

Abstract
hDlg, the human homologue of the Drosophila Discs-large (Dlg) tumor suppressor protein, is known to interact with the tumor suppressor protein APC and the human papillomavirus E6 transforming protein. In a two-hybrid screen, we identified a 322-aa serine/threonine kinase that binds to the PDZ2 domain of hDlg. The mRNA for this PDZ-binding kinase, or PBK, is most abundant in placenta and absent from adult brain tissue. The protein sequence of PBK has all the characteristic protein kinase subdomains and a C-terminal PDZ-binding T/SXV motif. In vitro, PBK binds specifically to PDZ2 of hDlg through its C-terminal T/SXV motif. PBK and hDlg are phosphorylated at mitosis in HeLa cells, and the mitotic phosphorylation of PBK is required for its kinase activity. In vitro, cdc2/cyclin B phosphorylates PBK. This evidence shows how PBK could link hDlg or other PDZ-containing proteins to signal transduction pathways regulating the cell cycle or cellular proliferation.
Multiple proteins, including some tumor suppressors, regulate the cell cycle machinery in a network of signaling pathways. hDlg, the human homologue of the Drosophila tumor suppressor Discs-large (Dlg), may be among these proteins. But how it interacts with signal transduction pathways is not fully understood.
hDlg is a membrane-associated guanylate kinase homologue (MAGUK) (1). Like other MAGUKs, it contains several protein–protein interaction domains: three PDZ domains, an SH3 domain, and a C-terminal guanylate kinase homology region that may act as a protein-binding domain (2–4). There are several MAGUKs in mammalian organisms. Some, like PSD-95 and Chapsyn-110, appear to be important for the function of synapses (5, 6). Others, such as ZO-1 and ZO-2, are part of the tight junction protein complex (7, 8). Yet another, p55, is a component of the erythrocyte membrane skeleton (9). Although all are homologues of the Drosophila tumor suppressor Dlg, it is unclear whether they also act as tumor suppressor proteins. A p55 mutation was identified in a leukemia patient, but it is not known whether this mutation was important for cellular transformation (10). ZO-1 protein levels are correlated with the differentiation level of breast and intestinal track tumors, but no causal effect of the lack of ZO-1 has been demonstrated in tumor formation (11, 12).
The identification of several proteins that bind to hDlg supports its involvement in cellular growth control. The adenomatous polyposis coli (APC) tumor suppressor protein binds the PDZ2 domain of hDlg in vitro, and the two proteins coimmunoprecipitate and colocalize in some cell types (13, 14). Furthermore, the PDZ2 domain of hDlg also interacts with the human papillomavirus E6 transforming protein and the 9ORF1 of the transforming adenovirus E4 [(15, 16), S.G., unpublished data]. Only proteins from cancer-causing strains of the human papillomavirus and adenovirus E4 have the PDZ2 binding motif, and this motif is essential for their transforming activity in cultured cells (15, 16).
In search of the signal transduction pathway in which hDlg could participate to regulate cellular proliferation, we performed a two-hybrid screen to find other interacting proteins. We discovered a PDZ-binding kinase (PBK) that interacts in vitro with the PDZ2 domain of hDlg and show that PBK is a cell cycle-regulated kinase that is phosphorylated and active only at mitosis.
Experimental Procedures
Two-Hybrid Screen.
The two-hybrid screen was performed by using the MatchMaker system (CLONTECH). The bait, full-length hDlg cDNA, was subcloned in pGBT9 and used to screen a HeLa cell cDNA library in pGAD-GH (CLONTECH). Around 750,000 independent clones were screened, of which 2,000 were His+ and 240 were positive for β-galactosidase activity. Two of those clones were partial cDNA sequences of PBK.
Cloning and Mutagenesis.
The full-length PBK cDNA was obtained by a set of PCR amplifications from the pGAD-GH HeLa cDNA library (CLONTECH). To clone the 5′ end, the first amplification used a sense primer within the vector sequence (5′-CTGTCACCTGGTTGGACGGAC-3′) and an antisense primer within the original PBK clone (5′-CGCAAGCCACACTTCAGC-3′). The secondary PCR used nested primers with the sense primer within the vector sequence (5′-TACCACTACAATGGATG) and the antisense primer within the original PBK clone (5′-CTCAGGGTCAGTCACAGTC-3′). The PBK sequence was confirmed by cloning the same full-length cDNA from the λExlox HeLa cDNA library (Novagen) and later by comparison to human expressed sequence tag (EST) sequences.
To generate constructs expressing PBK with a mutant C terminus, the PBK cDNA sequence was amplified by PCR by using a wild-type 5′ primer sequence (5′-GCGGGATCCATGGAAGGGATCAGTAAATTCAAG-3′) and an antisense primer in which the appropriate bases were substituted (V322A: 5′-GCGACTAGTCAGGCATCTGTTTCCAGAGCTTCAAC-3′, T320A: 5′-GCGACTAGTCACTAGACATCTGCTTCCAGAGCTTC-3′, base changes are underlined). For the deletion mutant, the same sense primer was used with an antisense primer that inserted a stop codon after base 936 (Δ: 5′-GCGACTAGTCATGCAGCAGAAGGACGATC-3′).
The construct used to express inactive PBK with mutations of lysines 64 and 65 to alanines was generated with the QuickChange Site-Directed Mutagenesis kit (Stratagene) and the primers:
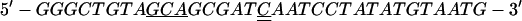 |
and
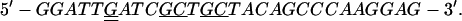 |
The underlined bases show where the sequences differ from that of wild-type kinase. The doubly underlined bases are silent mutations that remove a VspI restriction site to facilitate screening of mutant sequences. All the constructs were verified by sequencing (Sequenase Ver. 2.0, United States Biochemical).
Recombinant Protein Expression.
The pGEX expression system was used to express recombinant proteins in Escherichia coli. The coding sequences were subcloned in frame downstream of glutathione S-transferase (GST) in a pGEX-2T vector. The various hDlg constructs have been described elsewhere (1, 17). The proteins were expressed in the E. coli 71/18 bacterial strain and purified as described (18).
The proteins used in in vitro kinase assays were expressed in Sf9 cells by using the pAcGHLT B transfer vector and the BaculoGold baculovirus expression system (PharMingen). The vector was modified to remove the protein kinase A labeling site, the polyhistidine tag, and the thrombin cleavage site. Wild-type and inactive mutant PBK cDNAs were subcloned in frame downstream of GST. Baculovirus generation and protein expression were performed as recommended by the manufacturer, harvesting the cells 72 h after infection. The baculoviruses used for the expression cdc2 and cyclin B were a generous gift from Frederic Yarm and Ray Erikson (Harvard University).
To isolate active PBK from Sf9 cells, the cells were treated with 100 nM okadaic acid for 3 h before harvesting (19, 20). The cells were lysed in lysis buffer (10 mM Tris⋅Cl, pH 7.5/130 mM NaCl/1% Triton X-100/10 mM NaF/10 mM Na3PO4/10 mM Na4P2O7/1 mM Pefabloc/1 μg/ml pepstatin/1 μg/ml leupeptin). The cleared lysates were incubated with glutathione Sepharose-4b (Sigma), and the beads were washed with lysis buffer and then with storage buffer (50 mM Tris⋅Cl, pH 7.5/150 mM NaCl/10 mM NaF/1 mM Na3VO4/1 mM ETDA/1 mM DTT). Bound proteins were eluted in 50 mM Tris⋅Cl, pH 7.5, and 5 mM reduced glutathione, then dialyzed against storage buffer. A sample was precipitated with acetone and resuspended in 1% SDS to determine the protein concentration by using the BCA protein determination kit (Pierce).
The TNT SP6 coupled reticulocyte lysate expression system (Promega) was used to produce radiolabeled proteins for in vitro binding assays and blot overlay assays. The relevant cDNAs were subcloned into the pNB40 vector (21). 35S-methionine and 14C-leucine were obtained from Amersham Pharmacia.
In Vitro Binding Assays and Blot Overlay Assays.
For in vitro binding assays, fusion proteins (2 μg) on beads were incubated 2 h with 10 μl of reticulocyte lysate containing the 14C-labeled kinase with 40 μl RIPA buffer (50 mM Tris⋅Cl, pH 7.5/150 mM NaCl/1% Nonidet P-40/0.5% deoxycholate/0.1% SDS/1 mM EDTA/1 mM DTT). The beads were washed in RIPA buffer, and the bead pellet was resuspended in Laemmli sample buffer for SDS/PAGE analysis and autoradiography.
For blot overlay assays, equivalent amounts of each GST-PBK fusion protein were run on a 10% Laemmli gel and then transferred to nitrocellulose. The blots were probed with 35S-labeled hDlg or GST-PDZ1–2, washed extensively, and dried for autoradiography as described (22).
Antibody Production, Immunoprecipitations, and Immunoblots.
A rabbit polyclonal antibody against PBK was generated as described (23) (Serasource, Royalston, MA) by using the E. coli expressed full-length PBK released from GST with thrombin, further purified on a Mono-Q column, and dialyzed into PBS. The anti-hDlg antibody has been described elsewhere (17).
For immunoprecipitations, about 106 HeLa cells were lysed in 1 ml of Nonidet P-40 lysis buffer (50 mM Tris⋅Cl, pH 7.5/150 mM NaCl/0.5% Nonidet P-40/10 mM β-glycerophosphate/10 mM NaF/10 mM Na3PO4/10 mM Na4P2O7/1 mM Na3VO4/1 mM Pefabloc/1 μg/ml pepstatin/1 μg/ml leupeptin/1 mM EDTA/1 mM DTT). The cellular debris were sedimented, and lysates (about 100 μl) with equal amounts of total protein were incubated with the appropriate serum. The resulting immunocomplexes were recovered with 10 μl of protein A-Sepharose CL-4B beads (Pharmacia Biotech), washed in Nonidet P-40 washing buffer (lysis buffer without β-glycerophosphate, Na3PO4, or Na4P2O7), then resuspended in Laemmli sample buffer for SDS/PAGE analysis and immunoblotting. Alternatively, the complexes were washed once more in kinase assay buffer (see below) and resuspended for kinase assays.
For immunoblots, anti-PBK serum was used at 1:500, and anti-hDlg serum (directed against residues 200–960) was used at 1:1,000. The primary antibodies were detected with an alkaline phosphatase conjugated goat anti-rabbit IgG antibody (1:1,000, Zymed) and NBT/BCIP detection reagents (Sigma).
In Vitro Kinase Assays.
Proteins purified from Sf9 cells (1 or 2 μg) or immunoprecipitates containing endogenous PBK from about 105 HeLa cells were used in in vitro kinase assays. For autophosphorylation assays, the kinase was incubated in 30 μl kinase assay buffer (50 mM Tris⋅Cl, pH 7.5/150 mM NaCl/10 mM MgCl2/10 mM NaF/1 mM Na3VO4/1 mM EDTA/1 mM DTT) supplemented with 5 μCi of (32P-γ)-ATP. For substrate phosphorylation assays, 2 μg of myelin basic protein, histone (Sigma) or GST-hDlg, and 100 μM nonradiolabeled ATP were also added. After 45 min at 30°C or 2 h on ice, the reactions were stopped by addition of Laemmli sample buffer. The samples were resolved on 10% tricine gels and analyzed by autoradiography.
For phosphorylation of PBK by cdc2/cyclin B, 2 μg of GST, or wild-type or inactive mutant GST-PBK, was incubated 45 min at 30°C with 5 units of cdc2/cyclin B (New England Biolabs) in the recommended cdc2/cyclin B assay buffer supplemented with 100 μM ATP and 5 μCi of (32P-γ)-ATP for radiolabeling.
Cell Culture and Cell Cycle Synchronization.
HeLa cells were grown in DMEM low glucose supplemented with 4 mM l-glutamine and 7.5% FBS (Life Technologies) under a 5% CO2 atmosphere at 37°C. To synchronize cells at S-phase, we performed a double thymidine block (24). Nine hours after releasing the cells from the second thymidine block, the mitotic cells were isolated by gently pipetting media over them. To isolate G1-phase cells, mitotic cells were replated and incubated another 1.5 h. By visual inspection, over 95% of the cells were then postmitotic.
Results
Two-Hybrid Screen and Cloning of PBK.
A two-hybrid screen with full-length hDlg as bait identified many hDlg-interacting clones, two of which encoded the C-terminal half of a protein kinase (Fig. 1). The 5′ end of the PBK cDNA was cloned by PCR from the pGAD-GH HeLa cDNA library to complete the coding sequence. The entire sequence was confirmed by sequencing additional clones isolated from a phage λExlox HeLa cDNA library. Several EST sequences in the human est and unigene databases corroborate the PBK sequence from HeLa cell cDNA libraries (Fig. 1). Mouse, zebrafish, and Drosophila homologues were also found in the est and unigene databases (Fig. 1).
Figure 1.

The protein sequences of human PBK and its homologues from other species. Shaded residues show identity or conservation among at least two homologues. Boxed residues are conserved among most known kinases. Asterisks (*) mark residues that have a conserved hydrophobic character, size, or charge in the various kinase subdomains (identified with roman numerals above the sequences). The original clone of PBK encodes residues 185–334 of human PBK in this alignment. The sequence of PBK has been deposited in GenBank (accession no. AF189722).
The human PBK cDNA encodes a 322-aa protein (calculated molecular mass, 35 kDa) containing all the conserved subdomains and most of the nearly invariant residues found in kinases (Fig. 1). Its active site sequence corresponds to the consensus for serine/threonine or dual specificity kinases (D-X-K-X-X-N, residues 174 to 179 of the alignment in Fig. 1). Another feature of the human PBK sequence is the ETDV motif at the C terminus, which matches the binding specificity of the hDlg PDZ1 and PDZ2 domain determined by peptide screens (25).
Most of the mouse PBK amino acid sequence can be deduced from overlapping EST sequences (Fig. 1). Only a short piece of the sequence is still undetermined, corresponding to residues 232 to 258 of the alignment in Fig. 1. The mouse protein sequence shows 89% identity with the human sequence and also contains all of the appropriate kinase subdomains and conserved residues. All five identical ESTs encoding the mouse protein C terminus code for a C-terminal SSKH motif.
As expected, the zebrafish and Drosophila homologues are more distantly related to human PBK than the mouse homologue, but still contain all the kinase domain features. The C-terminal sequences for both of these proteins have not been determined. The amino acid conservation across the four homologues (shaded residues in Fig. 1) is not restricted to kinase subdomains. Even the most distantly related homologue, from Drosophila, shows much higher identity with human PBK (35% over the entire available sequence, 44% over the sequence confirmed by multiple EST sequences) than any other known non-PBK kinase.
Tissue Distribution of the PBK mRNA.
Unlike hDlg, which is ubiquitously expressed (1), the tissue distribution of PBK mRNA varies. It is most abundant in placenta and is also present in heart muscle and pancreas, and at low levels in skeletal muscle, kidney, liver, and lung (Fig. 2). No signal was detected from adult brain mRNA by Northern blot (Fig. 2) or by PCR amplification from a human adult brain cDNA library (data not shown). Thus, even although PBK and hDlg are both present in many tissues, their overall distribution differs.
Figure 2.

The PBK mRNA is present in several human tissues and most abundant in placenta. Autoradiograph of a human multiple tissue mRNA blot (CLONTECH) probed by hybridization (26) with radiolabeled 966bp PBK cDNA ORF. The tissue source is indicated above each lane.
In Vitro Interaction of PBK with hDlg.
Because human PBK has a C-terminal T/SXV motif corresponding to the consensus sequence known to interact with hDlg, we examined whether the two proteins could interact in vitro as well as in the two-hybrid system. PBK bound to a GST-hDlg fusion protein but not to GST alone. Furthermore, the three PDZ domains of hDlg were sufficient for PBK binding, whereas other domains of hDlg did not bind PBK in vitro (Fig. 3A). The binding of PBK's C-terminal domain specifically required PDZ2 of hDlg, and it did not interact with PDZ1 or PDZ3 (Fig. 3B). The C-terminal motif of PBK was essential for its interaction with hDlg in vitro. Full-length hDlg or its PDZ1–2 domains bound to wild-type PBK but not to a mutant form of PBK lacking its C-terminal 10 residues (Fig. 3C). PBK mutants in which Thr-320 or Val-322 in the C-terminal motif were mutated to Ala-320 or Ala-322, respectively, showed greatly reduced binding to full-length hDlg and to hDlg's PDZ1–2 domains (Fig. 3C).
Figure 3.

The C-terminal ETDV motif of PBK interacts specifically with PDZ2 of hDlg in vitro. (A and B) Autoradiographs of in vitro binding assays. (A) 14C-labeled PBK (43 kDa) coprecipitated with bead-bound GST fusions of full-length hDlg (GST-hDlg) and of the three PDZ domains of hDlg (GST-PDZ1–2-3). PBK did not bind to GST or fusions of GST with the N-terminal, SH3, or guanylate kinase homology domains of hDlg (GST-N-term, GST-SH3, and GST-SH3 + GUK). (B) Radiolabeled C terminus of PBK (20kDa) bound to GST-fusion proteins containing PDZ2 of hDlg (GST-hDlg, GST-PDZ1–2-3, GST-PDZ1–2, and GST-PDZ2) but not to fusions of the other PDZ domains of hDlg (GST-PDZ1, GST-PDZ3). (C) Autoradiograph of blot overlay assays. GST fusions of PBK with wild-type C-terminal motif (WT), with a deletion of the last 10 amino acids (Δ), or with single residue substitutions within the C-terminal motif (T320A and V322A) were separated on a gel and transferred to nitrocellulose. Blots were probed with radiolabeled hDlg (Left) or with a radiolabeled GST fusion of PDZ1–2 of hDlg (Right).
Cell Cycle-Dependent Phosphorylation of PBK.
Kinase assays with recombinant PBK or with PBK immunoprecipitated from asynchronous HeLa cells resulted in no detectable activity (data not shown). Therefore, we reasoned that PBK might be regulated in a cell cycle-dependent fashion. In fact, we observed that in M-phase HeLa cells, a subset of both PBK and hDlg molecules converted to slow migrating species by SDS/PAGE, whereas there was no change in asynchronous (less than 5% mitotic) cells or in S-phase and G1-phase cells (Fig. 4).
Figure 4.

hDlg and PBK have slower migrating species that appear only at mitosis. Lysates of asynchronous (A) or synchronized HeLa cells at G1-phase (G1), S-phase (S), or mitosis (M) were separated by SDS/PAGE, then transferred to nitrocellulose. The blots were then probed with anti-hDlg (Left) or anti-PBK antibodies (Right). In both cases, slower migrating species of the proteins appear at mitosis. There was half as much of the asynchronous cell lysate as of the other lysates.
The change in PBK mobility at mitosis was attributable to phosphorylation. Simply omitting phosphatase inhibitors in lysate preparation was sufficient to cause the slower migrating species of PBK to disappear (Fig. 5).
Figure 5.

hDlg and PBK are phosphorylated at mitosis. HeLa cell lysates were prepared in the presence (+) or absence (−) of phosphatase inhibitors (inh.) or calf intestinal phosphatase (CIP, Life Technologies, Grand Island, NY). The lysates were separated by SDS/PAGE and transferred to nitrocellulose. The blots were probed with anti-hDlg (Upper) or anti-PBK antibodies (Lower).
In the case of hDlg, the absence of phosphatase inhibitors in the lysis buffer did not affect the pattern of bands observed on the immunoblot of S-phase or M-phase lysates, but treatment with calf intestinal phosphatase caused the fastest migrating species to shift upward in both S-phase and mitotic cell lysates (Fig. 5). The slower migrating species of hDlg at mitosis was unaffected. This suggests that some isoforms of hDlg are phosphorylated at both S-phase and mitosis. It does not exclude the possibility that other isoforms are specifically phosphorylated at mitosis because calf intestinal phosphatase may not be able to remove all phosphate groups from hDlg.
PBK Is a Mitotically Active Serine/Threonine Kinase.
We examined whether PBK that was phosphorylated at mitosis was enzymatically active. We found that a protein band comigrating with PBK became phosphorylated in an immunocomplex assay (Fig. 6). Because this band was absent in immunoprecipitates made by using preimmune serum, we concluded that PBK itself was phosphorylated. This phosphorylation was cell cycle dependent because PBK immunoprecipitated from asynchronous cells, S-phase cells, or G1-phase cells was not phosphorylated (Fig. 6; G1, data not shown).
Figure 6.

Only PBK from mitotic HeLa cells becomes phosphorylated in an immunocomplex kinase assay. Preimmune serum or immune serum precipitates from asynchronous cell lysate (A) and from lysates of cells synchronized at S-phase (S) or M-phase (M) were assayed for kinase activity in kinase assay buffer supplemented with (32P-γ)-ATP. The immunocomplexes were separated by SDS/PAGE and transferred to nitrocellulose. The blot was probed with anti-PBK antibody (Lower) to verify that PBK was immunoprecipitated in the presence of serum. PBK isolated from mitotic cells appears as a doublet. An autoradiograph of the same blot (Upper) shows that only PBK isolated from mitotic cells was phosphorylated.
To confirm that the observed activity was caused by PBK and not another kinase contaminant, we produced and isolated recombinant wild-type and mutant (K64 and K65 mutated to A64 and A65) PBK from mitotic insect cells by using a method specifically designed to isolate active mitotic kinases (19, 20). Sf9 cells expressing GST-PBK fusion proteins were treated with 100 nM okadaic acid for 3 h before their lysis. Okadaic acid, a phosphatase inhibitor, induces premature mitosis in insect cells. As expected, PBK fusion proteins isolated from Sf9 cells treated with okadaic acid migrated more slowly than those isolated from nontreated cells (data not shown).
The wild-type and mutant PBK were tested for activity with no substrate (autophosphorylation) and with myelin basic protein or histone as substrates. In all three cases, only wild-type kinase isolated from okadaic acid-treated Sf9 cells was active (Fig. 7). Because no activity was detected with PBK bearing mutations at lysines 64 and 65, the activity observed in these assays can be attributed only to PBK (Fig. 7).
Figure 7.

PBK is activated by a mitotic phosphorylation that can be induced by treating Sf9 cells with okadaic acid. Autoradiograph of an in vitro kinase assay: GST fusions of PBK (WT) or mutant PBK where lysines 64 and 65 were mutated to alanines (Mut) were purified from Sf9 cells treated (+ OA) or not (no OA) with 100 nM okadaic acid before lysis. These fusion proteins were used in in vitro autophosphorylation assays (GST-PBK, Left) or assays with myelin basic protein (MBP, Center) or histone (Right) as substrates.
The ability of PBK to phosphorylate myelin basic protein and histone in vitro together with the sequence of its active site implied that it is a Ser/Thr kinase. This expectation was confirmed by phosphoamino acid analysis (Fig. 8): autophosphorylation and histone phosphorylation occurred primarily on threonines, whereas myelin basic protein phosphorylation occurred on both threonines and serines.
Figure 8.

PBK is a serine/threonine kinase. GST-PBK, myelin basic protein (MBP), and histone were phosphorylated in vitro by GST-PBK isolated from okadaic acid-treated Sf9 cells. Autoradiographs of the cellulose plates (EM Science) after separating the proteins on 10% tricine gel, transfer to a poly(vinylidene difluoride) membrane, and phosphoamino acid analysis by two-dimensional electrophoresis (27). Phosphoamino acids from histone (Left), GST-PBK (Center), and MBP (Right) are shown. The traced outlines show where the nonradiolabeled phosphoamino acid standards migrated (phosphoserine, S, phosphothreonine, T, and phosphotyrosine, Y).
PBK Is a Substrate of Cdc2/Cyclin B in Vitro.
Finally, we attempted to reproduce the mitotic phosphorylation and activation of PBK by coinfecting Sf9 cells with viruses expressing human cdc2 and human cyclin B. Like PBK isolated from okadaic acid-treated cells, PBK isolated from cells coexpressing cdc2 and cyclin B was active. Because no histone phosphorylation was detected when kinase-deficient PBK is isolated from these cells, the phosphorylation observed was caused by PBK itself and not by cdc2/cyclin B copurifying with PBK (Fig. 9A).
Figure 9.

cdc2/cyclin B phosphorylation is important but not sufficient to activate PBK. (A) Autoradiograph of an in vitro kinase assay blot with GST fusions of PBK (WT) or a mutant of PBK (K64A-K65A, Mut) purified from cell treated or not with okadaic acid and coinfected or not with cdc2/cyclin B. Wild-type PBK isolated from cells treated with okadaic acid or from cells also expressing human cdc2/cyclin B can phosphorylate histone. (B) Autoradiograph of the blot of an in vitro kinase assay where GST and GST fusions of PBK (WT) or a mutant of PBK (K64A-K65A, Mut) purified from Sf9 cells not treated with okadaic acid were used as substrates for cdc2/cyclin B. Both wild-type and mutant PBK GST fusions are substrates of cdc2/cyclin B, but GST alone is not. (C) Autoradiograph of an autophosphorylation assay. GST fusions of PBK (WT) or a mutant of PBK (K64A and K65A, Mut) were purified from Sf9 cells treated (+ OA) or not treated (no OA) with 100 nM okadaic acid before lysis. These fusion proteins were phosphorylated or not by cdc2/cyclin B by using nonradiolabeled ATP. The GST-PBK fusions were then isolated by using glutathione-Sepharose beads and washed several times. PBK was finally assayed for autophosphorylation activity in the presence of radiolabeled ATP, separated on a 10% tricine gel and transferred to nitrocellulose.
Wild-type and inactive PBK were both phosphorylated by recombinant human cdc2/cyclin B in vitro (Fig. 9B). In a similar assay, nonradiolabeled ATP was used to phosphorylate wild-type and mutant PBK with recombinant cdc2/cyclin B. Then, we pulled down GST-PBK with glutathione-Sepharose beads and tested it for autophosphorylation activity (Fig. 9D). In vitro phosphorylation of PBK by cdc2/cyclin B was not sufficient to activate PBK. Again, only wild-type PBK isolated from okadaic acid-treated Sf9 cells was active. The in vitro phosphorylation of PBK by cdc2/cyclin may be quantitatively or qualitatively different from that achieved in vivo, or PBK activation may also require other posttranslational modifications.
Discussion
Cloning of PBK and Its Interaction with PDZ Domain-Containing Proteins.
Our results show that a 35-kDa PDZ-binding protein cloned in a two-hybrid screen is a cell cycle-regulated Ser/Thr kinase. In vitro assays confirmed the potential associations and sites of interaction between PBK and hDlg, although in vivo association could not be confirmed by immunoprecipitation (data not shown). Failure of interacting proteins to coimmunoprecipitate is not unprecedented (28) (human CASK, a protein containing PDZ domains, and syndecan-2, a protein with a T/SXV motif, also interact in vitro and colocalize but do not coimmunoprecipitate).
Because our anti-PBK antibodies gave inconsistent staining results in cells, whether hDlg and PBK colocalize remains to be investigated.
Cell Cycle-Dependent Phosphorylation and Activation.
What is most interesting about PBK is that it is phosphorylated in a cell cycle-dependent manner at mitosis, and that this phosphorylation is required for its activation. Furthermore, it is most abundant in placenta, a highly proliferative tissue, and absent, or at very low levels, in normal adult brain tissue where there is virtually no cellular proliferation. Interestingly, one human PBK EST sequence (GenBank no. AI366737) is from a brain anaplastic oligodendroglioma, a rapidly proliferating brain tumor. These results suggest that PBK may have a role in the regulation of cellular proliferation and progression of the cell cycle.
We have shown that only PBK immunoprecipitated from mitotic HeLa cells is phosphorylated in an immunocomplex kinase assay. These data, combined with the fact that recombinant PBK is activated during the premature mitosis induced by treating insect cells with okadaic acid, lead us to believe that the activity observed in the immunocomplex is caused by the autophosphorylation of active PBK.
Furthermore, PBK is active when it is isolated from Sf9 cells coexpressing human cdc2/cyclin B. Yet in vitro cdc2/cyclin B-induced phosphorylation alone was not sufficient for activation of PBK. These results suggest that, whereas cdc2/cyclin B phosphorylation of PBK is important for its activation, PBK requires further posttranslational modifications or a cofactor to be activated.
PBK has a characteristic cdc2/cyclin B phosphorylation site (S/T-P-X-K/R) (29) at its N terminus, and this site is conserved across species, implying that it is important for the activity of the protein. Phosphorylation at this site could be required for a conformational change or a relocalization that allows PBK to be acted on by a second kinase in its activation loop.
Most cell cycle research focuses on the timing of cell cycle regulator activation, but such activation frequently depends on localization of the regulator. For example, the activity of Cdc14, a protein phosphatase that promotes the breakdown of cyclin and exit from mitosis in yeast cells, must be released from its sequestered position in the nucleolus so that it can dephosphorylate its targets as it disperses throughout the cell at anaphase (30). Similarly, Plk1, Polo-like kinase 1, must be localized to the spindle poles and the cytokinetic neck filaments for proper function during mitosis (31). Finally, the Caenorhabditis elegans LET-23 receptor tyrosine kinase relies on basolateral membrane localization in vulval precursor cells (32) for normal downstream regulation of vulval development and cellular differentiation. This basolateral membrane localization depends on an interaction between the LET-23 C-terminal TCL motif and the PDZ domain of LIN-7. Single amino acid substitutions in LET-23 or LIN-7 that prevent their association lead to LET-23 mislocalization and a highly penetrant vulvaless phenotype (32). The intracellular localization and function of the mitotic kinase PBK may depend similarly on its PDZ-mediated interaction with MAGUKs or other PDZ-containing structural proteins. It will be important to determine whether a mutation of the C-terminal motif of PBK affects its ability to be phosphorylated mitosis, but to date our attempts to obtain stable transfectants of PBK have, for technical reasons, not been successful. Further study of PBK's binding partners and function in the cell cycle should clarify the role of MAGUKs in tumor suppression and in the regulation of cellular proliferation.
Acknowledgments
We thank the members of the Branton laboratory, especially Alain Viel and Eric Brandin, for helpful discussion, as well as Nana Coleman, Sunny Cheung, and Sarah Henrickson for technical assistance. We are grateful to Frederic R. Yarm and Raymond L. Erikson for providing baculovirus for the expression of cdc2 and cyclin B. This work was supported by a National Institutes of Health grant (GM57314–02, D.B. and R.A.L.). S.G. was supported by a National Science and Engineering Research Council 1967 Scholarship (Canada).
Abbreviations
- GST
glutathione S-transferase
- MAGUK
membrane-associated guanylate kinase homologue
- PBK
PDZ-binding kinase
- EST
expressed sequence tag
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF189722).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.090102397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.090102397
References
- 1.Lue R A, Marfatia S M, Branton D, Chishti A H. Proc Natl Acad Sci USA. 1994;91:9818–9822. doi: 10.1073/pnas.91.21.9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim E, Naisbitt S, Hsueh Y-P, Rao A, Rothschild A, Craig A M, Sheng M. J Cell Biol. 1997;136:669–678. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satoh K, Yanai H, Senda T, Kohu K, Nakamura T, Okumura N, Matsumine A, Kobayashi S, Toyoshima K, Akiyama T. Genes Cells. 1997;2:415–424. doi: 10.1046/j.1365-2443.1997.1310329.x. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi M, Hata Y, Hirao K, Toyoda A, Irie M, Takai Y. J Biol Chem. 1997;18:11943–11951. doi: 10.1074/jbc.272.18.11943. [DOI] [PubMed] [Google Scholar]
- 5.Cho K O, Hunt C A, Kennedy M B. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 6.Kim E, Cho K O, Rothschild A, Sheng M. Neuron. 1996;17:103–113. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- 7.Willott E, Balda M S, Fanning A S, Jameson B, Van Itallie C, Anderson J M. Proc Natl Acad Sci USA. 1993;90:7834–7838. doi: 10.1073/pnas.90.16.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jesaitis L A, Goodenough D A. J Cell Biol. 1994;124:949–961. doi: 10.1083/jcb.124.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marfatia S M, Lue R A, Branton D, Chishti A H. J Biol Chem. 1994;269:8631–8634. [PubMed] [Google Scholar]
- 10.Ruff P, Chishti A H, Grimm E, Bischoff D, Kim A C. Leuk Res. 1999;23:247–250. doi: 10.1016/s0145-2126(98)00164-7. [DOI] [PubMed] [Google Scholar]
- 11.Hoover K B, Liao S Y, Bryant P J. Am J Pathol. 1998;153:1767–1773. doi: 10.1016/S0002-9440(10)65691-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura Y, Shiozaki H, Hirao M, Maeno Y, Doki Y, Inoue M, Monden T, Ando-Akatsuka Y, Furuse M, Tsukita S, et al. Am J Pathol. 1997;151:45–54. [PMC free article] [PubMed] [Google Scholar]
- 13.Matsumine A, Ogai A, Senda T, Okumura N, Satoh K, Baeg G-H, Kawahara T, Kobayashi S, Okada M, Toyoshima K, et al. Science. 1996;272:1020–1023. doi: 10.1126/science.272.5264.1020. [DOI] [PubMed] [Google Scholar]
- 14.Senda T, Iino S, Matsushita K, Matsumine A, Kobayashi S, Akiyama T. Neuroscience. 1998;83:857–866. doi: 10.1016/s0306-4522(97)00459-4. [DOI] [PubMed] [Google Scholar]
- 15.Kiyono T, Hiraiwa A, Fujita M, Hayashi Y, Akiyama T, Ishibashi M. Proc Natl Acad Sci USA. 1997;94:11612–11616. doi: 10.1073/pnas.94.21.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S S, Weiss R S, Javier R T. Proc Natl Acad Sci USA. 1997;94:6670–6675. doi: 10.1073/pnas.94.13.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lue R A, Brandin E, Chan E P, Branton D. J Cell Biol. 1996;135:1125–1137. doi: 10.1083/jcb.135.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith D B, Johnson K S. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 19.Kumagai A, Dunphy W G. Science. 1996;273:1377–1380. doi: 10.1126/science.273.5280.1377. [DOI] [PubMed] [Google Scholar]
- 20.Qian Y W, Erikson E, Maller J L. Science. 1998;282:1701–1704. doi: 10.1126/science.282.5394.1701. [DOI] [PubMed] [Google Scholar]
- 21.Brown N H, Kafatos F C. J Mol Biol. 1988;203:425–437. doi: 10.1016/0022-2836(88)90010-1. [DOI] [PubMed] [Google Scholar]
- 22.Chan Y, Kunkel L M. FEBS Lett. 1997;410:153–159. doi: 10.1016/s0014-5793(97)00454-7. [DOI] [PubMed] [Google Scholar]
- 23.Sigel M B, Sinha Y N, VanderLaan W P. Methods Enzymol. 1983;93:3–12. doi: 10.1016/s0076-6879(83)93031-8. [DOI] [PubMed] [Google Scholar]
- 24.Stein G S, Stein J L, Lian J B, Last T J, Owen T, McCabe L. In: Cell Biology: A Laboratory Handbook. Celis J E, editor. Vol. 1. San Diego: Academic; 1994. pp. 282–287. [Google Scholar]
- 25.Songyang Z, Fanning A S, Fu C, Xu J, Marfatia S M, Chishti A H, Crompton A, Chan A C, Anderson J M, Cantley L C. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 27.Boyle W J, van der Geer P, Hunter T. Methods Enzymol. 1991;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- 28.Cohen A R, Woods D F, Marfatia S M, Walther Z, Chishti A H, Anderson J M, Wood D F. J Cell Biol. 1998;142:129–138. doi: 10.1083/jcb.142.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreno S, Nurse P. Cell. 1990;61:549–551. doi: 10.1016/0092-8674(90)90463-o. [DOI] [PubMed] [Google Scholar]
- 30.Visintin R, Hwang E S, Amon A. Nature (London) 1999;398:818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- 31.Lee K S, Grenfell T Z, Yarm F R, Erikson R L. Proc Natl Acad Sci USA. 1998;95:9301–9306. doi: 10.1073/pnas.95.16.9301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaech S M, Whitfield C W, Kim S K. Cell. 1998;94:761–771. doi: 10.1016/s0092-8674(00)81735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]