

Abstract
We report the x-ray crystallographic structures of the bisphosphonate N-[methyl(4-phenylbutyl)]-3-aminopropyl-1-hydroxy-1,1-bisphosphonate (BPH-210), a potent analog of pamidronate (Aredia), bound to farnesyl diphosphate synthase (FPPS) from Trypanosoma brucei as well as to geranylgeranyl diphosphate synthase from Saccharomyces cerevisiae. BPH-210 binds to FPPS, together with 3 Mg2+, with its long, hydrophobic phenylbutyl sidechain being located in the same binding pocket that is occupied by allylic diphosphates and other bisphosphonates. Binding is overwhelmingly entropy driven, as determined by isothermal titration calorimetry. The structure is of interest since it explains the lack of potency of longer chain analogs against FPPS, since these would be expected to have a steric clash with an aromatic ring at the distal end of the binding site. Unlike shorter chain FPPS inhibitors, such as pamidronate, BPH-210 is also found to be a potent inhibitor of human geranylgeranyl diphosphate synthase. In this case, the bisphosphonate binds only to the GGPP product inhibitory site, with only 1 (chain A) or 0 (chain B) Mg2+, and ΔS is much smaller and ΔH is ~6k cal more negative than in the case of FPPS binding. Overall, these results are of general interest since they show that some bisphosphonates can bind to more than one trans-prenyl synthase enzyme which, in some cases, can be expected to enhance their overall activity in vitro and in vivo.
INTRODUCTION
Bisphosphonates such as pamidronate (1, Aredia), risedronate (2, Actonel), zoledronate (3, Zometa) and ibandronate (4, Boniva) are used to treat a variety of bone resorption diseases 1,2, and there is also current interest in the use of bisphosphonates in immunotherapy of cancer since they activate γδ T cells (containing the Vγ2Vδ2 T cell receptor) of the immune system to kill tumor cells3,4. In earlier work 5, we proposed that these so-called nitrogen-containing bisphosphonates acted as cationic transition state/reactive intermediate analogs, binding to the allylic substrate binding site in the enzyme farnesyl diphosphate synthase (EC 2.5.1.10). This proposal turned out to be correct, and there are currently several published x-ray crystallographic structures of bisphosphonates bound to FPPSs from a variety of organisms, including Escherichia coli 6, human 7,8, Trypanosoma cruzi 9, Trypanosoma brucei 10, and Cryptosporidium parvum 11. The vast majority of published structures have focused on commercially available bisphosphonates, such as those shown below. However, another particularly potent class (in bone resorption) of bisphosphonates which have not yet been investigated structurally are the “aryl-x” bisphosphonates, such as 5 (BPH-210, using our previous nomenclature), a very potent inhibitor from Novartis 12, containing a long (phenylbutyl) side-chain. This compound is of interest since in addition to being very active as a bone anti-resorptive agent, it is also one of the most potent bisphosphonate inhibitors of bacterial (E. coli) cell growth 13, as well as having low μM activity against T. brucei (the causative agent of African sleeping sickness), with an IC50 of 250 nM (Ki = 21 nM) against T. brucei FPPS (Yin, F., Cao, R., et al., unpublished result) and an IC50 = 2.4 μM in T. brucei cell growth inhibition (Croft, SL., et al., personal communication). Moreover, BPH-210 has activity against P. falciparum 14, the causative agent of one form of malaria. There is, therefore, interest in determining how this molecule binds to FPPS from one or more of these organisms, and here we report the first x-ray crystallographic structure of BPH-210 bound to FPPS, from T. brucei. We also show that, unlike most other bisphosphonates (e.g. 1-4), BPH-210 is also a relatively potent inhibitor of geranylgeranyl diphosphate synthase (GGPPS). In recent work, the structures of GGPPS from two species, human and Saccharomyces cerevisiae, have been reported15,16 and it has been shown that, unlike FPPS, there are three possible bisphosphonate binding sites, with the most potent GGPPS inhibitors binding to the GGPPS product inhibitory site15,16. We investigate here the binding of BPH-210 to the S. cerevisiae enzyme. In addition, we have also determined the thermodynamics of binding of BPH-210 to both FPPS and GGPPS. While ΔG values are similar, ΔH and ΔS vary considerably, although in both cases, binding is entropy driven.
MATERIALS AND METHODS
Crystallization and data collection for T. brucei FPPS·BPH-210
Protein expression and crystallization were based on the crystallization conditions reported by Mao et al. 10,17. To obtain inhibitor bound crystals, protein at 5.55 mg/mL in 10 mM Hepes, pH 7.4, 1 mM MgCl2 and 10 mM mercaptoethanol was mixed with 2.5 mM BPH-210 plus 2.5 mM MgCl2, then incubated overnight on ice before setting up the drops. Crystals were grown at room temperature in hanging drops by mixing 1 μL of protein/bisphosphonate solution and 1 μL of precipitant, consisting of 10% (v/v) MPD and 100 mM ammonium acetate, pH 5.75. Prior to data collection, crystals were mounted in a cryo-loop and flash-frozen in liquid nitrogen after addition of 40% (v/v) MPD as a cryoprotectant. Diffraction data were obtained at 100 K using an ADSC Q4 CCD detector at the Advanced Photon Source, beamline 22BM (λ=1.0 Å). Diffraction data were processed and scaled by using the program HKL2000 18. The crystals belonged to the P3121 space group, with unit cell parameters of a = b = 92.214 Å and c = 177.747 Å. Each asymmetric unit contained two FPPS molecules. Data collection statistics are shown in Table 1.
Table 1.
Data collection and refinement statistics for BPH-210, N-[methyl(4-phenylbutyl)]-3-aminopropyl-1-hydroxy-1,1-bisphosphonate, bound to T. brucei FPPS (2P1C) and S. cerevisiae GGPPS (2Z7H).
| PDB number | 2P1C | 2Z7H |
| Data collection | ||
| Space group | P3121 | P212121 |
| Unit cell | ||
| α=β (°) | 90, | 90 |
| γ (°) | 120 | 90 |
| a (Å) | 92.124 | 47.41 |
| b (Å) | 92.124 | 116.64 |
| c (Å) | 177.747 | 128.14 |
| X-ray source | APS-22BMa | NSSRC-BL13B1c |
| Resolution (Å) | 30-2.37 (2.45–2.37) | 30-2.08 (2.15–2.08) |
| No. of reflection observed | 315,360 | 213,350 |
| Unique | 34,615 (2,457) | 77,404 (7152) |
| Completeness (%) | 95.7 (69.7) | 94.3 (89.5) |
| R-merge | 0.087 (0.664) | 0.045 (0.287) |
| I/σI | 24.3 | 30.9 |
| Multiplicity | 9.1 (5.1) | 5.2 (4.7) |
| Refinement statistics | ||
| Resolution range (Å) | 30.0–2.45 (2.54–2.45) | 30.0–2.08 (2.15–2.08) |
| R-work/R-free (%) | 26.6/29.9 | 19.5/27.1 |
| RMSD | ||
| Bond lengths | 0.006 | 0.015 |
| Bond angles | 1.100 | 1.600 |
| No. of atoms | ||
| Protein | 5,704 | 4955 |
| Bisphosphonates | 48 | 48 |
| Magnesium ion | 6 | 1 |
| Solvent (water) | 306 | 633 |
| B average (Å2) of protein | 53.70 | 42.9 |
| B average (Å2) of solvents | 51.77 | 60.3 |
| B average (Å2) of ligands (bisphosphonates, Mg2+) | 45.46 | 57.3 |
| Ramachandran plot (%) | ||
| Most favored | 91.6 | 96.1 |
| Additionally allowed | 8.4 | 3.9 |
| Generously allowed | 0 | 0 |
Advanced Photon Source at the Argonne National Laboratory
Values in parentheses are for the highest resolution shell
National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan)
Structure determination of T. brucei FPPS· BPH-210
The crystal structure of T. brucei FPPS· BPH-210 was determined by using the molecular replacement method using the program Molrep19. The previously solved T. brucei FPPS structure (PDB: 2EWG) 10 minus the ligand was used as a starting model. The 2Fo-Fc difference Fourier map showed clear electron densities for most amino acid residues, including those in the substrate binding site. Bisphosphonate density was obvious. Iterative rounds of refinement using CNS 20 and rebuilding using Coot 21 were then carried out. Rfree, computed with 3% randomly selected reflections was used as a quality monitor23. Solvent molecules were finally added and verified from the electron density map. This yielded R/Rfree values of 0.266/0.299. The quality of the refined model was assessed by using the Procheck22 program. The Ramachandran plot for the structure was of good quality. Additional statistics for the final model (PDB: 2P1C) are shown in Table 1.
Crystallization and data collection for S. cerevisiae GGPPS• BPH-210
Crystals of GGPPS• BPH-210 were prepared as described previously16. Basically, native GGPPS crystals were prepared by using the hanging drop method by mixing 2 μL of GGPPS solution with 2 μL of precipitant solution containing 0.08 M CH3COONa, pH 4.6, 16% PEG 4000, 6–10% glycerol, and 6–10% 1, 2-propanediol. Crystals were then soaked in a cryoprotectant solution containing 2.5 mM MgCl2, 2.5 mM BPH-210, 0.08 M CH3COONa, pH 4.6, 20% PEG 4000, 10% glycerol, and 10% 1, 2-propanediol, for 3–12 h. X-ray diffraction data were collected at beam line BL13B1 of the National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan). Diffraction data were processed and scaled by using the program HKL200018. Data collection statistics are shown in Table 1. Prior to use in structural refinements, 5% randomly selected reflections were set aside for calculating Rfree as a quality monitor23.
Structure determination of S. cerevisiae GGPPS• BPH-210
The structure of GGPPS• BPH-210 was determined by using native GGPPS (PDB: 2DH4) as the model for molecular replacement. For GGPPS• BPH-210, the 2Fo-Fc difference Fourier map showed clear electron densities for most amino acid residues, including those in the inhibitor binding site, but several loops and the C-terminal segments were disordered. The density for BPH-210 was obvious. Subsequent refinement with incorporation of the cofactors and water molecules at a 1.0σ map level yielded R and Rfree values of 0.195 and 0.271, respectively, at 2.08 Å resolution. Additional statistics for the final model (PDB: 2Z7H) are shown in Table 1.
Isothermal titration calorimetry (ITC)
ITC measurements were performed at 25°C using a MicroCal VP-ITC (MicroCal, Inc., Northampton, MA), basically as described previously24. For FPPS, we titrated 8 μL of a 0.3 mM ligand solution from a 250 μL syringe (rotating at 300 rpm) into the sample cell containing 1.42 mL of a 0.018 mM T. brucei FPPS solution. The buffer solution was 50 mM Hepes (pH 7.4) and 5 mM MgCl2. The duration of injection was set to 19.2 s, and the delay between injections was 240 s. The initial delay prior to the first injection was 60 s. To derive the heat associated with each injection, the area under each heat burst curve (microcalories per second versus seconds) was determined by integration (using Origin version 5.0 software; MicroCal, Inc., Northampton, MA). Fitting to a one-site binding model gave good accord with experiment. For GGPPS, the experimental conditions were basically the same except that 0.015 mM GGPPS and 0.8 mM BPH-210 were used. The buffer was 50 mM phosphate, pH 7.0, 1 mM MgCl2, and, again, fitting to a one-site binding model gave good accord with experiment. In this instance, we used the human enzyme since this gave higher GGPPS expression levels, and previously we showed that there was a very good correlation (R=0.9, p=0.0035) between the Ki values for inhibition of both species by bisphosphonates16.
RESULTS
Structure of FPPS· BPH-210
We show in Figure 1A the structure of BPH-210 bound to T. brucei FPPS. There are two FPPS molecules in the asymmetric unit and one molecule of BPH-210 binds per FPPS. There are 3 Mg2+ bound to each FPPS, as first observed with risedronate (2) bound to the E. coli protein by Hosfield et al. 6. The phosphonate groups make multiple contacts with these 3 Mg2+, which in turn interact with Asp residues in both the first and second DDXXD repeats, as well as with neighboring Arg 112 and Lys 212, 269, as shown in the ligand interaction diagram in Figure 1B. Very similar interactions are seen in the B chain, Figure 1C. 2Fo-Fc density maps are shown in Figure 1D, E. The phenylbutyl sidechain is involved in hydrophobic contacts with, among others, A169, M106, T168 and T213, plus, the ammonium group of BPH-210 is close to the OH group in Y216. As can be seen more clearly in Figure 2A, the phenylbutyl side-chain essentially fully occupies the GPP/FPP binding site first identified by Tarshis et al. in the avian protein 25 and, as expected, the Mg2+ seen in those structures (PDB: 1UBW and 1UBX) are in close spatial proximity to the Mg2+ seen in the FPPS• BPH-210 structure. The positions of these Mg2+, as well as the two phosphonate groups, are also very close (~0.97 Å, rmsd) to those seen with other aromatic bisphosphonate-Mg2+ structures, such as in zoledronate (3), bound to human FPPS 7,8. In addition, the position of the positively charged (ammonium) center in BPH-210 is likewise very close to the cationic charge centers in, e.g. the minodronate and zoledronate (Figure 2B, 2C) FPPS structures10 (PDB: 2EWG and 1ZW5). Unlike these smaller bisphosphonates, however, BPH-210 has a very long side-chain, and as can be seen in Figure 2C, this extends to the end of the allylic binding site pocket, where it interacts with Y99, as evidenced by the blue disc feature on Tyr99 in the ligand interaction diagram, Figure 1B, C. The para-carbon on the phenyl ring on the ligand is also partially solvent exposed (blue feature on the ring), as shown in Figure 1B, C, similar to the partial solvent exposure of the distal isoprene group seen in the FPPS·FPP structure (PDB: 1UBX).
Fig. 1.

Structure and interactions in FPPS• BPH-210. (A) Schematic of T. brucei FPPS• BPH-210 dimer. (B) Ligplot interaction diagram show FPPS• BPH-210 interactions in chain A. (C) Ligplot interaction diagram of FPPS• BPH-210 interactions in chain B. (D) 2Fo-Fc electron density map for BPH-210 bound to FPPS in chain A (green contoured at 1σ, red at 3σ). (E) 2Fo-Fc electron density map for BPH-210 in chain B (green contoured at 1σ, red at 3σ).
Fig. 2.

Comparison of FPPS• BPH-210 and other FPPS structures. (A) superimposed structures of T. brucei FPPS• BPH-210 with avian FPPS containing GPP or FPP (PDB: 1UBX, 1UBW). BPH-210 is in yellow; GPP, green, FPP, blue. The 3 Mg2+ in the T. brucei FPPS structure are in dark blue, the Mg2+ in the avian FPPS structures are in pink. (B) T. brucei FPPS• BPH-210 structure as in A (same colors) superimposed on T. brucei minodronate (PDB: 2EWG)-FPPS structure (minodronate in blue, Mg2+ in this structure in pink). (C) T. brucei FPPS• BPH-210 structure (colors as in A) superimposed on human zoledronate FPPS complex; PDB: 1ZW5) (zoledronate, blue; Mg2+ in this structure in pink). Also shown is the close proximity between Y99 (red) in the T. brucei structure and F99 (purple) in the human structure.
The interaction between Tyr99 and the phenyl ring in BPH-210 is of interest since it suggests a role for this residue in the regulation of isoprenoid diphosphate chain length elongation. As shown in Figure 3, a ClustalW26 alignment of T. brucei FPPS with rat, human and avian FPPS shows that there are two conserved aromatic amino-acids 4 and 5 residues upstream of the conserved DDXXD repeats in all four proteins. In rat, human and avian FPPS, these amino-acids are both Phe, but in the T. brucei FPPS, they are His and Tyr. In the avian enzyme, mutation of Phe to Ala results in isoprenoid diphosphates having chain lengths > 15-carbons25, so these Phe residues (and by analogy His, Tyr in the T. brucei protein) appear to act as a “wall”, inhibiting chain elongation beyond C15 (FPP), a steric effect. A similar effect is also seen in the Toxoplasma gondii “FPPS”, in which there is a Phe→Cys substitution in the fifth amino acid upstream of the first DDXXD motif (Figure 3), resulting in the production of the C20 species, GGPP27. This steric effect is also reflected in the ability of different bisphosphonates to inhibit bone resorption, in rats12. Specifically, shorter chain species (having fewer CH2 groups attached to the phenyl ring) have slightly less activity than does BPH-210: 1 and 1.4 μg/kg, as opposed to 0.4 μg/kg, for bisphosphonates containing 3 and 2 CH2 spacer groups, respectively (as opposed to the 4 in BPH-210), due at least in part to decreased hydrophobic stabilization in the active site of the protein. However, a much larger effect is seen on chain elongation: for the analog of BPH-210 containing 5 CH2 groups, the ED50 for bone resorption increases from 0.4 μg/kg to 20 μg/kg, and for the species with 6 CH2 groups, the ED50 increases further, to 1500 μg/kg 12. The lack of potency of the longer chain species is likely to be due, at least in part, to steric interactions with the aromatic group(s) at the end of the binding site, plus, increased solvent accessibility of the phenyl group in the longer chain bisphosphonates would also contribute to a decrease in activity, due to unfavorable hydrophobic interactions.
Fig. 3.

Partial sequence alignment of rat, human, chicken, T. brucei, T. gondi, FPPSs. The “first DDXXD” repeat is shaded in red, the two aromatic amino-acids 4 and 5 residues upstream of the first DDXXD repeat (PhePhe in rat, human and chicken, HisTyr in T. brucei) that control to a significant extent, product specificity, and which are likely to interact with the phenyl ring in BPH-210, are shown in green.
Structure of GGPPS· BPH-210
We next investigated the interaction between BPH-210 and GGPPS. In previous work, it has been shown that small nitrogen-containing bisphosphonates have potent activity (low μM IC50 values, low nM Ki values) against FPPS, from a variety of species28–31. However, these compounds (such as risedronate, 2) have very little activity (typically ≥ 100 μM IC50 values) against GGPPS32. On the other hand, larger, more hydrophobic bisphosphonates can be potent GGPPS inhibitors16,32, although of course if they are sufficiently large, they will not bind to the smaller binding pocket in FPPS. Since BPH-210 is clearly much larger than 1-4, it seemed that it might also be a GGPPS inhibitor. This turns out to be the case, and we find an IC50= 4.17 μM for BPH-210 in GGPPS inhibition, corresponding to a Ki = 115 nM, to be compared with an IC50 of ~200 μM (Ki ~5.6 μM) for risedronate (2)32. This “dual” activity is of course of potential interest in the context of cell based activity, where inhibitors of both FPPS and GGPPS might lead to synergistic effects.
We thus crystallized and determined the x-ray crystallographic structure of BPH-210 bound to GGPPS. As with FPPS, there are two molecules in the unit cell (Figure 4A) and one molecule of BPH-210 binds to each GGPPS molecule. Interestingly, the bisphosphonate does not bind to the FPP substrate site seen previously16, rather, it binds to the GGPP product inhibitory site, first identified by Kavanagh et al. in human GGPPS15, and more recently in the yeast enzyme (PDB: 2Z4V), as can be seen in the superposition (PDB: 2Z7H, 2FVI) shown in Figure 4B. And, unlike the 3 Mg2+ seen in the FPPS structures (with bisphosphonates present), there is only 1 Mg2+ present in chain A, and 0 in chain B. Protein-ligand interactions are very similar in both chains, and are illustrated in Figures 4C, D; 2Fo-Fc density maps are in Figure 4E, F. As with FPPS, there is evidence for solvent exposure of the phenyl ring on the bisphosphonate, together with a far more pronounced solvent exposure of one of the bisphosphonate groups (which interacts with Arg69, Figure 4C,D), an interaction seen also in several other bisphosphonate-GGPPS structures16.
Fig. 4.

Structures and interactions in GGPPS·BPH-210. (A) Structure of GGPPS·BPH-210. (B) Close-up view of GGPPS·BPH-210 (in blue, PDB: 2Z7H) superimposed on human GGPPS with bound GGPPS (yellow; PDB: 2FVI). The Mg2+ in the S. cerevisae GGPPS·BPH-210 structure is in blue, the Mg2+ in the human GGPPS•GGPP structure, in red. (C) Ligplot interactions in chain A of the GGPPS·BPH-210 complex. (D) Ligplot interactions in chain B. (E) 2Fo-Fc electron density map for BPH-210 bound to GGPPS in chain A (green contoured at 1σ, red at 3σ). (F) 2Fo-Fc electron density map for BPH-210 in chain B (green contoured at 1σ, red at 3σ).
Protein-ligand interactions
We next consider the question of the nature of the interactions between BPH-210 and FPPS, and GGPPS. As can be seen in Figures 1B,C and 4C,D, there are a larger number of protein-ligand contacts in the FPPS• BPH-210 structure than in the GGPPS• BPH-210 structure (19, 19 versus 11, 12) and, while it is not possible to quantitate the strength of these interactions from the x-ray structures alone, the much larger number of contacts seen in the FPPS structure does suggest that binding of BPH-210 may be stronger in the case of FPPS than with GGPPS. There are also more hydrophobic interactions in the FPPS structure, and in the GGPPS• BPH-210 structure, one phosphonate is exposed. To assess the actual thermodynamics of binding, we used isothermal titration calorimetry. The interaction between BPH-210 and FPPS is overwhelmingly entropy driven, with ΔH = 4.0 kcal mole−1 and ΔS = 49.9 cal deg−1 mole−1 (TΔS = −14.9 kcal mole−1), as shown in Figure 5A. This is very similar to the result obtained previously with ibandronate (4) and T. brucei FPPS, where we found ΔH = 6.03 kcal mole−1 and ΔS = 50.9 cal deg−1 mole−1 (at pH = 7.4) 24. Very similar results were also seen in the human enzyme 7,8. As can also be seen in Figure 5A, we find 0.92 moles of BPH-210 bound per FPPS, in accord with the single site occupancy seen in the x-ray result, Figure 1.
Fig. 5.
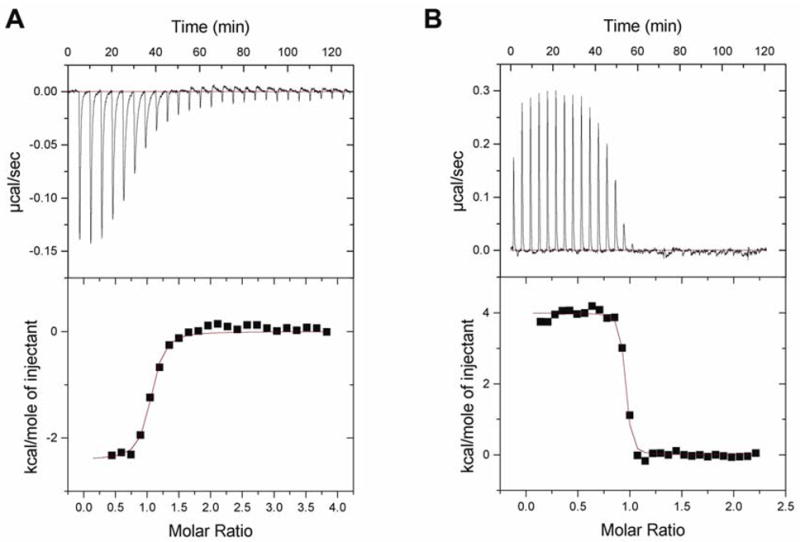
Isothermal titration calorimetry results. (A) ITC results at 300K for BPH-210 binding to T. brucei FPPS. Binding is overwhelmingly entropy driven (ΔH = 4.0 kcal mole−1, ΔS = 49.9 cal deg−1 mole, −TΔS = 14.9 kcal mole−1, Chi^2 =67712) and n=0.92. (B) ITC results at 300K for BPH-210 binding to H. sapiens GGPPS. Binding is enthalpy driven (ΔH = −2.41 kcal mole−1, ΔS = 23.07 cal deg−1 mole−1, −TΔS = 6.92 kcal mole−1, Chi^2 =7595) and n = 1.00.
As previously discussed in the case of ibandronate binding to FPPS, since the configurational entropy of BPH-210 can be expected to decrease on binding to FPPS, these results indicate the key importance of hydrophobic effects, that is, water molecules which are ordered around BPH-210 increase their entropy on movement of BPH-210 into the FPPS active site, and ordered water molecules in the active site also increase their entropy as they transfer to the bulk solvent, on ligand binding. On the other hand, in the case of BPH-210 binding to GGPPS, although binding is still entropy driven, binding is weaker and is exothermic, not endothermic, Figure 5B. For FPPS, we find that ΔG = −11 kcal mole−1 while for GGPPS, we find that ΔG = −9.3 kcal mole−1 (Figures 5A, B) with the TΔS term (−15 kcal vs. −6.9 kcal) clearly resulting in enhanced binding to FPPS over GGPPS. This result is also consistent with our Ki values (obtained of course under different experimental conditions and in the presence of isopentenyl diphosphate and either geranyldiphosphate or farnesyl diphosphate): Ki (BPH-210, T.brucei FPPS)=21 nM and Ki (BPH-210, human GGPPS)=115 nM (data not shown).
CONCLUSIONS
The results we have described above are of interest since they represent the first x-ray crystallographic structures of a potent farnesyl diphosphate synthase inhibitor which also inhibits geranylgeranyl diphosphate synthase. The bisphosphonate binds exclusively to the allylic site in FPPS, with its terminal phenyl ring having a face to face interaction with Y99, in the T. brucei protein. This binding motif and interaction pattern helps explain why this compound is a particularly potent inhibitor in bone resorption: shorter polymethylene side-chains can be expected to have poorer hydrophobic interactions in the FPPS active site, while larger ones lose almost all activity (n=4, ED50 = 0.4 μg/kg; n=5, ED50 = 20 μg/kg; n=6, ED50 = 1500 μg/kg, in bone resorption, where n is the number of methylene group spacers attached to the phenyl ring), due to repulsive steric interactions with F99 (in rat FPPS), located at the end of the allylic binding site, together with energetically unfavorable increased solvent-exposure with the longer-chain species. The close proximity of the phenyl ring in BPH-210 to residues that are likely to be involved in chain-length determination in some pathogenic species is also of interest since, with suitable functionalization, it may be possible to more specifically target these systems. In the case of GGPPS inhibition, the bisphosphonate binds exclusively to the GGPP product inhibitory site, not the FPP substrate site. There are fewer protein-ligand contacts seen in the GGPPS structure, and binding is weaker, as determined by both isothermal titration calorimetry and by Ki values, but in both enzymes, binding is still entropy driven. The ability of some bisphosphonates to inhibit both FPPS as well as GGPPS suggests that “dual function” bisphosphonates targeting more than one prenyltransferase may be worth pursuing further, since they may have enhanced activity in inhibiting protein prenylation, by inhibiting the sequential steps in the isoprenoid biosynthesis pathway.
Accession Codes
The atomic coordinates and structure factors for FPPS complexed with BPH-210 (2P1C) and GGPPS complexed with BPH-210 (2Z7H) have been deposited in the RCSB Protein Data Bank.
Acknowledgments
This work was supported by the United States Public Health Service (National Institutes of Health grant GM65307). We thank Roberto Docampo and Andrea Montalvetti for providing the T. brucei FPPS expression system and Hiroshi Sagami for providing the human GGPPS system. FPPS·BPH-210 data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22BM beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. GGPPS·BPH-210 data were collected at National Synchrotron Radiation Research Center, a national user facility supported by the National Science Council of Taiwan, ROC (grant NSC95-3112-B-001-015-Y to AH.-J.W.).
References
- 1.Russell RG. Bisphosphonates: from bench to bedside. Ann N Y Acad Sci. 2006;1068:367–401. doi: 10.1196/annals.1346.041. [DOI] [PubMed] [Google Scholar]
- 2.Green JR. Bisphosphonates: preclinical review. Oncologist. 2004;9 (Suppl 4):3–13. doi: 10.1634/theoncologist.9-90004-3. [DOI] [PubMed] [Google Scholar]
- 3.Kunzmann V, Bauer E, Wilhelm M. γδ T-cell stimulation by pamidronate. N Engl J Med. 1999;340(9):737–738. doi: 10.1056/NEJM199903043400914. [DOI] [PubMed] [Google Scholar]
- 4.Dieli F, Vermijlen D, Fulfaro F, Caccamo N, Meraviglia S, Cicero G, Roberts A, Buccheri S, D'Asaro M, Gebbia N, Salerno A, Eberl M, Hayday AC. Targeting human γδ T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007;67(15):7450–7457. doi: 10.1158/0008-5472.CAN-07-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin MB, Arnold W, Heath HT, 3rd, Urbina JA, Oldfield E. Nitrogen-containing bisphosphonates as carbocation transition state analogs for isoprenoid biosynthesis. Biochem Biophys Res Commun. 1999;263(3):754–758. doi: 10.1006/bbrc.1999.1404. [DOI] [PubMed] [Google Scholar]
- 6.Hosfield DJ, Zhang Y, Dougan DR, Broun A, Tari LW, Swanson RV, Finn J. Structural basis for bisphosphonate-mediated inhibition of isoprenoid biosynthesis. J Biol Chem. 2004;279(10):8526–8529. doi: 10.1074/jbc.C300511200. [DOI] [PubMed] [Google Scholar]
- 7.Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, Rogers MJ, Russell RG, Oppermann U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci USA. 2006;103(20):7829–7834. doi: 10.1073/pnas.0601643103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rondeau JM, Bitsch F, Bourgier E, Geiser M, Hemmig R, Kroemer M, Lehmann S, Ramage P, Rieffel S, Strauss A, Green JR, Jahnke W. Structural basis for the exceptional in vivo efficacy of bisphosphonate drugs. ChemMedChem. 2006;1(2):267–273. doi: 10.1002/cmdc.200500059. [DOI] [PubMed] [Google Scholar]
- 9.Gabelli SB, McLellan JS, Montalvetti A, Oldfield E, Docampo R, Amzel LM. Structure and mechanism of the farnesyl diphosphate synthase from Trypanosoma cruzi: implications for drug design. Proteins. 2006;62(1):80–88. doi: 10.1002/prot.20754. [DOI] [PubMed] [Google Scholar]
- 10.Mao J, Mukherjee S, Zhang Y, Cao R, Sanders JM, Song Y, Meints GA, Gao YG, Mukkamala D, Hudock MP, Oldfield E. Solid-state NMR, crystallographic, and computational investigation of bisphosphonates and farnesyl diphosphate synthase-bisphosphonate complexes. J Am Chem Soc. 2006;128(45):14485–14497. doi: 10.1021/ja061737c. [DOI] [PubMed] [Google Scholar]
- 11.Vedadi M, Lew J, Artz J, Amani M, Zhao YDA, Wasney GAGM, Hills T, Brokx S, Qiu W, Sharma S, Diassiti A, Alam Z, Melone M, Mulichak A, Wernimont A, Bray J, Loppnau P, Plotnikova O, Newberry K, Sundararajan E, Houston S, Walker J, Tempel W, Bochkarev A, Kozieradzki I, Edwards A, Arrowsmith C, Roos D, Kain K, Hui R. Cryptosporidium parvum putative polyprenyl pyrophosphate synthase (cgd4_2550) in complex with zoledronate PDB 2HER. 2006 [Google Scholar]
- 12.Widler L, Jaeggi KA, Glatt M, Muller K, Bachmann R, Bisping M, Born AR, Cortesi R, Guiglia G, Jeker H, Klein R, Ramseier U, Schmid J, Schreiber G, Seltenmeyer Y, Green JR. Highly potent geminal bisphosphonates. From pamidronate disodium (Aredia) to zoledronic acid (Zometa) J Med Chem. 2002;45(17):3721–3738. doi: 10.1021/jm020819i. [DOI] [PubMed] [Google Scholar]
- 13.Leon A, Liu L, Yang Y, Hudock MP, Hall P, Yin F, Studer D, Puan KJ, Morita CT, Oldfield E. Isoprenoid biosynthesis as a drug target: bisphosphonate inhibition of Escherichia coli K12 growth and synergistic effects of fosmidomycin. J Med Chem. 2006;49(25):7331–7341. doi: 10.1021/jm060492b. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh S, Chan JM, Lea CR, Meints GA, Lewis JC, Tovian ZS, Flessner RM, Loftus TC, Bruchhaus I, Kendrick H, Croft SL, Kemp RG, Kobayashi S, Nozaki T, Oldfield E. Effects of bisphosphonates on the growth of Entamoeba histolytica and Plasmodium species in vitro and in vivo. J Med Chem. 2004;47(1):175–187. doi: 10.1021/jm030084x. [DOI] [PubMed] [Google Scholar]
- 15.Kavanagh KL, Dunford JE, Bunkoczi G, Russell RG, Oppermann U. The crystal structure of human geranylgeranyl pyrophosphate synthase reveals a novel hexameric arrangement and inhibitory product binding. J Biol Chem. 2006;281(31):22004–22012. doi: 10.1074/jbc.M602603200. [DOI] [PubMed] [Google Scholar]
- 16.Guo RT, Cao R, Liang PH, Ko TP, Chang TH, Hudock MP, Jeng WY, Chen CK, Zhang Y, Song Y, Kuo CJ, Yin F, Oldfield E, Wang AH. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104(24):10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao J, Gao YG, Odeh S, Robinson H, Montalvetti A, Docampo R, Oldfield E. Crystallization and preliminary X-ray diffraction study of the farnesyl diphosphate synthase from Trypanosoma brucei. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 10):1863–1866. doi: 10.1107/S0907444904020633. [DOI] [PubMed] [Google Scholar]
- 18.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 12):1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 20.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 23.Brunger AT. Assessment of Phase Accuracy by Cross Validation - the Free R-Value - Methods and Applications. Acta Crystallogr D Biol Crystallogr. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- 24.Yin F, Cao R, Goddard A, Zhang Y, Oldfield E. Enthalpy versus entropy-driven binding of bisphosphonates to farnesyl diphosphate synthase. J Am Chem Soc. 2006;128(11):3524–3525. doi: 10.1021/ja0601639. [DOI] [PubMed] [Google Scholar]
- 25.Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD. Regulation of product chain length by isoprenyl diphosphate synthases. Proc Natl Acad Sci USA. 1996;93(26):15018–15023. doi: 10.1073/pnas.93.26.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ling Y, Li ZH, Miranda K, Oldfield E, Moreno SN. The farnesyl-diphosphate/geranylgeranyl-diphosphate synthase of Toxoplasma gondii is a bifunctional enzyme and a molecular target of bisphosphonates. J Biol Chem. 2007;282(42):30804–30816. doi: 10.1074/jbc.M703178200. [DOI] [PubMed] [Google Scholar]
- 28.Grove JE, Brown RJ, Watts DJ. The intracellular target for the antiresorptive aminobisphosphonate drugs in Dictyostelium discoideum is the enzyme farnesyl diphosphate synthase. J Bone Miner Res. 2000;15(5):971–981. doi: 10.1359/jbmr.2000.15.5.971. [DOI] [PubMed] [Google Scholar]
- 29.Montalvetti A, Fernandez A, Sanders JM, Ghosh S, Van Brussel E, Oldfield E, Docampo R. Farnesyl pyrophosphate synthase is an essential enzyme in Trypanosoma brucei. In vitro RNA interference and in vivo inhibition studies. J Biol Chem. 2003;278(19):17075–17083. doi: 10.1074/jbc.M210467200. [DOI] [PubMed] [Google Scholar]
- 30.Montalvetti A, Bailey BN, Martin MB, Severin GW, Oldfield E, Docampo R. Bisphosphonates are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase. J Biol Chem. 2001;276(36):33930–33937. doi: 10.1074/jbc.M103950200. [DOI] [PubMed] [Google Scholar]
- 31.Dunford JE, Thompson K, Coxon FP, Luckman SP, Hahn FM, Poulter CD, Ebetino FH, Rogers MJ. Structure-activity relationships for inhibition of farnesyl diphosphate synthase in vitro and inhibition of bone resorption in vivo by nitrogen-containing bisphosphonates. J Pharmacol Exp Ther. 2001;296(2):235–242. [PubMed] [Google Scholar]
- 32.Szabo CM, Matsumura Y, Fukura S, Martin MB, Sanders JM, Sengupta S, Cieslak JA, Loftus TC, Lea CR, Lee HJ, Koohang A, Coates RM, Sagami H, Oldfield E. Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates and diphosphates: a potential route to new bone antiresorption and antiparasitic agents. J Med Chem. 2002;45(11):2185–2196. doi: 10.1021/jm010412y. [DOI] [PubMed] [Google Scholar]