayed with the crystal structures of the Complex III dimer and Complex IV monomer as indicated. The face of Complex III that interacts with Complex IV is shown in Fig. 8.3B. The close proximity of the cytochrome c binding sites on Complexes III and IV is indicated. The organization of the supercomplex and cytochrome c interaction shown in Fig. 8.5 is based on this structure. Figure adapted from (Schafer et al., 2007)
Phospholipids, in particular CL, play an important role in the association of these individual complexes into a functional supercomplex. For S. cerevisiae Complexes III and IV behave kinetically in intact mitochondria or display on gel electrophoresis after extraction from mitochondria with mild detergents as an associated supercomplex. However, in mutants of S. cerevisiae lacking CL (null mutants in the CRDL gene) but containing highly elevated amounts of PG (a precursor to CL), these complexes behave kinetically (Zhang et al., 2005a) and move during gel electrophoresis (Zhang et al., 2002) as individual complexes. Importantly, use of a mutant strain in which CL content of yeast mitochondria could be exogenously regulated in vivo (Zhang et al., 2002) demonstrated a direct correlation between levels of CL and the ratio of supercomplex to individual complexes in the mutant membrane. CL-lacking mutant cells, which in glucose media (anaerobic growth conditions) did not exhibit any alterations in their phenotype, grew considerably slower and to a lower final density on non-fermentable carbon sources (require oxidative phosphorylation) than wild type cells. Cells with intermediate CL content displayed intermediate growth phenotypes demonstrating the direct dependence of the efficiency of the energetic system on the level of CL (Zhang et al., 2002).
Kinetic experiments for the first time provided evidences for the role of CL in the organization of respiratory chain supercomplexes in intact mitochondria not disrupted with detergent. The classical kinetic approach used was based on the assumption that if the diffusion rate of a mobile electron carrier (in this case cytochrome c) is faster than the rates of its reduction and oxidation during steady-state respiration, the mobile carrier behaves kinetically as a homogeneous pool (Boumans et al., 1998). In this case the overall respiration rate should show a hyperbolic relation to the rate of either the reduction or oxidation of the carrier (pool behavior) indicative of a mobile carrier freely diffusing between individual respiratory complexes (i.e., III and IV). However, the relation approaches linearity (non-pool behavior) as an increasing amount of the mobile carrier is restricted due to substrate channeling between associated respiratory complexes organization into a supercomplex (IV1III2IV1). A putative pool behavior of cytochrome c was studied by titration of the reduction rate of cytochrome c with the Complex III-specific inhibitor antimycin A (Boumans et al., 1998). In wild type yeast mitochondria containing normal levels of CL, cytochrome c did not show pool behavior consistent with the respiratory chain functioning as one unit in supercomplex organization with substrate channeling of cytochrome c between complexes III and IV (Boumans et al., 1998; Zhang et al., 2005a). However, in the case of the CL-lacking mutant cytochrome c exhibited pool behavior (Zhang et al., 2005a) consistent with the absence of respiratory chain supercomplex organization as previously shown in disrupted mitochondria (Zhang et al., 2002). These results further demonstrate an important role for CL in organization of not only the individual respiratory complexes but in higher order organization of these complexes into a “respirasome”.
High concentrations of detergents at low ionic strength easily dissociate yeast Complexes III and IV from the supercomplex (Schagger and Pfeiffer, 2000) suggesting their association is through hydrophobic interactions. A cavity in the Complex III formed by membrane-embedded TM helices of cytochrome c1 and cytochrome b with a lid on top of this cavity formed by subunits Qcr8 and Qcr6p has been suggested as a possible site of the interaction between Complexes III and IV (see Fig. 8.3B for the S. cerevisiae Complex III). CL together with PE fills this cavity and was proposed to act as a flexible amphipathic linkage between the above complexes (Pfeiffer et al., 2003). The structures of the supercomplexes from both bovine and S. cerevisiae are consistent with and support this lipid filled cavity of Complex III as the interface with Complex IV.
8.4.4 Disruption of Higher Complex Formation in Barth Syndrome and Apoptosis
The higher order organization of electron transfer complexes in multicellular organisms into respirasomes dependent on CL is supported by studies of mitochondria from patients with Barth syndrome. The human TAZ gene encoding Tafazzin, a phospholipid acyltranferase that is involved in remodeling of CL to its mature highly unsaturated fatty acid composition, provides structural uniformity within the CL pool (Xu et al., 2006). Mutations in the TAZ gene are associated with Barth syndrome, an X-linked genetic disorder characterized by cardiomyopathy, skeletal myopathy, neutropenia, and growth retardation (see (Hauff and Hatch, 2006; Schlame and Ren, 2006; Xu et al., 2006) for reviews). Mitochondria from Barth syndrome patients show a lower CL content, but more striking is the polydispersity in the normally very narrow acyl chain composition of CL. Lymphoblasts from patients with Barth syndrome show decreased stability of the I1III2IV1 supercomplex, where Complex IV is more dissociated from the supercomplex, suggesting that reduction in the mature species of CL results in unstable respiratory chain supercomplexes (McKenzie et al., 2006). The interaction between Complexes I and III was also affected resulting in decreased levels of the I1III2 supercomplex (McKenzie et al., 2006). Interestingly, S. cerevisiae contain an orthologue of the TAZ gene and disruption of the TAZ1 gene in S. cerevisiae affects the assembly and stability of Complex IV as well as its incorporation into the supercomplex (Brandner et al., 2005; Li et al., 2007).
The above abnormalities in molecular organization may be the molecular basis for or the result of abnormal architecture of mitochondrial cristae, which was observed using EM tomography in lymphoblast cells isolated from patients with Barth syndrome (Acehan et al., 2007). Although the authors pointed out that the absence of Tafazzin itself may cause a disruption of normal membrane adhesion due to localization of Tafazzin both in the inner and outer membrane surfaces encasing the inter-membrane space (Claypool et al., 2006), a direct relationship between cristae abnormality and the deficiency in CL was suggested. The inner membrane of mitochondria is organized in two morphologically distinct domains, the inner boundary membrane and the cristae membrane (Fig. 8.5A), which are connected by narrow tubular structures termed cristae junctions. The tomograms showed adhesion of two neighboring cristae membranes resulting in the disruption of the normal connection to the mitochondrial inner boundary membrane by the cristae junction structure, obliteration of the intra-cristae space, and stacking of the collapsed cristae. Cristae membranes appear to be enriched in Complexes I-V over the inner boundary membrane. However, there is normally dynamic redistribution of proteins between the inner boundary membrane and cristae membrane, which changes in accordance with changes in the physiological state of the cell (Vogel et al., 2006). Structural abnormalities in Barth syndrome mitochondria appear to block this dynamic. One possible explanation for the structural changes is that the abnormal spectrum of CL species may induce abnormalities in protein organization and aggregation. Large mitochondria with onion-like cristae structures were also present in lymphoblast cells isolated from patients with Barth syndrome. Similar onion-like organization of the inner membrane was demonstrated in S. cerevisiae mutants lacking small subunits of ATP synthase responsible for dimerization and oligomerization of Complex V (Paumard et al., 2002). Based on a decrease in membrane potential coupled with reduced oxidation of substrates by the respiratory chain in the mutant, disruption of microdomains organized both from the oligomers of Complex V and the respiratory chain supercomplexes was suggested (Bornhovd et al., 2006). Thus the change in the content of the mature species of CL may result in mis-assembly of respiratory chain supercomplexes, which in turn would result in distortion of microdomains formed by supercomplexes and changes in mitochondrial cristae morphology producing dysfunctional mitochondria.
Fig. 8.5.

Model for involvement of CL in the energy coupling processes or apoptosis in mitochondria. (A) Under normal conditions cristae membranes (CM) are separated from the remaining mitochondrial inner boundary membranes (IM) by the cristae junctions. CM are composed of microdomains enriched in CL (depicted in the bilayer as dimer of gray headgroups) and Complexes I, III, IV and V (F1F0ATP synthase) organized in I1III2IV1 (see Fig. 2.4) and V2 supercomplexes. The proton gradient (positive outward) generated by electron transfer from NADH to molecular oxygen via the supercomplex I1III2IV1 (with channeling of cytochrome c (Cytc) between Complex III and IV) is utilized to synthesize ATP. CL participates in the trapping of protons promoting localized protonic coupling between the respiratory supercomplex and Complex V located in close proximity to each other (figure adapted from (Papa et al., 2006)). (B) During early stages of apoptosis cristae junctions open allowing more equal distribution of CL between CM and IM with significant appearance of CL in the outer membrane (OM) via the contacts sites between the latter two membranes. Reduction of CL in the CM favors delocalization of the proton gradient and partial dissociation of Complex IV from the supercomplex. Binding of cytochrome c to CL disrupts electron flow between Complexes III and IV and induces increased production of reactive oxygen species by Complex III. Interaction of cytochrome c with CL converts the protein into a peroxidase, which catalyzes CL peroxidation (CLox), which further reduces CL levels (Basova et al., 2007)
CL also plays a direct structural and functional role in the regulation of the early steps of the apoptosis, which is dependent on the submitochondrial localization of CL. In the cristae membrane CL participates in organization of the respiratory chain complexes into supercomplexes as described above, which together with Complex V represent the main components of the cristae (Fig. 8.5A). Complex V was suggested to form oligomeric supercomplexes that are required for cristae formation (Dudkina et al., 2006; Giraud et al., 2002) and to create a scaffold to also keep respiratory supercomplexes in the cristae. Such an organization could trap supercomplexes within the cristae membrane microdomains and prevent them from diffusing to the inner boundary membrane via the cristae junctions thereby regulating dynamic redistribution of the components between sub-compartments of the inner membrane (Vogel et al., 2006). CL has been proposed to act as a proton sink at the membrane surface due to the abnormally high ionization constant (above 7.5) for one of its two phosphate groups (Kates et al., 1993). The ability of CL, enriched in the cristae, to trap protons and prevent escape of pumped protons to the bulk water phase would increase protonic coupling between respiratory supercomplexes and Complex V in cristae membrane microdomains (Haines and Dencher, 2002). Prevention of a delocalized proton electrochemical gradient would inhibit oxygen superoxide formation by the respiratory chain and the generation of other free radicals. In higher eukaryotes this would suppress induction of the mitochondrial pathway of apoptosis (Papa et al., 2006). During apoptosis CL levels increase in the outer mitochondrial membrane (Fig. 8.5B), which was proposed to be caused by redistribution of CL between the inner and outer leaflets of the inner membrane followed by movement of CL to the outer membrane via membrane contact sites between the two membranes (see (Lucken-Ardjomande and Martinou, 2005; Ott et al., 2007) for reviews). However, if compartmentalization of the inner membrane is taken into account, the redistribution of CL between cristae membranes and inner boundary membranes, due to disruption of cristae junctions during apoptosis (see below), might also play a critical role during induction of apoptosis. The reduction in cristae membrane CL might prevent the correct assembly of respiratory complexes and supercomplex formation (Fig. 8.5B).
CL can compete with Complex III for binding with cytochrome c in a dose dependent manner, and CL-bound cytochrome c does not effectively interact with respiratory supercomplexes (Basova et al., 2007). Thus, the increase in CL concentration in the outer leaflet of the inner boundary membrane would redistribute cytochrome c to the inner boundary membrane from the cristae making it unavailable for interaction with the supercomplexes (Fig. 8.5B). Interestingly, in mouse liver mitochondria under conditions where cells undergo apoptosis, cristae are remodeled and cristae junctions widen (Frezza et al., 2006; Scorrano et al., 2002). Cristae opening might increase the pool of cytochrome c available for binding with CL on the outer leaflet of the inner boundary membrane or the inner leaflet of the outer membrane. Interaction with CL causes structural changes in cytochrome c accompanied by a negative shift in its red-ox potential and conversion of the protein into a peroxidase, which catalyzes H2O2-dependent CL peroxidation (Basova et al., 2007). Reducing the amount of cytochrome c available as an electron acceptor from Complex III strongly promotes the production of superoxide by Complex III, which in turn would elevate H2O2 levels. CL peroxidation, which parallels the loss of CL during apoptosis, in turn might result in membrane detachment of cytochrome c and finally in its release into the cytosol through the permeabilized outer membrane (Ott et al., 2007) further activating downstream signals associated with apoptosis. Such a sequence of events may explain the paradox during progression of apoptosis where both an increase in outer membrane CL and reduction in total mitochondrial CL occur.
The exact mechanisms driving apoptotic redistribution of CL are still poorly understood although mitochondrial translocation and activity of tBid, a truncated form of the cytosolic protein Bid, seem to be associated with the apoptotic migration of CL (Lucken-Ardjomande and Martinou, 2005). In vitro experiments demonstrated that mitochondrial creatine kinase, which is localized to mitochondrial membrane contact sites, promotes segregation and clustering of CL in liposomes, suggesting that this phenomenon might occur at contact sites between the inner and outer mitochondrial membranes, creating nucleation sites for CL-domains capable of binding tBid and cytochrome c (Epand et al., 2007). Irrespective of the mechanisms leading to induction of mitochondrial-based apoptosis, it is clear that CL plays a central role in this process.
8.5 Concluding Remarks
Lipids have multiple functions in cells ranging from defining the bilayer permeability barrier of cell membranes and organelles to providing the matrix within which membrane proteins fold and function to being integral components of multisubunit protein complexes and supercomplexes. Diseases or physiological states that severely compromise membrane barrier function would be lethal which accounts for the lack of genetic disorders resulting in the lack of or extensive redistribution among major lipid classes. However, a large number of disorders directly involve lipids or most likely have a lipid involvement in the underlying molecular basis for the disease. Membrane proteins associated with some diseases express themselves as protein folding defects rather than defects in protein function and result in defects in trafficking and organelle targeting of proteins. Some membrane proteins may misfold in their proper location either spontaneously or in response to local changes in lipid composition. Mutation of structurally important residues could lead to a different organization of the mutant protein within the changing lipid environment as the protein moves through the same or altered organelle trafficking route. Studies on bacterial permeases clearly demonstrate that alterations in protein-lipid interactions can have dramatic effects during initial assembly and after stable assembly on the structure and function of membrane proteins. Therefore, lipid involvement in the underlying molecular mechanisms of protein folding disorders is certainly a distinct possibility.
Biochemical and genetic evidence has established a role for lipids as essential structural and functional components, equal to specific amino acid residues, in multicomponent molecular machines. In this regard CL is of particular clinical importance because of all major lipids CL levels are most dramatically affected by a number of pathological and physiological states. CL and changes in its levels are associated with diseases such as Barth syndrome and changes in cellular states brought about by ischemia/reperfusion, apoptosis, aging and oxidative damage. There are direct correlations between these changes in CL levels and the organization and function of mitochondrial respiratory components.
Therefore, defining the molecular basis for cellular dysfunction resulting from disrupted membrane associated processes must include a more extensive investigation of the role lipids play in these processes before a full understanding of normal function can be attained. Expansion of such basic information will uncover possible therapeutic treatments to modulate the seriousness of the genetic defects or pathophysiological states by affecting membrane lipid composition or distribution of individual lipid species thus opening new avenues for pharmacological intervention.
Acknowledgments
The authors thank Philip Heacock for assistance in preparation of the figures. Work of the authors presented in this review was supported by grants GM20478 and GM56389 from the US National Institutes of Health and funds from the John S. Dunn, Sr., Research Foundation awarded to W.D.
Abbreviations
- CL
cardiolipin
- CFTR
cystic fibrosis transmembrane conductance regulator
- Complex I
NADH:ubiquinone oxidoreductase
- Complex II
sucinate:ubiquinone oxidoreductase
- Complex III
cytochrome bc1 complex (ubiquinone:cytochrome c oxidoreductase)
- Complex IV
cytochrome c oxidase
- Complex V
F1F0-ATP synthase
- DGlcDG
diglucosyldiacylglycerol
- ER
endoplasmic reticulum
- GPI
glycosaminylated phosphatidylinositol
- LacY
lactose permease
- NKT
natural killer T
- NBD-1
first nucleotide binding domain
- MGlcDG
monoglucosyldiacylglycerol
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PI
phosphatidylinositol
- PS
phosphatidylserine
- TM
transmembrane domain
References
- Abramson J, Iwata S, Kaback HR. Lactose permease as a paradigm for membrane transport proteins (Review) Mol Membr Biol. 2004;21:227–236. doi: 10.1080/09687680410001716862. [DOI] [PubMed] [Google Scholar]
- Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest. 2007;87:40–48. doi: 10.1038/labinvest.3700480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, Aten J, Kuipers F, Serlie MJ, Wennekes T, Sethi JK, O’Rahilly S, Overkleeft HS. Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes. 2007;56:1341–1349. doi: 10.2337/db06-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemany R, Perona JS, Sanchez-Dominguez JM, Montero E, Canizares J, Bressani R, Escriba PV, Ruiz-Gutierrez V. G protein-coupled receptor systems and their lipid environment in health disorders during aging. Biochim Biophys Acta. 2007;1768:964–975. doi: 10.1016/j.bbamem.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Andersson K, Buschard K, Fredman P, Kaas A, Lidstrom AM, Madsbad S, Mortensen H, Jan-Eric M. Patients with insulin-dependent diabetes but not those with non-insulin-dependent diabetes have anti-sulfatide antibodies as determined with a new ELISA assay. Autoimmunity. 2002;35:463–468. doi: 10.1080/0891693021000047361. [DOI] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. Embo J. 2005;24:2096–2103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Hannan LA. Traffic jam: a compendium of human diseases that affect intracellular transport processes. Traffic. 2000;1:836–851. doi: 10.1034/j.1600-0854.2000.011104.x. [DOI] [PubMed] [Google Scholar]
- Basova LV, Kurnikov IV, Wang L, Ritov VB, Belikova NA, Vlasova II, Pacheco AA, Winnica DE, Peterson J, Bayir H, Waldeck DH, Kagan VE. Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry. 2007;46:3423–3434. doi: 10.1021/bi061854k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beja O, Bibi E. Multidrug resistance protein (Mdr)-alkaline phosphatase hybrids in Escherichia coli suggest a major revision in the topology of the C-terminal half of Mdr. J Biol Chem. 1995;270:12351–12354. doi: 10.1074/jbc.270.21.12351. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W. Phospholipid-assisted protein folding: phosphatidylethanolamine is required at a late step of the conformational maturation of the polytopic membrane protein lactose permease. Embo J. 1998;17:5255–5264. doi: 10.1093/emboj/17.18.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Dowhan W. Lipid-assisted protein folding. J Biol Chem. 1999;274:36827–36830. doi: 10.1074/jbc.274.52.36827. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Heacock PN, Dowhan W. A polytopic membrane protein displays a reversible topology dependent on membrane lipid composition. Embo J. 2002;21:2107–2116. doi: 10.1093/emboj/21.9.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Sun J, Kaback HR, Dowhan W. A phospholipid acts as a chaperone in assembly of a membrane transport protein. J Biol Chem. 1996;271:11615–11618. doi: 10.1074/jbc.271.20.11615. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Umeda M, Dowhan W. Phospholipid-assisted refolding of an integral membrane protein. Minimum structural features for phosphatidylethanolamine to act as a molecular chaperone. J Biol Chem. 1999;274:12339–12345. doi: 10.1074/jbc.274.18.12339. [DOI] [PubMed] [Google Scholar]
- Bogdanov M, Xie J, Heacock PN, Dowhan W. To flip or not to flip: protein-lipid charge interactions are a determinant of final membrane protein topology. J Cell Biol. 2008 doi: 10.1083/jcb.200803097. editorially accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M, Zhang W, Xie J, Dowhan W. Transmembrane protein topology mapping by the substituted cysteine accessibility method (SCAM(TM)): application to lipid-specific membrane protein topogenesis. Methods. 2005;36:148–171. doi: 10.1016/j.ymeth.2004.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhovd C, Vogel F, Neupert W, Reichert AS. Mitochondrial membrane potential is dependent on the oligomeric state of F1F0-ATP synthase supracomplexes. J Biol Chem. 2006;281:13990–13998. doi: 10.1074/jbc.M512334200. [DOI] [PubMed] [Google Scholar]
- Boumans H, Grivell LA, Berden JA. The respiratory chain in yeast behaves as a single functional unit. J Biol Chem. 1998;273:4872–4877. doi: 10.1074/jbc.273.9.4872. [DOI] [PubMed] [Google Scholar]
- Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol Biol Cell. 2005;16:5202–5214. doi: 10.1091/mbc.E05-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutkiewicz RR, Lin Y, Cho S, Hwang YK, Sriram V, Roberts TJ. CD1d-mediated antigen presentation to natural killer T (NKT) cells. Crit Rev Immunol. 2003;23:403–419. doi: 10.1615/critrevimmunol.v23.i56.30. [DOI] [PubMed] [Google Scholar]
- Buschard K, Blomqvist M, Osterbye T, Fredman P. Involvement of sulfatide in beta cells and type 1 and type 2 diabetes. Diabetologia. 2005;48:1957–1962. doi: 10.1007/s00125-005-1926-9. [DOI] [PubMed] [Google Scholar]
- Buschard K, Hanspers K, Fredman P, Reich EP. Treatment with sulfatide or its precursor, galactosylceramide, prevents diabetes in NOD mice. Autoimmunity. 2001;34:9–17. doi: 10.3109/08916930108994121. [DOI] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Chicco AJ, Sparagna GC. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol. 2007;292:C33–C44. doi: 10.1152/ajpcell.00243.2006. [DOI] [PubMed] [Google Scholar]
- Choo-Smith LP, Surewicz WK. The interaction between Alzheimer amyloid beta(1-40) peptide and ganglioside GM1-containing membranes. FEBS Lett. 1997;402:95–98. doi: 10.1016/s0014-5793(96)01504-9. [DOI] [PubMed] [Google Scholar]
- Claypool SM, McCaffery JM, Koehler CM. Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins. J Cell Biol. 2006;174:379–390. doi: 10.1083/jcb.200605043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe MJ. Cataract as a conformational disease - the Maillard reaction, alpha-crystallin and chemotherapy. Cell Mol Biol. 1998;44:1047–1050. [PubMed] [Google Scholar]
- De Silva AD, Park JJ, Matsuki N, Stanic AK, Brutkiewicz RR, Medof ME, Joyce S. Lipid protein interactions: the assembly of CD1d1 with cellular phospholipids occurs in the endoplasmic reticulum. J Immunol. 2002;168:723–733. doi: 10.4049/jimmunol.168.2.723. [DOI] [PubMed] [Google Scholar]
- Debnath D, Bhattacharya S, Chakrabarti A. Phospholipid assisted folding of a denatured heme protein: effect of phosphatidylethanolamine. Biochem Biophys Res Commun. 2003;301:979–984. doi: 10.1016/s0006-291x(03)00066-4. [DOI] [PubMed] [Google Scholar]
- Delgado-Partin VM, Dalbey RE. The proton motive force, acting on acidic residues, promotes translocation of amino-terminal domains of membrane proteins when the hydrophobicity of the translocation signal is low. J Biol Chem. 1998;273:9927–9934. doi: 10.1074/jbc.273.16.9927. [DOI] [PubMed] [Google Scholar]
- Dlugosz PJ, Billen LP, Annis MG, Zhu W, Zhang Z, Lin J, Leber B, Andrews DW. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 2006;25:2287–2296. doi: 10.1038/sj.emboj.7601126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol. 1998;8:189–194. doi: 10.1016/s0959-440x(98)80037-7. [DOI] [PubMed] [Google Scholar]
- Dowhan W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu Rev Biochem. 1997;66:99–232. doi: 10.1146/annurev.biochem.66.1.199. [DOI] [PubMed] [Google Scholar]
- Dowhan W, Bogdanov M, Mileykovskaya E. Functional roles of lipids in membranes. In: Vance DE, Vance JE, editors. Biochemistry of Lipids. Lipoproteins and Membranes. 5th Edition Elsevier Press; Amsterdam: 2008. pp. 1–37. [Google Scholar]
- Dowhan W, Mileykovskaya E, Bogdanov M. Diversity and versatility of lipid-protein interactions revealed by molecular genetic approaches. Biochim Biophys Acta. 2004;1666:19–39. doi: 10.1016/j.bbamem.2004.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews J. What’s in a number? Nat Rev Drug Discov. 2006;5:975. doi: 10.1038/nrd2205. [DOI] [PubMed] [Google Scholar]
- Dudkina NV, Sunderhaus S, Braun HP, Boekema EJ. Characterization of dimeric ATP synthase and cristae membrane ultrastructure from Saccharomyces and Polytomella mitochondria. FEBS Lett. 2006;580:3427–3432. doi: 10.1016/j.febslet.2006.04.097. [DOI] [PubMed] [Google Scholar]
- Dunlop J, Jones PC, Finbow ME. Membrane insertion and assembly of ductin: a polytopic channel with dual orientations. Embo J. 1995;14:3609–3616. doi: 10.1002/j.1460-2075.1995.tb00030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eidelman O, BarNoy S, Razin M, Zhang J, McPhie P, Lee G, Huang Z, Sorscher EJ, Pollard HB. Role for phospholipid interactions in the trafficking defect of Delta F508-CFTR. Biochemistry. 2002;41:11161–11170. doi: 10.1021/bi020289s. [DOI] [PubMed] [Google Scholar]
- Ellis RJ. Do molecular chaperones have to be proteins? Biochem Biophys Res Commun. 1997;238:687–692. doi: 10.1006/bbrc.1997.7339. [DOI] [PubMed] [Google Scholar]
- Elofsson A, von Heijne G. Membrane protein structure: prediction versus reality. Annu Rev Biochem. 2007;76:125–140. doi: 10.1146/annurev.biochem.76.052705.163539. [DOI] [PubMed] [Google Scholar]
- Epand RF, Tokarska-Schlattner M, Schlattner U, Wallimann T, Epand RM. Cardiolipin clusters and membrane domain formation induced by mitochondrial proteins. J Mol Biol. 2007;365:968–980. doi: 10.1016/j.jmb.2006.10.028. [DOI] [PubMed] [Google Scholar]
- Fadiel A, Eichenbaum KD, Hamza A, Tan O, Lee HH, Naftolin F. Modern pathology: protein mis-folding and mis-processing in complex disease. Curr Protein Pept Sci. 2007;8:29–37. doi: 10.2174/138920307779941532. [DOI] [PubMed] [Google Scholar]
- Fredman P, Mansson JE, Rynmark BM, Josefsen K, Ekblond A, Halldner L, Osterbye T, Horn T, Buschard K. The glycosphingolipid sulfatide in the islets of Langerhans in rat pancreas is processed through recycling: possible involvement in insulin trafficking. Glycobiology. 2000;10:39–50. doi: 10.1093/glycob/10.1.39. [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Gafvelin G, von Heijne G. Topological “frustration” in multispanning E. coli inner membrane proteins. Cell. 1994;77:401–412. doi: 10.1016/0092-8674(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Gelman MS, Kopito RR. Cystic fibrosis: premature degradation of mutant proteins as a molecular disease mechanism. Methods Mol Biol. 2003;232:27–37. doi: 10.1385/1-59259-394-1:27. [DOI] [PubMed] [Google Scholar]
- Genova ML, Bianchi C, Lenaz G. Supercomplex organization of the mitochondrial respiratory chain and the role of the Coenzyme Q pool: pathophysiological implications. Biofactors. 2005;25:5–20. doi: 10.1002/biof.5520250103. [DOI] [PubMed] [Google Scholar]
- Georges E, Tsuruo T, Ling V. Topology of P-glycoprotein as determined by epitope mapping of MRK-16 monoclonal antibody. J Biol Chem. 1993;268:1792–1798. [PubMed] [Google Scholar]
- Giraud MF, Paumard P, Soubannier V, Vaillier J, Arselin G, Salin B, Schaeffer J, Brethes D, di Rago JP, Velours J. Is there a relationship between the supramolecular organization of the mitochondrial ATP synthase and the formation of cristae? Biochim Biophys Acta. 2002;1555:174–180. doi: 10.1016/s0005-2728(02)00274-8. [DOI] [PubMed] [Google Scholar]
- Gouffi K, Gerard F, Santini CL, Wu LF. Dual topology of the Escherichia coli TatA protein. J Biol Chem. 2004;279:11608–11615. doi: 10.1074/jbc.M313187200. [DOI] [PubMed] [Google Scholar]
- Guan L, Kaback HR. Lessons from lactose permease. Annu Rev Biophys Biomol Struct. 2006;35:67–91. doi: 10.1146/annurev.biophys.35.040405.102005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumperz JE. The ins and outs of CD1 molecules: bringing lipids under immunological surveillance. Traffic. 2006;7:2–13. doi: 10.1111/j.1600-0854.2005.00364.x. [DOI] [PubMed] [Google Scholar]
- Haines TH, Dencher NA. Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett. 2002;528:35–39. doi: 10.1016/s0014-5793(02)03292-1. [DOI] [PubMed] [Google Scholar]
- Han X, Abendschein DR, Kelley JG, Gross RW. Diabetes-induced changes in specific lipid molecular species in rat myocardium. Biochem J. 2000;352:79–89. [PMC free article] [PubMed] [Google Scholar]
- Harrison RS, Sharpe PC, Singh Y, Fairlie DP. Amyloid peptides and proteins in review. Rev Physiol Biochem Pharmacol. 2007;159:1–77. doi: 10.1007/112_2007_0701. [DOI] [PubMed] [Google Scholar]
- Hauff KD, Hatch GM. Cardiolipin metabolism and Barth Syndrome. Prog Lipid Res. 2006;45:91–101. doi: 10.1016/j.plipres.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Hayden MR, Tyagi SC, Kerklo MM, Nicolls MR. Type 2 diabetes mellitus as a conformational disease. JOP. 2005;6:287–302. [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- Heinemeyer J, Braun HP, Boekema EJ, Kouril R. A structural model of the cytochrome C reductase/oxidase supercomplex from yeast mitochondria. J Biol Chem. 2007;282:12240–12248. doi: 10.1074/jbc.M610545200. [DOI] [PubMed] [Google Scholar]
- Hessa T, White SH, von Heijne G. Membrane insertion of a potassium-channel voltage sensor. Science. 2005;307:1427. doi: 10.1126/science.1109176. [DOI] [PubMed] [Google Scholar]
- Huggett J, Vaughan-Thomas A, Mason D. The open reading frame of the Na(+)- dependent glutamate transporter GLAST-1 is expressed in bone and a splice variant of this molecule is expressed in bone and brain. FEBS Lett. 2000;485:13–18. doi: 10.1016/s0014-5793(00)02175-x. [DOI] [PubMed] [Google Scholar]
- Hunte C. Specific protein-lipid interactions in membrane proteins. Biochem Soc Trans. 2005;33:938–942. doi: 10.1042/BST20050938. [DOI] [PubMed] [Google Scholar]
- Hunte C, Solmaz S, Palsdottir H, Wenz T. A Structural Perspective on Mechanism and Function of the Cytochrome bc (1) Complex. Results Probl Cell Differ. 2008;45:253–278. doi: 10.1007/400_2007_042. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Kida Y, Ikushiro S, Sakaguchi M. Manipulation of membrane protein topology on the endoplasmic reticulum by a specific ligand in living cells. J. Biochem. 2005;138:631–637. doi: 10.1093/jb/mvi157. [DOI] [PubMed] [Google Scholar]
- Jakes KS, Kienker PK, Slatin SL, Finkelstein A. Translocation of inserted foreign epitopes by a channel-forming protein. Proc Natl Acad Sci USA. 1998;95:4321–4326. doi: 10.1073/pnas.95.8.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jormakka M, Tornroth S, Byrne B, Iwata S. Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science. 2002;295:1863–1868. doi: 10.1126/science.1068186. [DOI] [PubMed] [Google Scholar]
- Joyce S. CD1d and natural T cells: how their properties jump-start the immune system. Cell Mol Life Sci. 2001;58:442–469. doi: 10.1007/PL00000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce S, Woods AS, Yewdell JW, Bennink JR, De Silva AD, Boesteanu A, Balk SP, Cotter RJ, Brutkiewicz RR. Natural ligand of mouse CD1d1: cellular glycosylpho-sphatidylinositol. Science. 1998;279:1541–1544. doi: 10.1126/science.279.5356.1541. [DOI] [PubMed] [Google Scholar]
- Kabayama K, Sato T, Kitamura F, Uemura S, Kang BW, Igarashi Y, Inokuchi J. TNFalpha-induced insulin resistance in adipocytes as a membrane microdomain disorder: involvement of ganglioside GM3. Glycobiology. 2005;15:21–29. doi: 10.1093/glycob/cwh135. [DOI] [PubMed] [Google Scholar]
- Kabayama K, Sato T, Saito K, Loberto N, Prinetti A, Sonnino S, Kinjo M, Igarashi Y, Inokuchi J. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc Natl Acad Sci USA. 2007;104:13678–13683. doi: 10.1073/pnas.0703650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Sakaguchi M, Kitamura A, Sato T, Mihara K, Hamasaki N. The tenth membrane region of band 3 is initially exposed to the luminal side of the endoplasmic reticulum and then integrated into a partially folded band 3 intermediate. Biochemistry. 2002;41:13973–13981. doi: 10.1021/bi026619q. [DOI] [PubMed] [Google Scholar]
- Kates M, Syz JY, Gosser D, Haines TH. pH-dissociation characteristics of cardiolipin and its 2′-deoxy analogue. Lipids. 1993;28:877–882. doi: 10.1007/BF02537494. [DOI] [PubMed] [Google Scholar]
- Kim PK, Annis MG, Dlugosz PJ, Leber B, Andrews DW. During apoptosis bcl-2 changes membrane topology at both the endoplasmic reticulum and mitochondria. Mol Cell. 2004;14:523–529. doi: 10.1016/s1097-2765(04)00263-1. [DOI] [PubMed] [Google Scholar]
- Koch M, Stronge VS, Shepherd D, Gadola SD, Mathew B, Ritter G, Fersht AR, Besra GS, Schmidt RR, Jones EY, Cerundolo V. The crystal structure of human CD1d with and without alpha-galactosylceramide. Nat Immunol. 2005;6:819–826. doi: 10.1038/ni1225. [DOI] [PubMed] [Google Scholar]
- Lambert C, Prange R. Dual topology of the hepatitis B virus large envelope protein: determinants influencing post-translational pre-S translocation. J Biol Chem. 2001;276:22265–22272. doi: 10.1074/jbc.M100956200. [DOI] [PubMed] [Google Scholar]
- Lange C, Nett JH, Trumpower BL, Hunte C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. Embo J. 2001;20:6591–6600. doi: 10.1093/emboj/20.23.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber B, Lin J, Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D. Membrane proteins which exhibit multiple topological orientations. Essays Biochem. 1996;31:49–60. [PubMed] [Google Scholar]
- Li G, Chen S, Thompson MN, Greenberg ML. New insights into the regulation of cardiolipin biosynthesis in yeast: implications for Barth syndrome. Biochim Biophys Acta. 2007;1771:432–441. doi: 10.1016/j.bbalip.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Lima PR, Baratti MO, Chiattone ML, Costa FF, Saad ST. Band 3Tambau: a de novo mutation in the AE1 gene associated with hereditary spherocytosis. Implications for anion exchange and insertion into the red blood cell membrane. Eur J Haematol. 2005;74:396–401. doi: 10.1111/j.1600-0609.2004.00405.x. [DOI] [PubMed] [Google Scholar]
- Lin JC, Liu HL. Protein conformational diseases: from mechanisms to drug designs. Curr Drug Discov Technol. 2006;3:145–153. doi: 10.2174/157016306778108866. [DOI] [PubMed] [Google Scholar]
- Linton KJ, Higgins CF. P-glycoprotein misfolds in Escherichia coli: evidence against alternating-topology models of the transport cycle. Mol Membr Biol. 2002;19:51–58. doi: 10.1080/09687680110103622. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. Membrane topology of a cysteine-less mutant of human P-glycoprotein. J Biol Chem. 1995;270:843–848. doi: 10.1074/jbc.270.2.843. [DOI] [PubMed] [Google Scholar]
- Lu Y, Turnbull IR, Bragin A, Carveth K, Verkman AS, Skach WR. Reorientation of aquaporin-1 topology during maturation in the endoplasmic reticulum. Mol Biol Cell. 2000;11:2973–2985. doi: 10.1091/mbc.11.9.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Martinou JC. Newcomers in the process of mitochondrial permeabilization. J Cell Sci. 2005;118:473–483. doi: 10.1242/jcs.01654. [DOI] [PubMed] [Google Scholar]
- McGinnes LW, Reitter JN, Gravel K, Morrison TG. Evidence for mixed membrane topology of the newcastle disease virus fusion protein. J Virol. 2003;77:1951–1963. doi: 10.1128/JVI.77.3.1951-1963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie M, Lazarou M, Thorburn DR, Ryan MT. Mitochondrial respiratory chain supercomplexes are destabilized in Barth Syndrome patients. J Mol Biol. 2006;361:462–469. doi: 10.1016/j.jmb.2006.06.057. [DOI] [PubMed] [Google Scholar]
- Mendoza JL, Thomas PJ. Building an understanding of cystic fibrosis on the foundation of ABC transporter structures. J Bioenerg Biomembr. 2007;39:499–505. doi: 10.1007/s10863-007-9117-7. [DOI] [PubMed] [Google Scholar]
- Milenkovic VM, Rivera A, Horling F, Weber BH. Insertion and topology of normal and mutant bestrophin-1 in the endoplasmic reticulum membrane. J Biol Chem. 2007;282:1313–1321. doi: 10.1074/jbc.M607383200. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E, Zhang M, Dowhan W. Cardiolipin in energy transducing membranes. Biochemistry (Mosc) 2005;70:154–158. doi: 10.1007/s10541-005-0095-2. [DOI] [PubMed] [Google Scholar]
- Molano A, Park SH, Chiu YH, Nosseir S, Bendelac A, Tsuji M. Cutting edge: the IgG response to the circumsporozoite protein is MHC class II-dependent and CD1d-independent: exploring the role of GPIs in NK T cell activation and antimalarial responses. J Immunol. 2000;164:5005–5009. doi: 10.4049/jimmunol.164.10.5005. [DOI] [PubMed] [Google Scholar]
- Monaco S, Zanusso G, Mazzucco S, Rizzuto N. Cerebral amyloidoses: molecular pathways and therapeutic challenges. Curr Med Chem. 2006;13:1903–1913. doi: 10.2174/092986706777585022. [DOI] [PubMed] [Google Scholar]
- Morillas M, Swietnicki W, Gambetti P, Surewicz WK. Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem. 1999;274:36859–36865. doi: 10.1074/jbc.274.52.36859. [DOI] [PubMed] [Google Scholar]
- Moss K, Helm A, Lu Y, Bragin A, Skach WR. Coupled translocation events generate topological heterogeneity at the endoplasmic reticulum membrane. Mol Biol Cell. 1998;9:2681–2697. doi: 10.1091/mbc.9.9.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugeta S, Beers MF. Processing of surfactant protein C requires a type II trans-membrane topology directed by juxtamembrane positively charged residues. J Biol Chem. 2003;278:47979–47986. doi: 10.1074/jbc.M308210200. [DOI] [PubMed] [Google Scholar]
- Mutter T, Dolinsky VW, Ma BJ, Taylor WA, Hatch GM. Thyroxine regulation of monolysocardiolipin acyltransferase activity in rat heart. Biochem J. 2000;346(Pt 2):403–406. [PMC free article] [PubMed] [Google Scholar]
- Nagamori S, Vazquez-Ibar JL, Weinglass AB, Kaback HR. In vitro synthesis of lactose permease to probe the mechanism of membrane insertion and folding. J Biol Chem. 2003;278:14820–14826. doi: 10.1074/jbc.M300332200. [DOI] [PubMed] [Google Scholar]
- Nilsson I, von Heijne G. Fine-tuning the topology of a polytopic membrane protein: role of positively and negatively charged amino acids. Cell. 1990;62:1135–1141. doi: 10.1016/0092-8674(90)90390-z. [DOI] [PubMed] [Google Scholar]
- Osterbye T, Jorgensen KH, Fredman P, Tranum-Jensen J, Kaas A, Brange J, Whittingham JL, Buschard K. Sulfatide promotes the folding of proinsulin, preserves insulin crystals, and mediates its monomerization. Glycobiology. 2001;11:473–479. doi: 10.1093/glycob/11.6.473. [DOI] [PubMed] [Google Scholar]
- Ott M, Zhivotovsky B, Orrenius S. Role of cardiolipin in cytochrome c release from mitochondria. Cell Death Differ. 2007;14:1243–1247. doi: 10.1038/sj.cdd.4402135. [DOI] [PubMed] [Google Scholar]
- Palsdottir H, Hunte C. Lipids in membrane protein structures. Biochim Biophys Acta. 2004;1666:2–18. doi: 10.1016/j.bbamem.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Papa S, Lorusso M, Di Paola M. Cooperativity and flexibility of the protonmotive activity of mitochondrial respiratory chain. Biochim Biophys Acta. 2006;1757:428–436. doi: 10.1016/j.bbabio.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Ruggiero FM. Cardiolipin-dependent decrease of cytochrome c oxidase activity in heart mitochondria from hypothyroid rats. Biochim Biophys Acta. 1997a;1319:5–8. doi: 10.1016/s0005-2728(97)00012-1. [DOI] [PubMed] [Google Scholar]
- Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997b;406:136–138. doi: 10.1016/s0014-5793(97)00264-0. [DOI] [PubMed] [Google Scholar]
- Parekh VV, Wilson MT, Van Kaer L. iNKT-cell responses to glycolipids. Crit Rev Immunol. 2005;25:183–213. doi: 10.1615/critrevimmunol.v25.i3.20. [DOI] [PubMed] [Google Scholar]
- Park JJ, Kang SJ, De Silva AD, Stanic AK, Casorati G, Hachey DL, Cresswell P, Joyce S. Lipid-protein interactions: biosynthetic assembly of CD1 with lipids in the endoplasmic reticulum is evolutionarily conserved. Proc Natl Acad Sci USA. 2004;101:1022–1026. doi: 10.1073/pnas.0307847100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brethes D, di Rago JP, Velours J. The ATP synthase is involved in generating mitochondrial cristae morphology. Embo J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosillo G, Di Venosa N, Ruggiero FM, Pistolese M, D’Agostino D, Tiravanti E, Fiore T, Paradies G. Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim Biophys Acta. 2005;1710:78–86. doi: 10.1016/j.bbabio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, Paradies G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta. 2007;1767:1260–1267. doi: 10.1016/j.bbabio.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. Faseb J. 2003;17:714–716. doi: 10.1096/fj.02-0729fje. [DOI] [PubMed] [Google Scholar]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schagger H. Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem. 2003;278:52873–52880. doi: 10.1074/jbc.M308366200. [DOI] [PubMed] [Google Scholar]
- Porcelli SA. Cutting glycolipids down to size. Nat Immunol. 2001;2:191–192. doi: 10.1038/85234. [DOI] [PubMed] [Google Scholar]
- Qin L, Sharpe MA, Garavito RM, Ferguson-Miller S. Conserved lipid-binding sites in membrane proteins: a focus on cytochrome c oxidase. Curr Opin Struct Biol. 2007;17:444–450. doi: 10.1016/j.sbi.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu BH, Strickland EH, Thomas PJ. Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J Biol Chem. 1997;272:15739–15744. doi: 10.1074/jbc.272.25.15739. [DOI] [PubMed] [Google Scholar]
- Roberts TJ, Sriram V, Spence PM, Gui M, Hayakawa K, Bacik I, Bennink JR, Yewdell JW, Brutkiewicz RR. Recycling CD1d1 molecules present endogenous antigens processed in an endocytic compartment to NKT cells. J Immunol. 2002;168:5409–5414. doi: 10.4049/jimmunol.168.11.5409. [DOI] [PubMed] [Google Scholar]
- Rutz C, Rosenthal W, Schulein R. A single negatively charged residue affects the orientation of a membrane protein in the inner membrane of Escherichia coli only when it is located adjacent to a transmembrane domain. J Biol Chem. 1999;274:33757–33763. doi: 10.1074/jbc.274.47.33757. [DOI] [PubMed] [Google Scholar]
- Sahin-Toth M, Kaback HR, Friedlander M. Association between the amino- and carboxyl-terminal halves of lactose permease is specific and mediated by multiple trans-membrane domains. Biochemistry. 1996;35:2016–2021. doi: 10.1021/bi952496g. [DOI] [PubMed] [Google Scholar]
- Sambamurti K, Suram A, Venugopal C, Prakasam A, Zhou Y, Lahiri DK, Greig NH. A partial failure of membrane protein turnover may cause Alzheimer’s disease: a new hypothesis. Curr Alzheimer Res. 2006;3:81–90. doi: 10.2174/156720506775697142. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Myers JK. Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct. 2004;33:25–51. doi: 10.1146/annurev.biophys.33.110502.140348. [DOI] [PubMed] [Google Scholar]
- Sanghera N, Pinheiro TJ. Binding of prion protein to lipid membranes and implications for prion conversion. J Mol Biol. 2002;315:1241–1256. doi: 10.1006/jmbi.2001.5322. [DOI] [PubMed] [Google Scholar]
- Schafer E, Dencher NA, Vonck J, Parcej DN. Three-dimensional structure of the respiratory chain supercomplex I1III2IV1 from bovine heart mitochondria. Biochemistry. 2007;46:12579–12585. doi: 10.1021/bi700983h. [DOI] [PubMed] [Google Scholar]
- Schagger H, Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. Embo J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Lett. 2006;580:5450–5455. doi: 10.1016/j.febslet.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Schlame M, Rua D, Greenberg ML. The biosynthesis and functional role of cardiolipin. Prog Lipid Res. 2000;39:257–288. doi: 10.1016/s0163-7827(00)00005-9. [DOI] [PubMed] [Google Scholar]
- Schleiff E, Tien R, Salomon M, Soll J. Lipid composition of outer leaflet of chloroplast outer envelope determines topology of OEP7. Molecular Biology of the Cell. 2001;12:4090–4102. doi: 10.1091/mbc.12.12.4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer K, Fahy E, Subramaniam S, Dennis EA. The lipid maps initiative in lipidomics. Methods Enzymol. 2007;432:171–183. doi: 10.1016/S0076-6879(07)32007-7. [DOI] [PubMed] [Google Scholar]
- Schmidt TR, Jaradat SA, Goodman M, Lomax MI, Grossman LI. Molecular evolution of cytochrome c oxidase: rate variation among subunit VIa isoforms. Mol Biol Evol. 1997;14:595–601. doi: 10.1093/oxfordjournals.molbev.a025798. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- Sedlak E, Panda M, Dale MP, Weintraub ST, Robinson NC. Photolabeling of cardiolipin binding subunits within bovine heart cytochrome c oxidase. Biochemistry. 2006;45:746–754. doi: 10.1021/bi050870z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. Embo J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skach WR, Calayag MC, Lingappa VR. Evidence for an alternate model of human P-glycoprotein structure and biogenesis. J Biol Chem. 1993;268:6903–6908. [PubMed] [Google Scholar]
- Sun J, Wu J, Carrasco N, Kaback HR. Identification of the epitope for monoclonal antibody 4B1 which uncouples lactose and proton translocation in the lactose permease of Escherichia coli. Biochemistry. 1996;35:990–998. doi: 10.1021/bi952166w. [DOI] [PubMed] [Google Scholar]
- Taanman JW, Capaldi RA. Subunit VIa of yeast cytochrome c oxidase is not necessary for assembly of the enzyme complex but modulates the enzyme activity. Isolation and characterization of the nuclear-coded gene. J Biol Chem. 1993;268:18754–18761. [PubMed] [Google Scholar]
- van Klompenburg W, Nilsson I, von Heijne G, de Kruijff B. Anionic phospholipids are determinants of membrane protein topology. Embo J. 1997;16:4261–4266. doi: 10.1093/emboj/16.14.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G, Leeflang BR, Liebisch G, Schmitz G, Goni FM. The European lipidomics initiative: enabling technologies. Methods Enzymol. 2007;432:213–232. doi: 10.1016/S0076-6879(07)32009-0. [DOI] [PubMed] [Google Scholar]
- Vigh L, Escriba PV, Sonnleitner A, Sonnleitner M, Piotto S, Maresca B, Horvath I, Harwood JL. The significance of lipid composition for membrane activity: new concepts and ways of assessing function. Prog Lipid Res. 2005;44:303–344. doi: 10.1016/j.plipres.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Vogel F, Bornhovd C, Neupert W, Reichert AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol. 2006;175:237–247. doi: 10.1083/jcb.200605138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. Embo J. 1986;5:3021–3027. doi: 10.1002/j.1460-2075.1986.tb04601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature. 1989;341:456–458. doi: 10.1038/341456a0. [DOI] [PubMed] [Google Scholar]
- Wang X, Bogdanov M, Dowhan W. Topology of polytopic membrane protein subdomains is dictated by membrane phospholipid composition. Embo J. 2002;21:5673–5681. doi: 10.1093/emboj/cdf571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White SH, von Heijne G. Do protein-lipid interactions determine the recognition of transmembrane helices at the ER translocon? Biochem Soc Trans. 2005;33:1012–1015. doi: 10.1042/BST20051012. [DOI] [PubMed] [Google Scholar]
- Xia D, Esser L, Yu L, Yu CA. Structural basis for the mechanism of electron bifurcation at the quinol oxidation site of the cytochrome bc1 complex. Photosynth Res. 2007;92:17–34. doi: 10.1007/s11120-007-9155-3. [DOI] [PubMed] [Google Scholar]
- Xie J, Bogdanov M, Heacock P, Dowhan W. Phosphatidylethanolamine and monoglucosyldiacylglycerol are interchangeable in supporting topogenesis and function of the polytopic membrane protein lactose permease. J Biol Chem. 2006;281:19172–19178. doi: 10.1074/jbc.M602565200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006;281:39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K. GM1 ganglioside and the seeding of amyloid in Alzheimer’s disease: endogenous seed for Alzheimer amyloid. Neuroscientist. 2005;11:250–260. doi: 10.1177/1073858405275177. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer’s disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
- Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- Zhang JT. Sequence requirements for membrane assembly of polytopic membrane proteins: molecular dissection of the membrane insertion process and topogenesis of the human MDR3 P-glycoprotein. Mol Biol Cell. 1996;7:1709–1721. doi: 10.1091/mbc.7.11.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JT. The multi-structural feature of the multidrug resistance gene product P-glycoprotein: implications for its mechanism of action (hypothesis) Mol Membr Biol. 2001;18:145–152. doi: 10.1080/09687680110048831. [DOI] [PubMed] [Google Scholar]
- Zhang JT, Lee CH, Duthie M, Ling V. Topological determinants of internal transmembrane segments in P-glycoprotein sequences. J Biol Chem. 1995;270:1742–1746. doi: 10.1074/jbc.270.4.1742. [DOI] [PubMed] [Google Scholar]
- Zhang JT, Ling V. Study of membrane orientation and glycosylated extracellular loops of mouse P-glycoprotein by in vitro translation. J Biol Chem. 1991;266:18224–18232. [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W. Gluing the respiratory chain together. Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J Biol Chem. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- Zhang M, Mileykovskaya E, Dowhan W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J Biol Chem. 2005a;280:29403–29408. doi: 10.1074/jbc.M504955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Bogdanov M, Pi J, Pittard AJ, Dowhan W. Reversible topological organization within a polytopic membrane protein is governed by a change in membrane phospholipid composition. J Biol Chem. 2003;278:50128–50135. doi: 10.1074/jbc.M309840200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Campbell HA, King SC, Dowhan W. Phospholipids as determinants of membrane protein topology. Phosphatidylethanolamine is required for the proper topological organization of the gamma-aminobutyric acid permease (GabP) of Escherichia coli. J Biol Chem. 2005b;280:26032–26038. doi: 10.1074/jbc.M504929200. [DOI] [PubMed] [Google Scholar]
- Zhao H, Przybylska M, Wu IH, Zhang J, Siegel C, Komarnitsky S, Yew NS, Cheng SH. Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes. 2007;56:1210–1218. doi: 10.2337/db06-0719. [DOI] [PubMed] [Google Scholar]
- Zhu Q, von Dippe P, Xing W, Levy D. Membrane topology and cell surface targeting of microsomal epoxide hydrolase. Evidence for multiple topological orientations. J Biol Chem. 1999;274:27898–27904. doi: 10.1074/jbc.274.39.27898. [DOI] [PubMed] [Google Scholar]