

Abstract
KCNE1 is a single span membrane protein that modulates the voltage-gated potassium channel KCNQ1 (KV7.1) by slowing activation and enhancing channel conductance to generate the slow delayed rectifier current (IKs) that is critical for the repolarization phase of the cardiac action potential. Perturbation of channel function by inherited mutations in KCNE1 or KCNQ1 results in increased susceptibility to cardiac arrhythmias and sudden death with or without accompanying deafness. Here, we present the three-dimensional structure of KCNE1. The transmembrane domain (TMD) of KCNE1 is a curved α-helix and is flanked by intra- and extracellular domains comprised of α-helices joined by flexible linkers. Experimentally-restrained docking of the KCNE1 TMD to a closed state model of KCNQ1 suggests that KCNE1 slows channel activation by sitting on and restricting the movement of the S4-S5 linker that connects the voltage sensor to the pore domain. We postulate that this is an adhesive interaction that must be disrupted before the channel can be opened in response to membrane depolarization. Docking to open KCNQ1 indicates that the extracellular end of the KCNE1 TMD forms an interface with an intersubunit cleft in the channel that is associated with most known gain-of-function disease mutations. Binding of KCNE1 to this “gain-of-function cleft” may explain how it increases conductance and stabilizes the open state. These working models for the KCNE1/KCNQ1 complexes may be used to formulate testable hypotheses for the molecular bases of disease phenotypes associated with the dozens of known inherited mutations in KCNE1 and KCNQ1.
KCNE1 (previously called minK) belongs to the KCNE family of single-span membrane proteins that modulate the activity of several voltage-gated K+ channels, including KCNQ1 (KV7.1). In cardiac myocytes KCNE1 forms obligate complexes with KCNQ1 to generate the slowly activating cardiac delayed rectifier current IKs, a critical determinant of myocardial repolarization(1-3). In the cochlea, this same channel complex enables secretion of K+ into endolymph and is critical for hearing(4). KCNE1 modulates KCNQ1 function by slowing voltage-stimulated channel activation, increasing conductance, and eliminating channel inactivation(2). The relevance of KCNE1 to proper channel function is underscored by heritable mutations in KCNE1 that are linked to congenital long QT syndrome (LQTS) and deafness (5-9). Although a number of structures of K+ channels and cytosolic channel accessory proteins have been determined(10-12), as have structures for cytosolic domains of KCNQ family members(13;14), little information exists regarding the structure of KCNE1 or any other transmembrane channel regulatory subunit belonging to the KCNE family. Here, we report the three-dimensional structure of full length human KCNE1 in model membranes and propose a working structural model for how the transmembrane domain of this accessory protein contributes to modulation of KCNQ1.
Materials and Methods
Abbreviated materials and methods are presented in this section. Full details are provided in the Supporting Information.
Determination of the KCNE1 Structure
Human KCNE1 was expressed in E. coli, purified into LMPG micelles, and prepared for solution NMR spectroscopy at pH 6.5 and 45°C as described previously(15). Resonance assignments were previously completed (15) and deposited in the BioMagResBank (www.bmrb.wisc.edu) with accession number 15102. Backbone structural restraints were acquired in the form of backbone/Cβ resonance chemical shifts, residual dipolar couplings (RDC), and short-range nuclear Overhauser effects. Five different Ser-to-Cys single-cysteine forms of KCNE1 were nitroxide spin-labeled and used to measure paramagnetic relaxation enhancements (PRE) that were converted to amide proton-spin label distances(16). The mutant forms of PMP22 used for the PRE measurements (see Supporting Information) exhibited only minor changes in TROSY NMR peak positions, indicating little structural perturbation due to the mutations and subsequent chemical modification. Examples of the NMR data from which the PRE and RDC data were acquired are shown in Figure 1. The NMR restraints were used to calculate the 3-dimensional structure of KCNE1 using XPLOR-NIH(17). Two sets of acceptable structures were initially obtained in which the extramembrane segments of the protein were arranged around the TMD in a topologically quasi-mirror fashion. One of these two sets was deemed to be correct because of better agreement with 1H-15N residual dipolar couplings from the extramembrane domains. These particular couplings were not used in the primary structural determination because of the known flexibility of parts of the extramembrane domains(15). A table describing the statistics of the determined KCNE1 structure is provided in the Supporting Information. The coordinate file for a representative of the ensemble that best satisfies all the data was deposited in the Protein Databank (code 2k21) and is available in the Supporting Information.
Figure 1.

Examples of NMR data from which structural restraints were derived for KCNE1. All spectra were acquired for uniformly 15N-labeled KCNE1 in LMPG micelles at 40°C and pH 6.0. Bottom left. Example of data used to obtain paramagnetic relaxation enhancement-derived distances. 600 MHz TROSY spectra from KCNE1 are shown before (paramagnetic) and after (diamagnetic) reduction of a nitroxide spin label attached to the side chain of Cys64 in the Cys105Ala/Ser64Cys single cysteine mutant form of KCNE1. Bottom right. Superimposed 800 MHz TROSY and semi-TROSY spectra of wild type KCNE1 that was marginally aligned within a stretched 5% polyacrylamide gel. Not shown is the corresponding pair of spectra from an unaligned sample. Doublets from both pairs of spectra were used as illustrated in the Upper Panel to determine the 1H-15N residual dipolar couplings (for Leu63--left and Asp85—right) by subtracting the J coupling measured in the isotropic spectra from the J+D coupling observed in the aligned spectra. Additional technical details are presented in the Supporting Information.
Data-Restrained Docking of KCNE1 to the KCNQ1 Channel
Twenty low energy models each for both the open and closed states of the KCNQ1 channel were developed as described previously(18) using a combination of homology modeling and ROSETTA calculations, drawing heavily upon the Kv2.1 crystal structure(10;18) and the related ROSETTA models of Yarov-Yarovoy et al.(19) The 10 lowest energy NMR structures of the TMD (sites 45-71) were then docked in backbone-only mode into each of the 40 KCNQ1 structures using ROSETTA-DOCK(20) in conjunction with application of very loose experimental/topological restraints: (i) KCNE1 sites 55 and 56 are restrained to be located near the center of the bilayer based on their inaccessibility to reagent added from either side of the membrane(21). ii) KCNE1 has an N-terminal-extracellular/C-terminal-intracellular topology, such that it is reasonable to restrict its tilt with respect to the bilayer normal to <50 degrees. (iii) Based on single and double mutation structure-function complementarity, KCNE1 sites 58 and 59 are thought to be in proximity to sites 340 and 339 in KCNQ1, respectively(22). (iv) Based on single and double Cys-insertion mutagenesis followed by functional testing in the presence and absence of Cd(II), sites 51 and 54 of KCNE1 are believed to be proximal to site 331 of KCNQ1, while site 51 is also believed to be proximal to site 328 of KCNQ1(23). 80,000 candidate models were generated, which were then filtered using the above experimental restraints with more stringent cut-offs, followed by clustering of the structures that satisfied the restraints into structural families. This led to about 200 families each for the open and closed states. Representatives from each family were then subjected to additional ROSETTA-DOCK calculations, this time in residue side chain-inclusive mode(24). The resulting ca. 3,000 models were then re-filtered using more even more stringent cut-offs for the experimental restraints and also additional open state-specific restraints (see Supporting Information), which led to the 17 open state models and 28 closed state models (see Results). Single representative structures that reflected the mean from each state were selected and subjected to further energy minimization. The PDB coordinates for these models are provided in the Supporting Information.
The key assumptions that were made in developing the models of the KCNE1/KCNQ1 complexes as described above are (i) that the KCNQ1 open and closed state models are essentially correct, (ii) that the presence of KCNE1 does not perturb the structures of KCNQ1's open and closed states, (iii) that the conformation of the KCNE1 TMD observed in LMPG micelles is retained when bound to KCNQ1, and (iv) those experimental restraints that were applied to both the open and closed states are equally applicable to both states.
Results and Discussion
Structure of KCNE1
Using solution NMR we determined the backbone structure of human KCNE1 in LMPG micelles, a model membrane system that was previously deemed to be well-suited for studies of KCNE1 from both NMR and protein functional standpoints (15). KCNE1 is seen to be comprised of an α-helical transmembrane domain (TMD, residues 45 through 71) and flanking N- and C-termini that consist of a series of flexibly linked alpha helices, with the distal C-terminus (residues 107-129) being disordered (Figure 2). The observed conformation of KCNE1 appears to reflect the micellar environment in which the NMR studies of the protein were carried out, as has previously been observed for some other membrane proteins in micelles(25-27). The overall shape of the protein is roughly spherical (Figure 2A) and yet the various segments are not compacted as in a well-folded globular protein. Subsequent rigid body docking of the NMR-determined structure into a 36 angstrom sphere that likely approximates the dimensions of an LMPG micelle leads to a model in which most extramembrane helices are situated on or near the micelle surface (Figure 2C). This model reflects the amphipathicity of the helix spanning residues 12-23 and the presence of a net positive charge for the helix spanning 92-106, properties that confer affinity for the negatively charged surface of LMPG micelles. Flexible linkage of the extramembrane helices may allow structural adaptation to KCNE1's local environment that, under physiological conditions, will be defined primarily by how it interacts with KCNQ1.
Figure 2.

Structure of KCNE1. See Supporting Information for experimental details of structural determination. A. Ensemble of 10 lowest energy conformations determined by NMR (front view). Conformers were overlaid based on minimizing the RMSD over the entire molecule, excluding the disordered C-terminus. The N-terminus is orange, the transmembrane domain is red, and the C-terminus is pink. B. Representative single structure from the lowest energy ensemble (front view). C. Results of rigid body docking of the NMR-determined KCNE1 structure (back view, residues 11-107 only) into a 36 angstrom sphere that represents the expected dimensions of an LMPG micelle. The actual size and shape of LMPG micelles remains to be experimentally determined. D. Front and side views of the overlaid transmembrane domains of the 10 lowest energy structures. In this case the depicted overlay is based on minimizing the RMSD of the transmembrane domain only. The side chain for Thr58 is depicted in Van der Waals mode.
The observed disorder of the distal C-terminus (Figure 2) is consistent with previous NMR relaxation data(15) and with the observation that this intracellular segment can be removed without perturbing normal KCNE1 modulation of KCNQ1 function(28). However, disordered segments are often involved in protein-protein recognition(29) and the distal C-terminus of KCNE1 is known to undergo interactions with non-channel proteins in cells(30), which may explain the presence of disease-linked mutation sites (Val 109 and Pro127) in this domain(15).
The KCNE1 juxtamembrane C-terminal domain contributes to the modulation of KCNQ1 and is also important for preventing channel inactivation(9;28), whereas the N-terminal of KCNE1 alters the pH sensitivity and pharmacological profile of the channel(31;32). As illustrated in Figure 3A, it is possible that the flexibly-linked helices from either or both termini may form an interface that spans multiple subunits of the channel so as to preclude binding of more than two KCNE1 subunits to any single tetrameric channel. This suggests an explanation for the known 2:4 stoichiometry of the fully-assembled KCNE1/KCNQ1 channel complex(33;34). Elucidating the details of the interactions of the KCNE1 N- and C-terminal helices with KCNQ1 will require future investigations.
Figure 3.

A. Structurally-based schematic that may explain the known 2:4 stoichiometry of the fully assembled KCNE1:KCNQ1 complex (extramembrane view). The KCNQ1 tetramer is illustrated by the green shapes. The α-helical KCNE1 TMD is illustrated as a pink circle and the connecting lines and pink rectangles indicate hypothetical locations of the N- and/or C-termini from two KCNE1 subunits. B. Sequence of KCNE1, with the NMR-determined locations of α-helices indicated as red boxes.
The KCNE1 TMD was the most precisely determined domain of the structure (Figure 2D) and has the highest immediate interest because a number of determinant determinants for modulation of channel activation reside in this segment (35-37). That the TMD of full length KCNE1 in LMPG micelles is helical agrees with the conclusion from previous biophysical studies of the isolated TMD polypeptide (38-40). However, what is novel and important about the structure introduced here is that the KCNE1 TMD is seen to adopt a curved helical conformation (Figure 2D), the detection of which reflects the power of residual dipolar coupling and paramagnetic relaxation enhancement NMR measurements. While it cannot yet be ruled out that this curvature could arise from micelle-specific distortion, this seems unlikely because the diameter of LMPG micelles is expected to be similar to the span of native membranes as a result of the long (C14) saturated acyl chain of LMPG. Instead, the curvature likely arises from the backbone conformational balance between glycines located in the TMD and a number of beta-branched amino acids also located therein (Figure 3B). It is noteworthy that the side chain of Thr58, a residue that is central in the distinct modulatory effects of KCNE1 relative to its homolog KCNE3(22;36), is located at the apex of the outer face of the curved helix and is well-exposed to interact with KCNQ1 (Figure 2D). It should be noted that the results of the structural calculations did not support our earlier suggestion that the transmembrane helix of KCNE1 undergoes a break in helicity around sites 59-61(15). The previous conclusion was based largely on NMR relaxation data that indicate an increase in the local backbone dynamics for those sites. This apparent discord in data interpretation may be reconciled either on the basis of there being increased local mobility at sites 59-61 within an unbroken helix or, more likely, by the presence of conformational exchange between a curved unbroken helix and a less-populated structure in which there is a break at sites 59-61.
Mechanisms of KCNE1 Modulation of KCNQ1 Channel Function
To potentially gain insight into how KCNE1 slows channel activation and enhances conductance we used the ROSETTA program(20;24) to perform ensemble docking of the 10 lowest energy experimental KCNE1 TMD structures into each member of a previously-developed ensemble of 20 low energy KCNQ1 models(18) for both open and closed channel states. These channel models were developed in a manner that relied heavily upon the approach taken by Yarov-Yarovoy et al. in their studies of the homologous Kv1.2 potassium channel(19). As described in detail in the Supporting Information this first phase of docking resulted in 80,000 models for the complex that were then filtered to select models that satisfy experimentally-derived restraints for the vertical placement of KCNE1 with respect to the center of the membrane(21) and for Cβ-Cβ distances between specific KCNE1/KCNQ1 residue pairs(22;23). In addition, we also employed open state-specific interresidue distance restraints from recent disulfide mapping studies (41;42). These experimentally-derived restraint filters resulted in the selection of precisely-converged structure ensembles for the closed and open states as depicted in Figure 4, which shows a representative pair of complexes that resemble the average for the open and closed state KCNE1/KCNQ1 ensembles. For both models it is observed that KCNE1 forms direct contacts with the S5-P-S6 pore domain and sits in a cleft between this pore domain and an adjacent voltage sensor. These findings are consistent with prior work that demonstrated the S5-P-S6 domain to be critical for the biochemical interaction between KCNQ1 and KCNE1(22;36;43). However, the cleft occupied by KCNE1 in the closed state is different from the one occupied in the open state. Videos were prepared that show the structural trajectory for the transition from the closed to open channel state, as based on simple linear extrapolation of atomic positions (Supporting Videos 1 and 2). While these videos do not represent an attempt to elucidate the transition state for the interconversion between open and closed conformations, the fact that the geometrically simplest path for converting between states does not require any stereochemical abominations, such as atoms or chains passing through each other, is reassuring.
Figure 4.
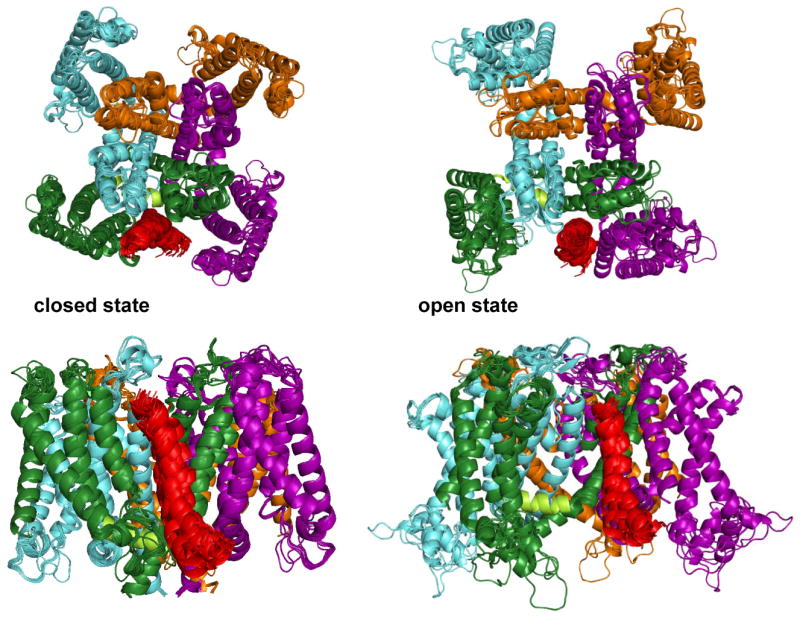
Ribbon diagram depictions of the 17 open state models and 28 closed state models of the KCNE1/KCNQ1 complex that were generated by high resolution (side chain-inclusive) docking and found to be consistent with a series of experimental restraints. The transmembrane domain of KCNE1 (residues 45-71) is shown in red, while the four KCNQ1 subunits are blue, orange, purple, and green.
The KCNE1/KCNQ1 models are best regarded as being medium resolution in nature, both because of the imperfect precision of structures that satisfy the experimental restraints used in docking (see Figure 4) and also because of uncertainty regarding some of the assumptions made in the restrained docking calculations (see end of Methods section). Therefore, a description of these models in terms of specific residue-residue contacts does not appear to be merited. However, even at moderate resolution, the general locations of the KCNE1 TMD with respect to KCNQ1suggest a compelling explanation mechanism for key aspects of KCNE1 function. In the closed state model (Figure 5) the intracellular end of the KCNE1 TMD sits on the S5 end of the critical S4-S5 linker of KCNQ1 that connects the voltage sensor to the pore domain and is thought to press downward on the cytosolic end of the S6 helix to hold the channel gate in a closed position(19;44). At the same time, the curved nature of KCNE1 enables the upper end of the TMD to form extensive contacts with a cleft formed between the upper part of S3 on the voltage sensor of one KCNQ1 subunit and the S5/S6 segments of another. We propose that this extensive contact surface between KCNE1 and KCNQ1 creates an adhesive interface between the two proteins that must be disrupted following membrane depolarization before the S4-S5 linker can be drawn back to allow the S6 gate to spring open. This offers a plausible explanation for how KCNE1 delays KCNQ1 channel activation.
Figure 5.

Details of the Open and Closed KCNE1/KCNQ1 Complexes. Ribbon diagrams of representative models for the closed and open state KCNE1/KCNQ1 complexes, as generated by experimentally-restrained docking calculations. The transmembrane domain of KCNE1 is shown in red. The S4-S5 linker is depicted as a light green helix in the green subunit. The side chains of the four gain-of-channel-function disease mutation sites in a selected GOF cleft are depicted in Van der Waals mode (Ser140, Val141, Ile274, Ala300).
When the KCNQ1-KCNE1 complex transitions to the open state, the closed state contacts between the KCNE1 TMD and KCNQ1 are completely disrupted as KCNQ1 rearranges itself around KCNE1, which itself undergoes only modest changes in position, tilt, and rotation (see Supporting Figure 2 and Videos 1-2). In the open state, the N-terminus of the KCNE1 TMD sits in a cleft that is vertically situated along the bilayer normal at roughly the same level as the selectivity filter and is in contact with three different KCNQ1subunits: S1 from one subunit, S5 and the end of the pore helix from a second subunit, and S6 from a third subunit. Notably, this cleft is the location for 4 out of 5 known KCNQ1 gain-of-function (GOF) disease-associated mutations (Ser140Gly, Val141Met, Ile274Val, Ala300Thr)(6;45-47). Moreover, for at least three out of 4 of these mutations the GOF channel phenotype is observed only when KCNE1 is present (see Discussion in(15)), a condition that is explained by our open channel complex model. In this model the central/C-terminus of the KCNE1 TMD sits at an interface between the cytosolic end of S1 in one subunit and S5 in another. Placement of the N-terminal end of the KCNE1 TMD in this “GOF cleft” likely stabilizes the open state of the channel and enhances conductance. It is also noteworthy that residues near the middle of KCNE1's TMD are in contact with the end of the “elbow” formed by the terminus of S4 and the ensuing S4-S5 linking helix, an interaction that may also stabilize the open channel state and possibly explains KCNE1-induced changes in state-dependent accessibility of the S4 segment to chemical modifiers(48;49).
In summary, determination of the KCNE1 structure using solution NMR and subsequent experimentally-restrained docking of the TMD into the KCNQ1 potassium channel offers a working model for how the TMD of KCNE1 plays a central role in modulating KCNQ1 function. These models and their underlying assumptions will, of course, have to tested by future experimental work. Moreover, the working models also do not directly address the nature of the transition state between the open and closed forms of the channel, which may be the point at which certain residues in KCNE1 and KCNQ1 play their key roles in channel modulation (c.f.(36)). Our models also do not address the how the juxtamembrane C-terminal domain domain of KCNE1 works in concert with the TMD to modulate KCNQ1 function(28;50), a matter that will also require additional studies. However, as the first semi-comprehensive attempt to describe the structural basis for KCNE1's modulation of KCNQ1 it is hoped that the proposed working models for KCNE1/KCNQ1 interactions will provide a lucid basis for generating testable hypotheses regarding both the mechanisms of KCNQ1 channel regulation and the nature of the molecular defects that lead to long QT syndrome and the other disorders known to result from dysfunction of KCNE1 and KCNQ1.
Supplementary Material
Full methodological details and a structural quality table are presented in the supporting information, as well as a schematic that outlines the restrained docking protocol used in this work and also a figure of the superimposed closed and open state KCNE1/KCNQ1 complexes. Supporting videos are also provided that show extracellular and membrane views of the interconversion between open and closed state KCNE1/KCNQ1 models. Finally, protein databank-format (PDB) files are provided for the open and closed state KCNE1/KCNQ1 models. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations
- GOF
gain-of-function
- LMPG
lyso-myristoylphosphatidylglycerol
- LQTS
long-QT syndrome
- NMR
nuclear magnetic resonance spectroscopy
- PRE
paramagnetic relaxation enhancement
- RDC
residual dipolar coupling
- RMDS
root mean-squared deviation
- TMD
transmembrane domain
- TROSY
transverse relaxation-optimized spectroscopy
Footnotes
This work was supported by NIH RO1 grants DC007416 (C.R.S.), by HL077188 (A.L.G.), by an American Heart Association fellowship (C.T., 0625586B), and by the Chinese National Science Research Plan 2006CB910204 (C.T.).
Coordinates for the structure of KCNE1 have been deposited in the Protein Databank www.pdb.org (code 2k21).
References
- 1.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 2.McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47:787–821. doi: 10.1016/j.neuropharm.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 4.Romey G, Attali B, Chouabe C, Abitbol I, Guillemare E, Barhanin J, Lazdunski M. Molecular mechanism and functional significance of the MinK control of the KvLQT1 channel activity. J Biol Chem. 1997;272:16713–16716. doi: 10.1074/jbc.272.27.16713. [DOI] [PubMed] [Google Scholar]
- 5.Abbott GW, Goldstein SA. Disease-associated mutations in KCNE potassium channel subunits (MiRPs) reveal promiscuous disruption of multiple currents and conservation of mechanism. FASEB J. 2002;16:390–400. doi: 10.1096/fj.01-0520hyp. [DOI] [PubMed] [Google Scholar]
- 6.Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y, Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J, Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 7.Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 8.Peters TA, Monnens LA, Cremers CW, Curfs JH. Genetic disorders of transporters/channels in the inner ear and their relation to the kidney. Pediatr Nephrol. 2004;19:1194–1201. doi: 10.1007/s00467-004-1626-6. [DOI] [PubMed] [Google Scholar]
- 9.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 10.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 11.Mackinnon R. Nobel Lecture. Potassium channels and the atomic basis of selective ion conduction. Biosci Rep. 2004;24:75–100. doi: 10.1007/s10540-004-7190-2. [DOI] [PubMed] [Google Scholar]
- 12.Torres YP, Morera FJ, Carvacho I, Latorre R. A marriage of convenience: beta-subunits and voltage-dependent K+ channels. J Biol Chem. 2007;282:24485–24489. doi: 10.1074/jbc.R700022200. [DOI] [PubMed] [Google Scholar]
- 13.Howard RJ, Clark KA, Holton JM, Minor DL., Jr Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron. 2007;53:663–675. doi: 10.1016/j.neuron.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiener R, Haitin Y, Shamgar L, Fernandez-Alonso MC, Martos A, Chomsky-Hecht O, Rivas G, Attali B, Hirsch JA. The KCNQ1 (Kv7.1) COOH Terminus, a Multitiered Scaffold for Subunit Assembly and Protein Interaction. J Biol Chem. 2008;283:5815–5830. doi: 10.1074/jbc.M707541200. [DOI] [PubMed] [Google Scholar]
- 15.Tian C, Vanoye CG, Kang C, Welch RC, Kim HJ, George AL, Jr, Sanders CR. Preparation, functional characterization, and NMR studies of human KCNE1, a voltage-gated potassium channel accessory subunit associated with deafness and long QT syndrome. Biochemistry. 2007;46:11459–11472. doi: 10.1021/bi700705j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 17.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 18.Smith JA, Vanoye CG, George AL, Jr, Meiler J, Sanders CR. Structural models for the KCNQ1 voltage-gated potassium channel. Biochemistry. 2007;46:14141–14152. doi: 10.1021/bi701597s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yarov-Yarovoy V, Baker D, Catterall WA. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc Natl Acad Sci U S A. 2006;103:7292–7297. doi: 10.1073/pnas.0602350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 21.Tai KK, Goldstein SA. The conduction pore of a cardiac potassium channel. Nature. 1998;391:605–608. doi: 10.1038/35416. [DOI] [PubMed] [Google Scholar]
- 22.Panaghie G, Tai KK, Abbott GW. Interaction of KCNE subunits with the KCNQ1 K+ channel pore. J Physiol. 2006;570:455–467. doi: 10.1113/jphysiol.2005.100644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tapper AR, George AL., Jr Location and orientation of minK within the I(Ks) potassium channel complex. J Biol Chem. 2001;276:38249–38254. doi: 10.1074/jbc.M103956200. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Schueler-Furman O, Baker D. Improved side-chain modeling for protein-protein docking. Protein Sci. 2005;14:1328–1339. doi: 10.1110/ps.041222905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choowongkomon K, Carlin CR, Sonnichsen FD. A structural model for the membrane-bound form of the juxtamembrane domain of the epidermal growth factor receptor. J Biol Chem. 2005;280:24043–24052. doi: 10.1074/jbc.M502698200. [DOI] [PubMed] [Google Scholar]
- 26.Chou JJ, Kaufman JD, Stahl SJ, Wingfield PT, Bax A. Micelle-induced curvature in a water-insoluble HIV-1 Env peptide revealed by NMR dipolar coupling measurement in stretched polyacrylamide gel. J Am Chem Soc. 2002;124:2450–2451. doi: 10.1021/ja017875d. [DOI] [PubMed] [Google Scholar]
- 27.Lee SY, Lee A, Chen J, Mackinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tapper AR, George AL., Jr MinK subdomains that mediate modulation of and association with KvLQT1. J Gen Physiol. 2000;116:379–390. doi: 10.1085/jgp.116.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 30.Furukawa T, Ono Y, Tsuchiya H, Katayama Y, Bang ML, Labeit D, Labeit S, Inagaki N, Gregorio CC. Specific interaction of the potassium channel beta-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J Mol Biol. 2001;313:775–784. doi: 10.1006/jmbi.2001.5053. [DOI] [PubMed] [Google Scholar]
- 31.Heitzmann D, Koren V, Wagner M, Sterner C, Reichold M, Tegtmeier I, Volk T, Warth R. KCNE beta subunits determine pH sensitivity of KCNQ1 potassium channels. Cell Physiol Biochem. 2007;19:21–32. doi: 10.1159/000099189. [DOI] [PubMed] [Google Scholar]
- 32.Peretz A, Schottelndreier H, haron-Shamgar LB, Attali B. Modulation of homomeric and heteromeric KCNQ1 channels by external acidification. J Physiol. 2002;545:751–766. doi: 10.1113/jphysiol.2002.028381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen H, Kim LA, Rajan S, Xu S, Goldstein SA. Charybdotoxin binding in the I(Ks) pore demonstrates two MinK subunits in each channel complex. Neuron. 2003;40:15–23. doi: 10.1016/s0896-6273(03)00570-1. [DOI] [PubMed] [Google Scholar]
- 34.Morin TJ, Kobertz WR. Counting membrane-embedded KCNE beta-subunits in functioning K+ channel complexes. Proc Natl Acad Sci U S A. 2008;105:1478–1482. doi: 10.1073/pnas.0710366105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen H, Goldstein SA. Serial perturbation of MinK in IKs implies an alpha-helical transmembrane span traversing the channel corpus. Biophys J. 2007;93:2332–2340. doi: 10.1529/biophysj.107.109702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melman YF, Krumerman A, McDonald TV. A single transmembrane site in the KCNE-encoded proteins controls the specificity of KvLQT1 channel gating. J Biol Chem. 2002;277:25187–25194. doi: 10.1074/jbc.M200564200. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Zheng R, Melman YF, McDonald TV. C-Terminal Interactions Between KCNE1 and KCNQ1. Biophys J. 2008;94:1382. [Google Scholar]
- 38.Ben Efraim I, Bach D, Shai Y. Spectroscopic and functional characterization of the putative transmembrane segment of the minK potassium channel. Biochemistry. 1993:2371–2377. doi: 10.1021/bi00060a031. [DOI] [PubMed] [Google Scholar]
- 39.Mercer EA, Abbott GW, Brazier SP, Ramesh B, Haris PI, Srai SK. Synthetic putative transmembrane region of minimal potassium channel protein (minK) adopts an alpha-helical conformation in phospholipid membranes. Biochem J. 1997;325(Pt 2):475–479. doi: 10.1042/bj3250475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aggeli A, Boden N, Cheng YL, Findlay JB, Knowles PF, Kovatchev P, Turnbull PJ. Peptides modeled on the transmembrane region of the slow voltage-gated IsK potassium channel: structural characterization of peptide assemblies in the beta-strand conformation. Biochemistry. 1996;35:16213–16221. doi: 10.1021/bi960891g. [DOI] [PubMed] [Google Scholar]
- 41.Chung DY, Chan PJ, Karlin A, Marx SO, Liu G, Kass RS. Cysteine Substitution Reveals Novel Inter-subunit Interactions In The Iks Potassium Channel. Biophysical Journal. 2008;94:82. [Google Scholar]
- 42.Xu X, Jiang M, Hsu KL, Cheng CS, Zhang M, Lyu PC, Tseng GY. KCNQ1 and KCNE1 Make State-dependent Contacts in Their Extracellular Domains. Biophysical Journal. 2008;94:1745. doi: 10.1085/jgp.200809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melman YF, Um SY, Krumerman A, Kagan A, McDonald TV. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron. 2004;42:927–937. doi: 10.1016/j.neuron.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Panaghie G, Abbott GW. The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J Gen Physiol. 2007;129:121–133. doi: 10.1085/jgp.200609612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL, Jr, Schwartz PJ. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–367. doi: 10.1161/CIRCULATIONAHA.106.658021. [DOI] [PubMed] [Google Scholar]
- 46.Bianchi L, Priori SG, Napolitano C, Surewicz KA, Dennis AT, Memmi M, Schwartz PJ, Brown AM. Mechanisms of I(Ks) suppression in LQT1 mutants. Am J Physiol Heart Circ Physiol. 2000;279:H3003–H3011. doi: 10.1152/ajpheart.2000.279.6.H3003. [DOI] [PubMed] [Google Scholar]
- 47.Hong K, Piper DR, az-Valdecantos A, Brugada J, Oliva A, Burashnikov E, Santos-de-Soto J, Grueso-Montero J, az-Enfante E, Brugada P, Sachse F, Sanguinetti MC, Brugada R. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005;68:433–440. doi: 10.1016/j.cardiores.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 48.Nakajo K, Kubo Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J Gen Physiol. 2007;130:269–281. doi: 10.1085/jgp.200709805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rocheleau JM, Kobertz WR. KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. J Gen Physiol. 2008;131:59–68. doi: 10.1085/jgp.200709816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rocheleau JM, Gage SD, Kobertz WR. Secondary structure of a KCNE cytoplasmic domain. J Gen Physiol. 2006;128:721–729. doi: 10.1085/jgp.200609657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.