

Abstract
Type IV pili (T4P) are retractile appendages that contribute to the virulence of bacterial pathogens. PilF is a Pseudomonas aeruginosa lipoprotein that is essential for T4P biogenesis. Phenotypic characterization of a pilF mutant confirmed that T4P-mediated functions are abrogated: T4P were no longer present on the cell surface, twitching motility was abolished, and the mutant was resistant to infection by T4P retraction-dependent bacteriophage. The results of cellular fractionation studies indicated that PilF is the outer membrane pilotin required for the localization and multimerization of the secretin, PilQ. Mutation of the putative PilF lipidation site untethered the protein from the outer membrane, causing secretin assembly in both inner and outer membranes. T4P-mediated twitching motility and bacteriophage susceptibility were moderately decreased in the lipidation site mutant, while cell surface piliation was substantially reduced. The tethering of PilF to the outer membrane promotes the correct localization of PilQ and appears to be required for the formation of stable T4P. Our 2.0-Å structure of PilF revealed a superhelical arrangement of six tetratricopeptide protein-protein interaction motifs that may mediate the contacts with PilQ during secretin assembly. An alignment of pseudomonad PilF sequences revealed three highly conserved surfaces that may be involved in PilF function.
Type IV pili (T4P) are adhesive cell surface appendages formed by a broad range of bacterial species and consist of multiprotein complexes that span the cell envelope. In gram-negative bacteria, T4P are thought to polymerize at the inner membrane and are extruded from the cell through an oligomeric secretin pore located in the outer membrane (7, 35). A single pilus fiber is 5 to 8 nm in diameter and can be extended out of the cell for several micrometers (61). Once attached to a surface by its distal tip, a single pilus can mediate a retractile force in excess of 100 pN (33), making the retraction complex one of the most powerful molecular motors assayed to date. The cellular organization of T4P proteins in gram-positive bacteria has yet to be clearly defined, although functionally equivalent proteins are presumably clustered at the single membrane (46, 64). T4P have been extensively studied in Pseudomonas, Neisseria, Escherichia coli, and Vibrio cholerae, where they act as key virulence factors during infection by mediating attachment to host cells (19, 36, 59, 60). The extension and retraction of the pilus fiber have also been implicated in a number of other functions, including twitching motility (52), cell-cell interactions (16), natural competence (63), biofilm formation (10), and infection by pilus-specific bacteriophage (4).
PilF, a predicted lipoprotein, is essential for T4P biogenesis in Pseudomonas aeruginosa (65). The pilF gene was initially identified in a transposon mutagenesis screen for pilus-associated twitching motility genes (65). Orthologues of PilF in Neisseria meningitidis (PilW) and Myxococcus xanthus (Tgl) have overall sequence identities to PilF of 26% and 21%, respectively. Both PilW and Tgl have been shown to localize to the outer membrane, where they assist in the assembly of the outer membrane secretin (8, 48). The inactivation of the pilT gene encoding the pilus retraction ATPase in a pilW mutant background restored T4P production in Neisseria. However, the T4P that were produced in the double mutant were fewer in number, prone to shearing, and functionally aberrant (8). The pilW pilT mutant phenotype suggests that PilW may play a secondary role in the stabilization of the pilus fiber and/or the intermembrane scaffold through which the pilus passes. In a recent publication describing a 2.2-Å-resolution structure of P. aeruginosa PilF (27), the authors suggested that PilF is localized to the cytoplasmic face of the inner membrane. Here we show that PilF is an outer membrane lipoprotein whose specific role in P. aeruginosa T4P biogenesis is to promote the insertion and multimerization of PilQ in the outer membrane to form the secretin pore. An independent, higher-resolution, 2.0-Å structure of PilF has been determined and used to identify the location of highly conserved residues on the surface of PilF which may be involved in mediating protein-protein interactions with PilQ and/or with proteins forming the T4P scaffold.
MATERIALS AND METHODS
Bacterial strains.
The bacterial strains and vectors used in this study are described in Table 1. Antibiotics were used at the following concentrations where appropriate: 50 μg/ml kanamycin for E. coli, 15 μg/ml gentamicin for E. coli, and 30 μg/ml gentamicin for P. aeruginosa. Genetic constructs were introduced into chemically competent E. coli cells by heat shock transformation. Constructs were introduced into P. aeruginosa cells by electroporation of cells resuspended in a 0.3 M sucrose solution at 8 kV/cm with an exponential decay constant of 4 ms.
TABLE 1.
Summary of strains and plasmids used
| Strain or vector | Characteristics | Source or reference |
|---|---|---|
| Strain | ||
| NovaBlue singles competent cells | endA1 hsdR17 (rK12− mK12+) supE44 thi-1 recA1 gyrA96 relA1 lac F′[proA+B+lacIqZΔM15::Tn10] (Tetr) | Novagen |
| E. coli BL21-CodonPlus(DE3)-RP | F−ompT hsdS(rB− mB−) dcm+ Tetrgal λ(DE3) endA Hte [argU proL Camr] | Novagen |
| E. coli B834(DE3) | F−ompT hsdSB(rB− mB−) gal dcm met(DE3) | Novagen |
| P. aeruginosa mPAO1 | Wild type | 24 |
| P. aeruginosa mPAO1 pilF::Tn5 | Transposon insertion mutant | 24 |
| Vector | ||
| pSTBLUE-1 | Blunt-ended cloning acceptor vector | Novagen |
| pET28a | T7 expression system inducible by IPTG | Novagen |
| pET28a-pilF | PAO1 pilF construct | This work |
| pUCP20Gm | Shuttle vector with SmaI-flanked gentamicin cassette inserted into ScaI site within bla | 10 |
| pUCP20Gm-pilF | PAO1 pilF construct | This work |
| pUCP20Gm-pilF C18G | Cys18 of PilF putative lipidation site replaced by Gly | This work |
PilF expression constructs and site-directed mutagenesis.
P. aeruginosa mPAO1 genomic DNA was isolated by using Bio-Rad InstaGene matrix. pilF was amplified by PCR using primers designed to remove the first 21 codons, corresponding to the signal sequence, and to insert NdeI and BamHI restriction sites at the N and C termini, respectively. The PCR product was initially ligated into pSTBLUE-1 by using a pSTBLUE-1 Perfectly Blunt cloning kit (Novagen). pilF was subcloned into pET28a at the corresponding NdeI and BamHI restriction sites to produce the pET28a-pilF construct. PilF with a thrombin-cleavable His6 N-terminal purification tag could thus be produced by using the isopropyl β-d-1-thiogalactopyranoside (IPTG)-inducible T7 expression system.
The pUCP20Gm-pilF complementation vector was made by PCR amplification of the full pilF sequence from P. aeruginosa mPAO1 genomic DNA. The addition of BamHI and HindIII restriction sites to the N and C termini, respectively, allowed the product to be inserted into the pUCP20Gm vector. A QuikChange site-directed mutagenesis kit (Stratagene) was used to mutate the conserved lipidation site Cys18 to Gly.
Surface protein isolation.
Surface proteins were sheared by using a protocol adapted from Castric (9). Each bacterial strain was streaked onto five Davis minimal medium 1.5% agar plates in a grid pattern and incubated for 48 h at 37°C. Cells were gently scraped off the agar surface with a sterile microscope slide into 2 ml of sterile PBS. Five minutes of vortexing was used to shear surface proteins. The suspension was divided equally into two 1.5-ml microcentrifuge tubes and centrifuged for 5 min at 4,600 × g at room temperature to pellet cells. The supernatant was transferred to new tubes and spun for an additional 25 min at 21,130 × g to remove the remaining cells. To precipitate sheared proteins, 1 M MgCl2 was added, to a final concentration of 0.1 M, and the solution incubated overnight at 4°C. Precipitated proteins were collected by centrifugation for 30 min at 21,130 × g. The pellet was resuspended in 100 μl phosphate-buffered saline and mixed with 4× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading dye (200 mM Tris-HCl, pH 6.8, 8% [wt/vol] SDS, 0.4% [wt/vol] bromophenol blue, and 40% [vol/vol] glycerol) at a 3:1 ratio and boiled for 10 min. Samples were loaded on a 16% SDS-PAGE gel with a prestained PageRuler protein ladder (Fermentas). Gels were stained with Coomassie blue dye.
Twitching assay.
Twitching motility was evaluated as described by Semmler et al. (52). Cells were stab inoculated into Luria-Bertani (LB) 1.0% agar plates and incubated for 24 h at 37°C. The agar was carefully removed, and adherent cells stained with 1% crystal violet at room temperature for 10 min. Unbound dye was rinsed off with water. The area of the twitching zone was measured by using ImageJ (National Institutes of Health). The average area was calculated from a minimum of 23 replicates.
Phage sensitivity assay.
P. aeruginosa strains to be tested were streaked as a single line on LB plates with the appropriate antibiotic, and 2 to 3 μl of PO4 phage (titer, approximately 108 PFU per ml) were applied at the center of each streak and grown overnight at 37°C. Sensitivity was defined as the lack of growth in the area of contact with phage (52).
Membrane fractionation.
The protocol used for membrane fractionation was adapted from Hancock and Nikaido (20). One liter of LB broth was inoculated with 10 ml of overnight culture and grown at 37°C with shaking at 180 rpm to an optical density at 600 nm of 0.1. Cells were harvested by centrifugation (7,000 × g, 4°C, 25 min). Two liters of cells were combined and washed with 10 ml of buffer A (30 mM Tris, pH 8.0). The cells were then resuspended in 3 ml buffer A with 20% (wt/vol) sucrose, 5 μl DNase I (Fermentas), 5 μl RNase (Sigma-Aldrich), 2 mg lysozyme, and 1 mM phenylmethylsulfonyl fluoride. Resuspended cells were lysed by two passes through a French press at 15,000 lb/in2. Cellular debris was removed by centrifugation (1,000 × g, 4°C, 10 min). Three milliliters of buffer A was added to the supernatant, and the mixture was applied to the top of a discontinuous sucrose gradient containing 6 ml of 15% (wt/vol) sucrose layered on 1 ml of 70% (wt/vol) sucrose. The gradient was centrifuged at 183,000 × g for 1 h at 4°C. The top 11 ml of the gradient, containing the soluble lysate, was removed, and the bottom 2 ml containing the membrane fraction was applied to a second discontinuous sucrose gradient of 3.5 ml of 52% (wt/vol), 3.5 ml of 58% (wt/vol), 2 ml of 64% (wt/vol), and 1 ml of 70% (wt/vol) sucrose and centrifuged at 183,000 × g at 4°C for at least 14 h. Membrane-containing fractions located at the interfaces between sucrose solutions of different densities were collected by puncturing the centrifuge tube with a 1.5-in.-long, 18-gauge syringe needle.
Whole-cell lysate analysis.
Whole-cell lysate was prepared by growing 5-ml cultures of each strain in LB broth overnight at 37°C with shaking at 180 rpm. Cells were harvested by centrifugation (1,300 × g, 4°C, 10 min). The supernatant was removed, and the pellet weighed and then resuspended in double-distilled water to a concentration of 0.1 mg/μl. Cells were lysed by the addition of 2× SDS-PAGE sample buffer, boiled at 100°C for 10 min, and analyzed by Western blotting. The relative expression levels were compared by densitometry with ImageJ (NIH).
Protein purification and antibody production.
PilF protein for antibody production was expressed and purified as follows. The pET28a-pilF expression vector was transformed into E. coli BL21 Codon Plus cells (Strategene). A 10-ml overnight culture was used to inoculate 1 liter of LB broth, and the culture was grown at 37°C to an optical density at 600 nm of 0.4 to 0.6. Protein expression was induced by adding IPTG to a final concentration of 1.0 mM. After 5 h, the cells were harvested by centrifugation (7,025 × g, 4°C, 20 min) and the cell pellet resuspended in 50 ml buffer B (20 mM Tris, pH 7.9, 500 mM NaCl) before being lysed by five sonication cycles of 60-s pulses followed by 60 s of cooling on ice. Centrifugation (22,658 × g, 4°C, 20 min) was used to remove cellular debris. The protein was bound to a 5-ml Ni-nitrilotriacetic acid resin column preequilibrated with buffer B and washed with 30 ml buffer B with 5 mM imidazole followed by 20 ml buffer B with 40 mM imidazole, and the bound protein was eluted in buffer B with 100 mM imidazole. The eluted solution was then dialyzed overnight at 4°C against 2 liters of buffer B. The purified PilF protein was used to generate specific antisera in rabbits at the Hospital for Sick Children Animal Facility using their standard operating procedures. Prior to use, the polyclonal antisera were purified by the method described by Salamitou et al. (49). Antisera against mature full-length PilQ were produced in the same manner (unpublished data).
Western blot analysis.
Samples from the membrane fractionation experiment were mixed with 4× SDS-PAGE sample buffer at a ratio of 3:1. Whole-cell lysates were mixed with 2× SDS-PAGE sample buffer at a ratio of 1:1. All samples were boiled for 10 min, and 25 μl of each sample was separated on a 12% SDS-PAGE gel and transferred to polyvinylidene difluoride or nitrocellulose. Proteins of interest were detected by using rabbit polyclonal antibodies to PilF and PilQ. OprF was detected by using a rabbit monoclonal antibody generously provided by Robert Hancock. The secondary anti-rabbit antibody conjugated to alkaline phosphatase was used per the manufacturer's instructions (Bio-Rad), and the blots developed with 5-bromo-4-chloro-3′-indolylphosphate (BCIP) p-toluidine salt and nitroblue tetrazolium chloride (Pierce).
Protein purification and structure determination.
Selenomethioninyl-incorporating protein for structural determination was expressed from the pET28a-pilF construct by using the B834 Met− E. coli auxotroph (Novagen) according to the protocol outlined by Lee et al. (32). The selenomethioninyl protein was purified as described above for the native protein, with the addition of 2 mM dithiothreitol to all buffers to prevent oxidation of the selenomethionine. The results of matrix-assisted laser desorption ionization mass spectroscopy and circular dichroism confirmed that the full incorporation of four selenomethionines did not change the thermal stability or protein secondary structure. Purified protein was concentrated to ∼16 mg/ml as determined by the Bradford assay (3). The initial screening for crystallization conditions was performed by using the native sulfur-containing protein and commercially available sparse-matrix screens. The optimized crystallization conditions were subsequently used to crystallize the selenomethioninyl protein. Crystals were grown using the hanging-drop vapor diffusion method by mixing 1.5 μl of protein and precipitating solution (95 mM tri-sodium citrate, pH 5.6, 19% [vol/vol] isopropanol, 19% [wt/vol] polyethylene glycol 4000, 5% [vol/vol] glycerol) on a glass cover slide and equilibrating against 200 μl of the same precipitating solution at 16°C. Crystals (∼0.55 by 0.27 by 0.05 mm) grew within 5 to 7 days. Prior to data collection, the crystals were washed with mother liquor containing 15% (vol/vol) glycerol and flash frozen in liquid nitrogen in preparation for transportation to the synchrotron. Diffraction data to 2.0-Å resolution were collected at the selenium peak and at a remote wavelength at Beam line X12-C at the National Synchrotron Light Source, Brookhaven National Laboratory. The data were processed and reduced by using d*TREK (43) and DREAR (2). The peak data were used to solve the structure by using BnP (68). SOLVE/RESOLVE (58) and ARP/wARP (42) were subsequently used to autobuild the initial model, which was refined against the remote data set by using CNS (5). Rounds of torsion angle, conjugate gradient, and B-factor refinement were alternated with manual rebuilding in COOT (15) to yield a final model with an R and Rfree of 20.9 and 24.8%, respectively. The data collection and refinement statistics are presented in Table 2.
TABLE 2.
Data collection, phasing, and refinement statistics
| Parametera | Peak | Remote |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.980301 | 0.95 |
| No. of measured reflections | 317,815 | 322,971 |
| No. of unique reflections | 45,717 | 45,900 |
| Redundancy | 6.95 (5.80) | 7.04 (6.64) |
| Resolution (Å)b | 30.86-2.00 (2.07-2.00) | 28.26-2.00 (2.07-2.00) |
| Rmergec (%)b | 6.2 (52.8) | 6.1 (59.7) |
| Completeness (%)b | 99.6 (97.7) | 99.8 (100) |
| Average I/σ(I)b | 10.9 (2.4) | 10.7 (2.0) |
| Phasing | ||
| Phasing power | 1.96 | |
| Figure of merit | 0.374 | |
| Figure of merit after solvent flattening | 0.938 | |
| Refinement | ||
| Protein atoms | 3,574 | |
| Water molecules | 317 | |
| R/Rfree (%)d | 20.9/24.8 | |
| Mean B-factor (Å2) | 47.63 | |
| RMS deviation from ideal | ||
| Bond length (Å) | 0.01 | |
| Bond angles (°) | 1.36 | |
| Residues in Ramachandran plot (%)e | ||
| Favored | 95.9 | |
| Allowed | 4.1 | |
| Outliers | 0 | |
| Estimated coordinate errorf | 0.34 |
Space group, P21212; unit cell dimensions (Å, °), a = 137.2, b = 69.1, c = 70.2, α = β = γ = 90.0.
Values in parentheses are for the outer resolution shell, 2.07 to 2.00 Å.
Rmerge = ∑I−<I>/∑I, where I is the measured intensity for symmetry-related reflections and <I> is the mean intensity for the reflection.
Rcryst = ∑(Fobs−Fcalc)/∑(Fobs); Rfree is Rcryst for the 5% cross-validated test data.
According to the Ramachandran plot in PROCHECK (31).
Calculated using the method of Read (47).
Sequence alignments and structural analysis.
PilF sequences from P. aeruginosa PAO1, Pseudomonas entomophila L48, Pseudomonas fluorescens PfO-1, Pseudomonas putida F1, Pseudomonas stutzeri A1501, and Pseudomonas syringae DC3000 were aligned with ClustalW2 (30), using the default settings (data not shown). The structural alignments of PilF and PilW (PDB ID 2VQ2) were performed by using the Combinatorial Extension (CE) server (54). Individual tetratricopeptides (TPRs) of PilF were aligned with those in the Hsp70/Hsp90 organizing protein (HOP) (50), cyclophilin 40 (56), protein phosphatase 5 (PP5) (69), and O-linked GlcNAc transferase (OGT) (25) with DaliLite (22). The surface accessibility of PilF residues was determined by using PISA (28) and compared to values corresponding to fully solvated residues as defined by Miller and colleagues (37).
Protein structure accession number.
The refined structure was deposited in the Protein Data Bank (PDB ID 2HO1).
RESULTS
Effect of pilF mutation on PilQ stability/expression.
Based on the previously reported functional linkage of PilF and PilQ homologues in other bacteria, we examined whether the levels of PilF affected PilQ protein levels or oligomerization status (Fig. 1). Comparison of the wild-type mPAO1 strain with a pilF::Tn5 mutant showed that the loss of PilF expression abolished multimerization of PilQ, as PilQ is only detected at the molecular weight corresponding to a monomer. Complementation of the pilF::Tn5 mutant with pUCP20Gm-pilF restored PilF expression and the multimerization of PilQ (Fig. 1). The amount of PilF expressed from the multicopy complementation vector was approximately twofold greater than the amount expressed by the mPAO1 wild-type strain, but the amount of PilQ and the distribution between its monomer and multimer forms in the complemented mutant appeared similar to those in the wild type (Fig. 1). Sequence analysis suggested that PilF is a typical lipoprotein, with a canonical lipobox containing a lipidation site at Cys18. We therefore generated a Cys18Gly mutant (C18G) in the pUCP20Gm-pilF vector and used this construct to complement the pilF::Tn5 mutant. Western blot analysis of the whole-cell levels of PilF and of PilQ monomers and multimers showed that the C18G mutant was expressed at levels comparable to the levels of the unmodified PilF protein and that the amount and distribution of PilQ between the monomer and multimer forms was comparable to those observed for the pilF::Tn5 mutant complemented with the wild-type PilF protein.
FIG. 1.
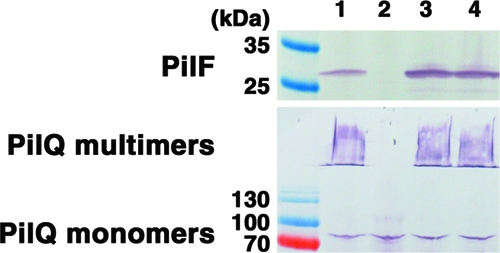
Loss of PilF disrupts PilQ oligomerization. Results of Western blot analysis of PilF and PilQ expression in whole-cell lysates of wild-type P. aeruginosa strain mPAO1 (lane 1), its pilF::Tn5 mutant (lane 2), the pilF::Tn5 mutant complemented with PilF (lane 3), and the C18G lipidation mutant (lane 4). The amount of sample loaded in each lane was normalized by cell mass.
PilF localization and effects of mutation on PilQ assembly and localization.
Previous studies of N. meningitidis and M. xanthus have shown that their PilF orthologs (PilW and Tgl) are localized to the outer membrane (8, 48). To supplement the biochemical characterization, Tgl localization in the outer membrane was also elegantly demonstrated by in vivo extracellular complementation of an M. xanthus tgl mutant (41). In contrast, Kim and colleagues (27) have proposed that P. aeruginosa PilF is localized to the inner membrane. We therefore sought to determine if the cellular localization of PilF in P. aeruginosa is different from that of its orthologs PilW and Tgl. Whole-cell lysates of P. aeruginosa were fractionated on discontinuous sucrose density gradients (20), using the well-characterized outer membrane protein OprF (66) as a control to monitor accurate separation of the inner and outer membranes (Fig. 2).
FIG. 2.

Subcellular localization of PilF and PilQ. Results of Western blot analysis of PilF, PilQ, and OprF in the sucrose gradient fractionations of wild-type P. aeruginosa strain mPAO1 (first panel), the pilF::Tn5 mutant (second panel), the pilF::Tn5 mutant complemented with PilF (+F; third panel), and the C18G lipidation mutant (fourth panel). In each panel, the inner membrane (lane 1), outer membrane fractions I and II (lanes 2 and 3), and the soluble lysate (lane 4) were analyzed.
Our results show that in strain mPAO1, PilF is highly enriched in the outer membrane fractions (Fig. 2). PilQ monomers were observed predominantly in the inner membrane, while SDS- and heat-resistant multimers were present only in the outer membrane fractions of wild-type P. aeruginosa (Fig. 2). PilF and the PilQ monomers detected in the soluble lysate may represent intermediates that have yet to become membrane associated or proteins released during cell lysis prior to sucrose gradient fractionation. In the absence of PilF, PilQ multimers were no longer detectable in any fraction and monomers were only found in the inner membrane (Fig. 2). Complementation of the pilF::Tn5 mutant with the pUCP20Gm-pilF vector restored the expression of PilF, which was enriched in the outer membrane fractions. The PilQ outer membrane localization and multimerization were also restored to wild-type levels (Fig. 2). In contrast, sucrose density gradient analysis of the pilF::Tn5 mutant complemented with the PilF C18G mutant showed a loss of association with the outer membrane, as the PilF C18G protein was only found in the soluble fraction (Fig. 2). In this strain, PilQ multimers were detected not only in the outer membrane but also in the inner membrane and the soluble lysate (Fig. 2).
The role of PilF in surface piliation and T4P function.
T4P expression and function were assayed in the wild type strain, the pilF::Tn5 mutant, and the mutants complemented with either wild type or C18G pilF. Although absent in the pilF::Tn5 mutant, twitching motility was restored by complementation with either wild-type pilF or the C18G variant (Fig. 3A). Mutation of the putative lipidation site caused a statistically significant decrease in the size of the twitching motility zones (Fig. 3B) that was not due to a defect in the growth of the C18G-complemented strain (data not shown).
FIG. 3.
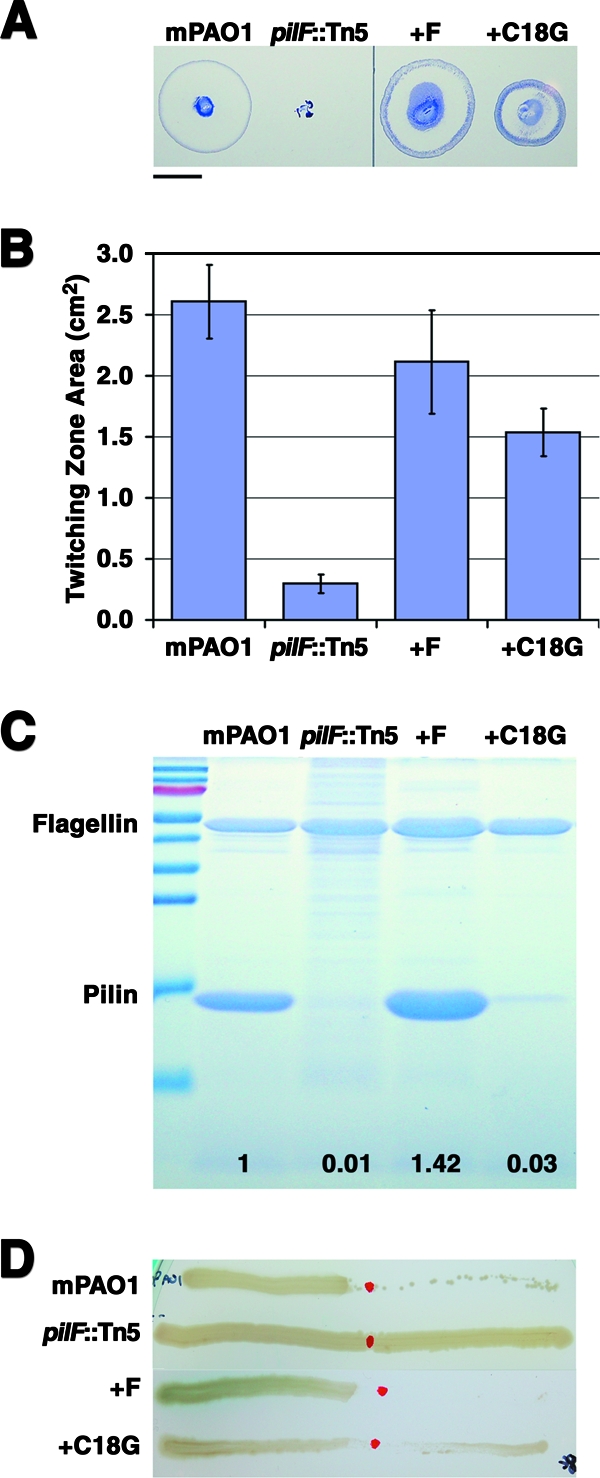
Functional characterization of the pilF::Tn5 mutant. Characterization of T4P expression and function of wild-type P. aeruginosa strain mPAO1, the pilF::Tn5 mutant, the pilF::Tn5 mutant complemented with PilF (+F), and the C18G lipidation mutant. (A) Representative plates showing twitching motility movement of cells away from the point of inoculation detected by using crystal violet dye. Bar, 1 cm. (B) Average twitching zone areas quantified from at least 23 replicates. All values are significantly different from each other at a P value of <0.0001. Error bars show standard deviations. (C) Sheared surface proteins were analyzed by Coomassie-stained 16% SDS-PAGE. The positions of the pilin and flagellin bands are indicated. Values at the bottom represent the ratio of pilin relative to the level in mPAO1 after normalization to the amount of flagellin present. The molecular mass ladder (top to bottom) is 130, 100, 70, 55, 45, 35, 25, 15, and 10 kDa. (D) The strains were tested for sensitivity to PO4 bacteriophage lysis. Sensitivity was defined as the absence of growth on contact with PO4 phage that had been spotted on the center of the plate.
As the degree of twitching motility can vary based on the expression level of T4P and the ability of surface-exposed T4P to retract, proteins were sheared from the cell surface and analyzed by SDS-PAGE to determine if the observed differences were caused by changes in the level of surface piliation. Flagellin was used as a loading control, as the expression of the flagellum is independent of T4P (35). No pili were recovered from the surface of the pilF::Tn5 mutant, but piliation was restored to greater than wild-type levels when the mutant was complemented with wild-type pilF (Fig. 3C). Surprisingly, on complementation of the pilF::Tn5 mutant with the C18G protein, the surface piliation was only 2% of that observed when the mutant was complemented with wild-type pilF, despite the ability of this strain to twitch to at least 50% of wild-type levels. The poor correlation between the level of surface piliation in complemented strains and the size of their twitching motility zones has been noted previously (1).
As twitching motility depends on T4P retraction, each strain was assayed for sensitivity to lysis by the pilus-specific bacteriophage PO4, which kills cells expressing retractile pili (4) (Fig. 3D). Consistent with the observation that the pilF::Tn5 mutant produces no T4P, it was resistant to killing by PO4 bacteriophage. Complementation of the pilF::Tn5 mutant with wild-type pilF restored the sensitivity to phage, while the phenotype of the C18G-expressing strain appeared to be intermediate between those of the wild-type mPAO1 and the pilF::Tn5 mutant.
PilF structure.
The structure of mature PilF, minus its signal sequence and lipidation site, was determined at a 2.0-Å resolution using selenomethionine incorporation and the single anomalous diffraction method (Table 2). The refined structure has a final R and Rfree of 20.9% and 24.8%, respectively. Two molecules of PilF were found in the asymmetric unit (Fig. 4). The quality of the electron density prevented us from modeling the His6 purification tag and the first 9 and 10 N-terminal residues of molecules A and B, respectively, and resulted in the truncation of the following residues to alanine: residues R33, E65, E122, D226, and K236 in molecule A and Q49, E92, E122, R225, D226, L233, and K236 in molecule B. Each molecule was found to contain 13 antiparallel α-helices arranged in a bilayered right-handed superhelix (Fig. 4). Helices 1 to 12 form six consecutive TPR-like repeats. The hydrophobic surface of the last TPR-like repeat is shielded from the solvent by its interaction with helix 13. The TPR motif is a 34-amino-acid repeat with a highly degenerate sequence that has been found to participate in protein-protein interactions in multiprotein complexes (13). The topology of PilF has been described in detail previously (27). Although the root mean square (RMS) deviation of main chain atoms between our 2.0-Å structure (PDB ID 2HO1; released July 2006) and the 2.2-Å structure of Kim et al. (27) (PDB ID 2FI7; released June 2006) show that the proteins are highly similar (RMS deviation = 0.58 Å), our structure has fewer geometric violations as judged by the smaller percentage of rotamer and Ramachandran outliers, 1.7 versus 8.3% and 0 versus 0.5%, respectively, and a greater percentage of residues in the favored region of the Ramachandran plot (96 versus 93.9%). We also find no evidence for a cis-peptide for residue 237 of molecule A.
FIG. 4.
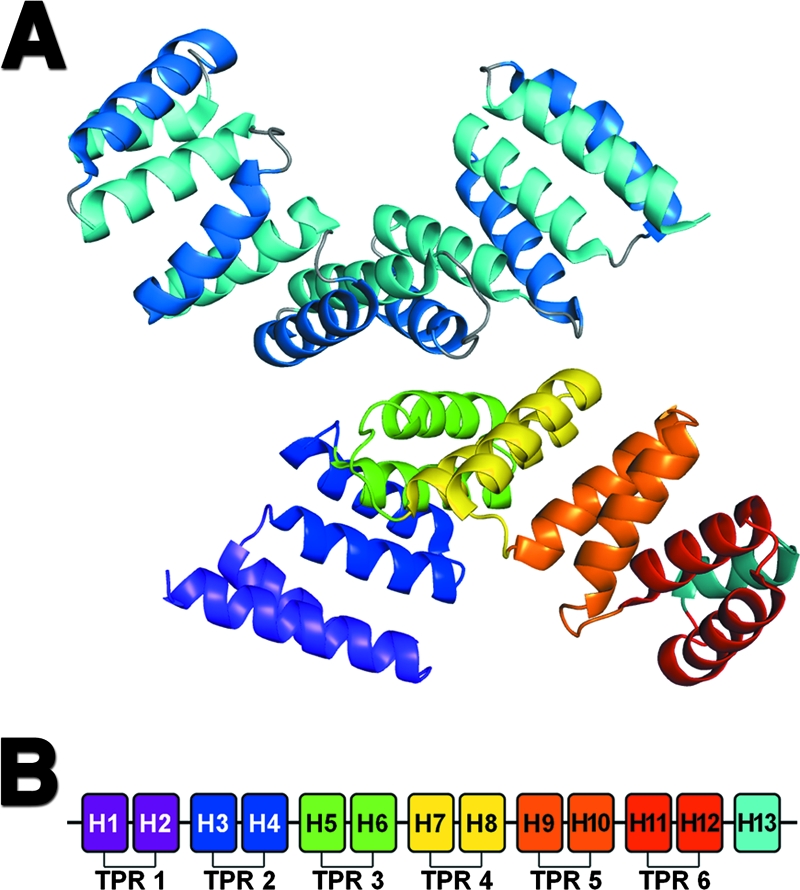
Crystallographic structure of PilF. (A) Ribbon diagram showing the arrangement of the two molecules of PilF present in the crystallographic asymmetric unit. The top molecule has been colored to show the bilayered arrangement of helices. Helices in the inner concave and outer convex surfaces are colored in light and dark blue, respectively. The second (bottom) molecule has been colored to show the six TPR motifs found in PilF. The TPR motifs are colored from purple at the N terminus to red at the C terminus. The final solvation helix is shown in light blue. (B) Schematic representation of the 13 helices found in PilF. The six TPR motifs and C-terminal solvation helix are colored as described for the second (bottom) molecule in panel A.
Structural homologues of PilF.
Previous structural homology searches using the DALI server (23, 27) showed that PilF was structurally related to members of the TPR-containing superfamily, including the Ras-related C3 botulinum toxin p67phox (29), PP5 (69), and FK506 binding proteins (67), as well as the TPR domain of OGT (25) and HOP (50). Our search identified additional structural homologues, including PilW, the PilF orthologue in N. meningitidis (62), and the P. aeruginosa and Yersinia pestis type III secretion chaperone proteins PscG (45) and YscG (55), respectively. The structures of PscG and YscG in complex with their binding partners are of particular interest, as they provide insight into PilF function (see Discussion). The structural similarity between PilF and PilW was not unexpected given the 26% sequence identity between the two proteins and the comparable roles that they play in T4P assembly in P. aeruginosa and N. meningitidis, respectively. Comparison of the PilF and PilW structures revealed a Cα RMS deviation of 2.6 Å for 210 of the 250 residues. However, superposition of the pairs of helices in each TPR motif revealed that the individual motifs are highly similar between the two structures, with an average Cα displacement between TPR motifs of 0.69 ± 0.17 Å (mean ± standard deviation). Examination of the superimposed structures suggested that the largest structural difference occurred at the C terminus, as the positions of TPR motif 6 and helix 13 relative to the rest of the structure were different in the two proteins. This observation prompted us to align the two proteins using subsets of the TPR motifs. The alignment of TPR motifs 1 to 4, comprising residues 31 to 170 in PilF, gave a Cα RMS deviation of 1.2 Å for the aligned residues and showed a clear difference in the orientation of TPR motifs 5 and 6, as well as helix 13, in the two structures (Fig. 5). The superposition of the two proteins using their C-terminal regions, comprising residues 171 to 249, gave a Cα RMS deviation of 1.6 Å for the aligned residues and again revealed a significant displacement of the proteins' N-terminal regions (data not shown). The observed differences suggest that a rigid body movement occurs between the N- and C-terminal regions of PilW relative to PilF. This relative difference is not the consequence of the disulfide bond in PilW between residues 115 and 150, which links TPR motifs 3 and 4. The differences observed could be due to the sequence variations between the two proteins, which will affect the relative packing of the TPR motifs, and/or the consequence of the extensive number of intermolecular crystal contacts in the hexagonal lattice of PilW. Significantly more intermolecular crystal contacts are found at the C terminus of PilW than are observed in the PilF crystal structure.
FIG. 5.
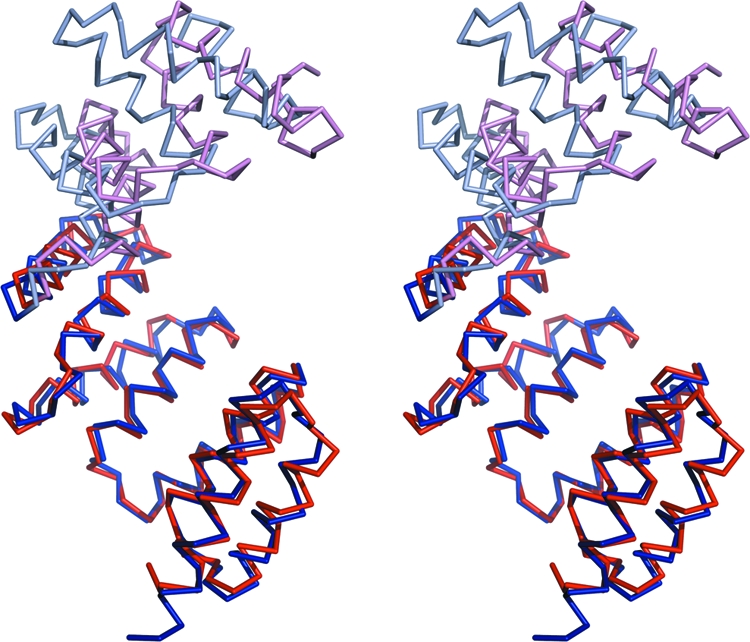
Structural differences between PilF and PilW. Stereo view of the Cα trace of PilW superimposed on PilF. The proteins were superimposed using TPR motifs 1 to 4, comprising residues 34 to 170 in PilF. TPR motifs 1 to 4 of PilF and PilW are drawn in dark blue and dark red, respectively. The remaining regions of PilF and PilW are drawn in light blue and light red, respectively.
Identification of potential protein-protein interaction surfaces.
Using bioinformatics methods, we have defined surfaces that are likely to be integral to PilF function based on their absolute conservation in a number of pseudomonads and a solvent-accessible surface area in excess of 10% of the residue's total surface area (37). The sequence comparison was restricted to other pseudomonads, as the PilF proteins in this genus share between 55 and 65% sequence identity with P. aeruginosa PilF and are anticipated to be able to cross-complement. Of the 95 strictly conserved amino acid residues in PilF, 87 were modeled in our structure, and 38 of these residues were more than 90% buried. As described above, the quality of the electron density prevented the N-terminal region of the protein from being modeled. Twenty-three of the buried residues correspond to positions critical for the assembly of the TPR motif (13). Thirty-five of the remaining 49 conserved residues were found to be more than 10% exposed to solvent and to cluster into three distinct regions on the surface of PilF (Fig. 6A and B). The predominantly hydrophobic N-terminal cluster (Fig. 6A and B, region 1) has a surface area of 597 Å2 and is composed of residues G32, R33, A36, A39, Y40, Q42, L43, G46, Y47, and K62. The cluster running through the central concave groove of PilF (Fig. 6A and B, region 2) is composed of the mainly charged or polar residues Q82, E86, L89, R106, N110, D134, Y137, P138, E139, R140, R142, L170, Q174, and P175 and forms an 878-Å2 surface. The third cluster (Fig. 6A and B, region 3) is found at the C terminus, contains residues E180, A209, R210, Q234, R237, L238, Y239, P240, G241, E244, and Y245, and is 777 Å2 in size.
FIG. 6.
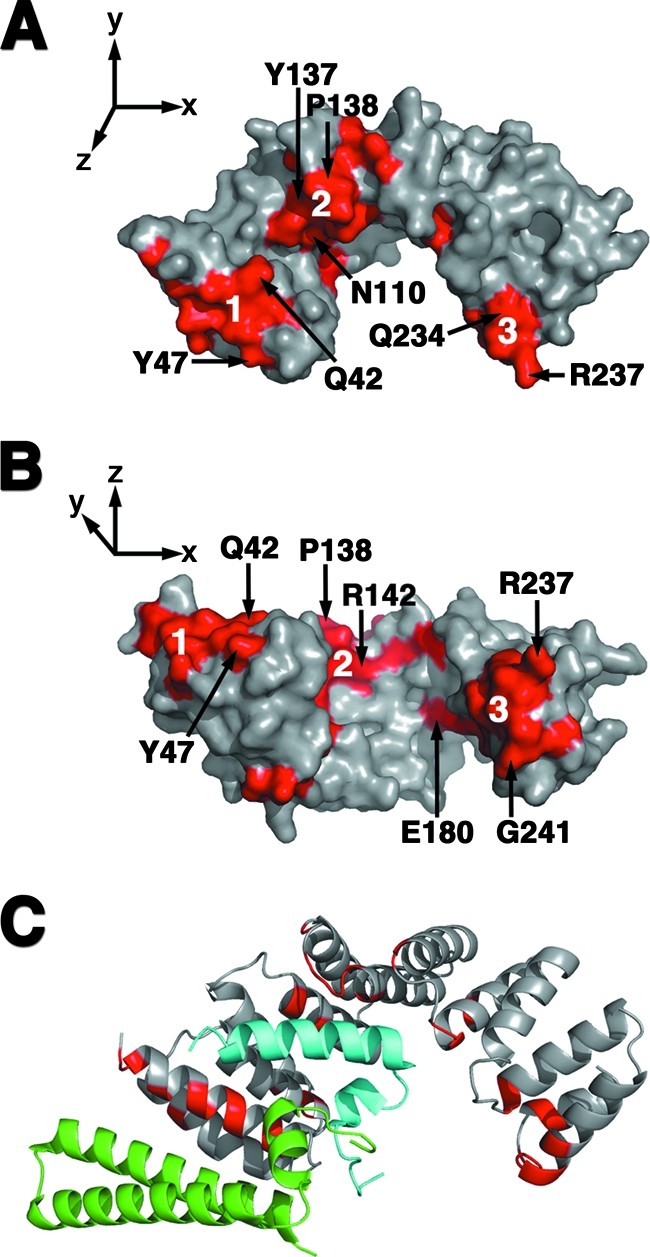
Potential PilF interaction interfaces. (A and B) The three conserved surfaces of PilF are highlighted in red on these surface representations of the PilF molecule. The molecule in panel B is drawn orthogonally to the representation in panel A. Representative residues from each conserved surface are labeled. The N-terminal, central groove, and C-terminal clusters are labeled 1, 2, and 3, respectively. (C) The substrates of PscG were overlaid on the monomer of PilF after alignment of the PscG and PilF TPR motifs. PilF is drawn in the same orientation as in Panel A. Residues that belong to the three conserved clusters on the surface of PilF are shown in red on the ribbon diagram. The substrates PscE and PscF are colored in green and light blue, respectively.
DISCUSSION
While over 50 proteins are involved in the synthesis, function, and regulation of P. aeruginosa T4P, PilF belongs to a subset of proteins that are essential for pilus biogenesis. Mutation of pilF in P. aeruginosa resulted in a strain that lacks surface piliation (65), but the mechanism underlying this phenotype was not identified. In this study, we have demonstrated that PilF is an outer membrane protein required for the multimerization and outer membrane localization of PilQ, the secretin oligomer through which the pilus extends out of the cell. Our results are in sharp contrast to the inner membrane localization and cytoplasmic function of PilF proposed by Kim et al. (27) but are consistent with the results of studies of the PilF orthologues PilW (8) and Tgl (48). Indeed, even in the absence of its lipidation site, PilF is exported through the inner membrane. Unlike E. coli, where an aspartic acid residue at position +2 relative to the lipidated Cys at position +1 is the main signal for inner membrane retention (57), a definitive pattern has not yet been delineated for P. aeruginosa. Narita and Tokuda (39) have proposed that Lys and Ser at positions +3 and +4, respectively, determine the inner membrane localization of lipoproteins in P. aeruginosa but have also identified exceptions to this rule given the low sequence conservation of residues at these two positions. As PilF has Val, Thr, and Ser at positions +2 to +4, respectively, a Ser at position 4 is not sufficient for inner membrane retention, suggesting that it is the combination of residues at these positions which determines protein localization within P. aeruginosa membranes.
We have shown that outer membrane tethering of PilF by lipidation promotes the transport of PilQ monomers from the inner membrane and the formation of highly stable multimers in the outer membrane. In the absence of pilF, PilQ monomers are retained in the inner membrane, do not form any stable secretins, and thus, prevent surface display of T4P. This finding contradicts those in Neisseria, where the deletion of pilW resulted in the loss of PilQ multimers but the PilQ monomers were still localized to the outer membrane (8). These data, coupled with the observation that surface piliation is restored in a pilW pilT double mutant (8), suggest a role for PilW in the multimerization and stabilization of the PilQ secretin, rather than in its localization as observed for PilF. Our data are more consistent with the results of studies of the type III secretins MxiD from Shigella flexneri (51) and YscC from Yersinia enterocolitica (6), which require the pilot proteins MxiM and YscW, respectively, to ensure their proper localization to and oligomerization in the outer membrane. As the assembly and/or outer membrane localization of secretins are functions that have been ascribed to lipoproteins referred to as “pilotins” (12, 53, 70), our results suggest that PilF assumes the pilotin role in the P. aeruginosa T4P system.
The outer membrane tethering of PilF appears to be important, but not essential, for T4P biogenesis. In contrast, lack of lipidation of the outer membrane secretin chaperones in Klebsiella general secretion (21), Erwinia type II secretion (53), and Yersinia type III secretion (6) abolishes function of the system in all of these by preventing outer membrane localization of the secretin. While some PilQ multimers are still present in the outer membrane of the C18G mutant, significantly fewer T4P are produced on the cell surface. Although detachment from the outer membrane does not affect PilF's ability to multimerize PilQ, the phenotype of the C18G mutant suggests that tethering via its lipid anchor promotes the correct localization of PilQ and proper orientation of the assembled secretins, which in turn permits the optimal display of pili on the cell surface. This hypothesis is consistent with the phenotype of the Neisseria pilW pilT double mutant, in which the T4P that were surface exposed were not functionally equivalent to wild-type T4P (8).
Complementation with the C18G mutant of PilF led to the assembly of some PilQ multimers in the inner membrane. If these inner membrane secretins are wild-type dodecamers, the central pore would have to be closed for the cells to be viable. Secretins purified from N. meningitidis have previously been observed to be closed by cryoelectron microscopy (11), implying that the opening of the secretin needs to be induced by specific interactions with other T4P protein(s) and/or with the pilus itself. In contrast, inner membrane insertion and oligomerization of the Klebsiella oxytoca secretin PulD, involved in type 2 secretion, has been observed in the absence of the PulS protein. In a pulS mutant, PulD forms open inner membrane oligomers, which causes leakage of cytoplasmic contents and induces the phage shock response (18). Interestingly, no phage shock response was induced when PulD (minus its signal sequence) was expressed in E. coli, even though it formed multimers in the inner membrane (17).
The functional unit of PilF in the cell has not yet been determined, but the observed crystallographic dimer is unlikely to be biologically relevant as the putative lipidation sites are found on opposite faces, which would preclude their simultaneous binding to the outer membrane. The contact surface between the PilF molecules buries an average area of 699.2 Å2 per molecule, less than the minimum of 856 Å2 found in 85% of protein dimers (44). Six of the 24 residues in molecule A and 5 of 22 residues in monomer B found at the interface are hydrophobic in nature and account for 14.1% and 22.3%, respectively, of the buried surface area. The remaining residues at the interface are polar or charged and form 15 hydrogen bonds and seven salt bridges between the two monomers. Analysis of the contact region by using PISA (28) suggests that it does not have the characteristics of a biological interaction surface. Our proposal that PilF functions as a monomer is consistent with the observation that PilW crystallizes with a monomer in the asymmetric unit (62).
The structure of PilF shows a superhelical arrangement of six TPR motifs, motifs often implicated in protein-protein interactions within multiprotein complexes (13, 34). While TPR motifs in eukaryotes have been extensively studied, a comparison with those in prokaryotes has yet to be undertaken as the structure of the motif is less well characterized. A structural alignment of the individual TPR motifs in PilF with those found in the eukaryotic synthetic consensus TPR motif (26) and the extensively studied TPR-containing proteins HOP (50), cyclophilin 40 (56), PP5 (69), and OGT (25) yields an average Cα root-mean-square deviation of 1.1 Å with similar positions involved in interhelical packing. Thus, the structure of PilF supports the hypothesis that the TPR structure is similar in both prokaryotic and eukaryotic TPRs, implying a shared evolutionary origin for this motif.
Kim et al. (27) identified PilF's central groove as the likely site of protein-protein interaction, and in particular, they implicated residues N110, N146, and E180. Trindade and colleagues' analysis of PilW identified the same residues, as well as residues N109 and Y137, as being potentially important for mediating protein-protein interactions (62). Our sequence alignments support the concept that these residues may play a role in PilF function, as N109, N110, Y137, and E180 are absolutely conserved in pseudomonads, while N146 is replaced by a Ser in P. syringae. However, examination of the location and solvent accessibility of these residues in our PilF structure suggests that N109 is not likely to be directly involved in protein-protein interactions as it is more than 90% buried. Interestingly, the results of more-recent studies of TPR-containing proteins have shown that both the inner concave and the outer convex surfaces may be involved in protein-protein interactions with different partners (14, 45). In addition to residues in extended conformations, TPRs have also been found to interact with regions of proteins containing defined secondary structure (45, 71). Therefore, it is possible that other surfaces of PilF, as well as the central concave groove, may be involved in binding either structured or extended regions of its partner(s). A sequence alignment of PilF in pseudomonads reveals three main clusters of conserved residues on the protein surface that are not involved in TPR packing (Fig. 6A and B). In agreement with the results of previous studies, residues N110, Y137, and E180 are located in the cluster of residues we identify within the concave groove of PilF (Fig. 6A and B, region 2). Reinforcing the importance of these regions, InterProSurf (40) and Sharp2 (38) predict them to be potential sites of protein-protein interaction based their high interface propensities. Furthermore, a structural alignment of PilF with the type 3 secretion chaperone (45) PscG shows that its binding partners, PscE and PscF, are easily accommodated by PilF and that, remarkably, the PscE and PscF binding sites overlap with two of the three highly conserved surfaces of PilF (Fig. 6C). In combination, these results strongly suggest that the residues that comprise these surfaces are involved in PilF function, and targeted mutagenesis to verify their importance is under way.
In conclusion, we have demonstrated that PilF is the outer membrane lipoprotein responsible for the multimerization and outer membrane localization of the PilQ secretin in P. aeruginosa T4P. Three surfaces that meet the criteria required for protein-protein interactions, conserved across pseudomonads, have been identified on our structure of PilF. We propose that these surfaces bind to structured and/or extended regions of PilF's partner(s) in the T4P system.
Acknowledgments
We thank Patrick Yip for technical assistance; Dante Neculai, Martin McMillan, and G. David Smith for help with various aspects of the data collection, structure determination, and validation; Robert Hancock for the gift of the OprF monoclonal antibody; and the Advanced Protein Technology Centre at The Hospital for Sick Children for assistance with DNA sequencing and mass spectrometry.
P.L.H. and L.L.B are the recipients of a Canada Research Chair and a Canadian Institute of Health Research (CIHR) New Investigator award, respectively. J.K., S.T., and S.-Y.K. were funded, in part, by graduate scholarships from the Natural Science and Engineering Research Council of Canada, the Canadian Cystic Fibrosis Foundation, CIHR, the Ontario Graduate Scholarship program, Ontario Student Opportunities Trust Fund, and The Hospital for Sick Children Foundation Student Scholarship program. L.M.S. was funded by a fellowship from the CIHR Strategic Training Program in Membrane Proteins Associated with Disease. Station X12-C at the National Synchrotron Light Source, Brookhaven National Laboratory, is supported by the United States Department of Energy.
Footnotes
Published ahead of print on 5 September 2008.
REFERENCES
- 1.Alm, R. A., and J. S. Mattick. 1996. Identification of two genes with prepilin-like leader sequences involved in type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J. Bacteriol. 1783809-3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blessing, B. H. 1987. Data reduction and error analysis for accurate single crystal diffraction intensities. Crystallogr. Rev. 13-58. [Google Scholar]
- 3.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72248-254. [DOI] [PubMed] [Google Scholar]
- 4.Bradley, D. E. 1974. The adsorption of Pseudomonas aeruginosa pilus-dependent bacteriophages to a host mutant with nonretractile pili. Virology 58149-163. [DOI] [PubMed] [Google Scholar]
- 5.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54905-921. [DOI] [PubMed] [Google Scholar]
- 6.Burghout, P., F. Beckers, E. de Wit, R. van Boxtel, G. R. Cornelis, J. Tommassen, and M. Koster. 2004. Role of the pilot protein YscW in the biogenesis of the YscC secretin in Yersinia enterocolitica. J. Bacteriol. 1865366-5375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burrows, L. L. 2005. Weapons of mass retraction. Mol. Microbiol. 57878-888. [DOI] [PubMed] [Google Scholar]
- 8.Carbonnelle, E., S. Helaine, L. Prouvensier, X. Nassif, and V. Pelicic. 2005. Type IV pilus biogenesis in Neisseria meningitidis: PilW is involved in a step occurring after pilus assembly, essential for fibre stability and function. Mol. Microbiol. 5554-64. [DOI] [PubMed] [Google Scholar]
- 9.Castric, P. 1995. pilO, a gene required for glycosylation of Pseudomonas aeruginosa 1244 pilin. Microbiology 1411247-1254. [DOI] [PubMed] [Google Scholar]
- 10.Chiang, P., and L. L. Burrows. 2003. Biofilm formation by hyperpiliated mutants of Pseudomonas aeruginosa. J. Bacteriol. 1852374-2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins, R. F., S. A. Frye, A. Kitmitto, R. C. Ford, T. Tonjum, and J. P. Derrick. 2004. Structure of the Neisseria meningitidis outer membrane PilQ secretin complex at 12 A resolution. J. Biol. Chem. 27939750-39756. [DOI] [PubMed] [Google Scholar]
- 12.Daefler, S., I. Guilvout, K. R. Hardie, A. P. Pugsley, and M. Russel. 1997. The C-terminal domain of the secretin PulD contains the binding site for its cognate chaperone, PulS, and confers PulS dependence on pIVf1 function. Mol. Microbiol. 24465-475. [DOI] [PubMed] [Google Scholar]
- 13.D'Andrea, L. D., and L. Regan. 2003. TPR proteins: the versatile helix. Trends Biochem. Sci. 28655-662. [DOI] [PubMed] [Google Scholar]
- 14.Edqvist, P. J., J. E. Broms, H. J. Betts, A. Forsberg, M. J. Pallen, and M. S. Francis. 2006. Tetratricopeptide repeats in the type III secretion chaperone, LcrH: their role in substrate binding and secretion. Mol. Microbiol. 5931-44. [DOI] [PubMed] [Google Scholar]
- 15.Emsley, P., and K. Cowtan. 2004. COOT: model-building tools for molecular graphics. Acta Crystallogr. D 602126-2132. [DOI] [PubMed] [Google Scholar]
- 16.Glessner, A., R. S. Smith, B. H. Iglewski, and J. B. Robinson. 1999. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of twitching motility. J. Bacteriol. 1811623-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guilvout, I., M. Chami, C. Berrier, A. Ghazi, A. Engel, A. P. Pugsley, and N. Bayan. 2008. In vitro multimerization and membrane insertion of bacterial outer membrane secretin PulD. J. Mol. Biol. 38213-23. [DOI] [PubMed] [Google Scholar]
- 18.Guilvout, I., M. Chami, A. Engel, A. P. Pugsley, and N. Bayan. 2006. Bacterial outer membrane secretin PulD assembles and inserts into the inner membrane in the absence of its pilotin. EMBO J. 255241-5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn, H. P. 1997. The type-4 pilus is the major virulence-associated adhesin of Pseudomonas aeruginosa: a review. Gene 19299-108. [DOI] [PubMed] [Google Scholar]
- 20.Hancock, R. E., and H. Nikaido. 1978. Outer membranes of gram-negative bacteria. XIX. Isolation from Pseudomonas aeruginosa PAO1 and use in reconstitution and definition of the permeability barrier. J. Bacteriol. 136381-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardie, K. R., A. Seydel, I. Guilvout, and A. P. Pugsley. 1996. The secretin-specific, chaperone-like protein of the general secretory pathway: separation of proteolytic protection and piloting functions. Mol. Microbiol. 22967-976. [DOI] [PubMed] [Google Scholar]
- 22.Holm, L., and J. Park. 2000. DaliLite workbench for protein structure comparison. Bioinformatics 16566-567. [DOI] [PubMed] [Google Scholar]
- 23.Holm, L., and C. Sander. 1996. Mapping the protein universe. Science 273595-603. [DOI] [PubMed] [Google Scholar]
- 24.Jacobs, M. A., A. Alwood, I. Thaipisuttikul, D. Spencer, E. Haugen, S. Ernst, O. Will, R. Kaul, C. Raymond, R. Levy, L. Chun-Rong, D. Guenthner, D. Bovee, M. V. Olson, and C. Manoil. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 10014339-14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jinek, M., J. Rehwinkel, B. D. Lazarus, E. Izaurralde, J. A. Hanover, and E. Conti. 2004. The superhelical TPR-repeat domain of O-linked GlcNAc transferase exhibits structural similarities to importin alpha. Nat. Struct. Mol. Biol. 111001-1007. [DOI] [PubMed] [Google Scholar]
- 26.Kajander, T., A. L. Cortajarena, S. Mochrie, and L. Regan. 2007. Structure and stability of designed TPR protein superhelices: unusual crystal packing and implications for natural TPR proteins. Acta Crystallogr. D 63800-811. [DOI] [PubMed] [Google Scholar]
- 27.Kim, K., J. Oh, D. Han, E. E. Kim, B. Lee, and Y. Kim. 2006. Crystal structure of PilF: functional implication in the type 4 pilus biogenesis in Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 3401028-1038. [DOI] [PubMed] [Google Scholar]
- 28.Krissinel, E., and K. Henrick. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372774-797. [DOI] [PubMed] [Google Scholar]
- 29.Lapouge, K., S. J. Smith, P. A. Walker, S. J. Gamblin, S. J. Smerdon, and K. Rittinger. 2000. Structure of the TPR domain of p67phox in complex with Rac.GTP. Mol. Cell 6899-907. [DOI] [PubMed] [Google Scholar]
- 30.Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson, and D. G. Higgins. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 232947-2948. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26283-291. [Google Scholar]
- 32.Lee, J. E., K. A. Cornell, M. K. Riscoe, and P. L. Howell. 2001. Structure of E. coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase reveals similarity to the purine nucleoside phosphorylases. Structure 9941-953. [DOI] [PubMed] [Google Scholar]
- 33.Maier, B. 2005. Using laser tweezers to measure twitching motility in Neisseria. Curr. Opin. Microbiol. 8344-349. [DOI] [PubMed] [Google Scholar]
- 34.Main, E. R., Y. Xiong, M. J. Cocco, L. D'Andrea, and L. Regan. 2003. Design of stable alpha-helical arrays from an idealized TPR motif. Structure 11497-508. [DOI] [PubMed] [Google Scholar]
- 35.Mattick, J. S. 2002. Type IV pili and twitching motility. Annu. Rev. Microbiol. 56289-314. [DOI] [PubMed] [Google Scholar]
- 36.Merz, A. J., and M. So. 2000. Interactions of pathogenic neisseriae with epithelial cell membranes. Annu. Rev. Cell Dev. Biol. 16423-457. [DOI] [PubMed] [Google Scholar]
- 37.Miller, S., J. Janin, A. M. Lesk, and C. Chothia. 1987. Interior and surface of monomeric proteins. J. Mol. Biol. 196641-656. [DOI] [PubMed] [Google Scholar]
- 38.Murakami, Y., and S. Jones. 2006. SHARP2: protein-protein interaction predictions using patch analysis. Bioinformatics 221794-1795. [DOI] [PubMed] [Google Scholar]
- 39.Narita, S., and H. Tokuda. 2007. Amino acids at positions 3 and 4 determine the membrane specificity of Pseudomonas aeruginosa lipoproteins. J. Biol. Chem. 28213372-13378. [DOI] [PubMed] [Google Scholar]
- 40.Negi, S. S., C. H. Schein, N. Oezguen, T. D. Power, and W. Braun. 2007. InterProSurf: a web server for predicting interacting sites on protein surfaces. Bioinformatics 233397-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nudleman, E., D. Wall, and D. Kaiser. 2005. Cell-to-cell transfer of bacterial outer membrane lipoproteins. Science 309125-127. [DOI] [PubMed] [Google Scholar]
- 42.Perrakis, A., R. Morris, and V. S. Lamzin. 1999. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6458-463. [DOI] [PubMed] [Google Scholar]
- 43.Pflugrath, J. W. 1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D 551718-1725. [DOI] [PubMed] [Google Scholar]
- 44.Ponstingl, H., K. Henrick, and J. M. Thornton. 2000. Discriminating between homodimeric and monomeric proteins in the crystalline state. Proteins 4147-57. [DOI] [PubMed] [Google Scholar]
- 45.Quinaud, M., S. Ple, V. Job, C. Contreras-Martel, J. P. Simorre, I. Attree, and A. Dessen. 2007. Structure of the heterotrimeric complex that regulates type III secretion needle formation. Proc. Natl. Acad. Sci. USA 1047803-7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rakotoarivonina, H., G. Jubelin, M. Hebraud, B. Gaillard-Martinie, E. Forano, and P. Mosoni. 2002. Adhesion to cellulose of the gram-positive bacterium Ruminococcus albus involves type IV pili. Microbiology 1481871-1880. [DOI] [PubMed] [Google Scholar]
- 47.Read, R. J. 1986. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A 42140-149. [Google Scholar]
- 48.Rodriguez-Soto, J. P., and D. Kaiser. 1997. Identification and localization of the Tgl protein, which is required for Myxococcus xanthus social motility. J. Bacteriol. 1794372-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salamitou, S., M. Lemaire, T. Fujino, H. Ohayon, P. Gounon, P. Beguin, and J. P. Aubert. 1994. Subcellular localization of Clostridium thermocellum ORF3p, a protein carrying a receptor for the docking sequence borne by the catalytic components of the cellulosome. J. Bacteriol. 1762828-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scheufler, C., A. Brinker, G. Bourenkov, S. Pegoraro, L. Moroder, H. Bartunik, F. U. Hartl, and I. Moarefi. 2000. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 101199-210. [DOI] [PubMed] [Google Scholar]
- 51.Schuch, R., and A. T. Maurelli. 2001. MxiM and MxiJ, base elements of the Mxi-Spa type III secretion system of Shigella, interact with and stabilize the MxiD secretin in the cell envelope. J. Bacteriol. 1836991-6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semmler, A. B., C. B. Whitchurch, and J. S. Mattick. 1999. A re-examination of twitching motility in Pseudomonas aeruginosa. Microbiology 1452863-2873. [DOI] [PubMed] [Google Scholar]
- 53.Shevchik, V. E., and G. Condemine. 1998. Functional characterization of the Erwinia chrysanthemi OutS protein, an element of a type II secretion system. Microbiology 1443219-3228. [DOI] [PubMed] [Google Scholar]
- 54.Shindyalov, I. N., and P. E. Bourne. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11739-747. [DOI] [PubMed] [Google Scholar]
- 55.Sun, P., J. E. Tropea, B. P. Austin, S. Cherry, and D. S. Waugh. 2008. Structural characterization of the Yersinia pestis type III secretion system needle protein YscF in complex with its heterodimeric chaperone YscE/YscG. J. Mol. Biol. 377819-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor, P., J. Dornan, A. Carrello, R. F. Minchin, T. Ratajczak, and M. D. Walkinshaw. 2001. Two structures of cyclophilin 40: folding and fidelity in the TPR domains. Structure 9431-438. [DOI] [PubMed] [Google Scholar]
- 57.Terada, M., T. Kuroda, S. I. Matsuyama, and H. Tokuda. 2001. Lipoprotein sorting signals evaluated as the LolA-dependent release of lipoproteins from the cytoplasmic membrane of Escherichia coli. J. Biol. Chem. 27647690-47694. [DOI] [PubMed] [Google Scholar]
- 58.Terwilliger, T. C. 2003. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D 5938-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thelin, K. H., and R. K. Taylor. 1996. Toxin-coregulated pilus, but not mannose-sensitive hemagglutinin, is required for colonization by Vibrio cholerae O1 El Tor biotype and O139 strains. Infect. Immun. 642853-2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tobe, T., and C. Sasakawa. 2002. Species-specific cell adhesion of enteropathogenic Escherichia coli is mediated by type IV bundle-forming pili. Cell. Microbiol. 429-42. [DOI] [PubMed] [Google Scholar]
- 61.Touhami, A., M. H. Jericho, J. M. Boyd, and T. J. Beveridge. 2006. Nanoscale characterization and determination of adhesion forces of Pseudomonas aeruginosa pili by using atomic force microscopy. J. Bacteriol. 188370-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trindade, M. B., V. Job, C. Contreras-Martel, V. Pelicic, and A. Dessen. 2008. Structure of a widely conserved type IV pilus biogenesis factor that affects the stability of secretin multimers. J. Mol. Biol. 3781031-1039. [DOI] [PubMed] [Google Scholar]
- 63.van Schaik, E. J., C. L. Giltner, G. F. Audette, D. W. Keizer, D. L. Bautista, C. M. Slupsky, B. D. Sykes, and R. T. Irvin. 2005. DNA binding: a novel function of Pseudomonas aeruginosa type IV pili. J. Bacteriol. 1871455-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Varga, J. J., V. Nguyen, D. K. O'Brien, K. Rodgers, R. A. Walker, and S. B. Melville. 2006. Type IV pili-dependent gliding motility in the gram-positive pathogen Clostridium perfringens and other clostridia. Mol. Microbiol. 62680-694. [DOI] [PubMed] [Google Scholar]
- 65.Watson, A. A., R. A. Alm, and J. S. Mattick. 1996. Identification of a gene, pilF, required for type 4 fimbrial biogenesis and twitching motility in Pseudomonas aeruginosa. Gene 18049-56. [DOI] [PubMed] [Google Scholar]
- 66.Woodruff, W. A., and R. E. Hancock. 1988. Construction and characterization of Pseudomonas aeruginosa protein F-deficient mutants after in vitro and in vivo insertion mutagenesis of the cloned gene. J. Bacteriol. 1702592-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu, B., P. Li, Y. Liu, Z. Lou, Y. Ding, C. Shu, S. Ye, M. Bartlam, B. Shen, and Z. Rao. 2004. 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc. Natl. Acad. Sci. USA 1018348-8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu, H., C. M. Weeks, and H. A. Hauptman. 2005. Optimizing statistical Shake-and-Bake for Se-atom substructure determination. Acta Crystallogr. D 61976-981. [DOI] [PubMed] [Google Scholar]
- 69.Yang, J., S. M. Roe, M. J. Cliff, M. A. Williams, J. E. Ladbury, P. T. Cohen, and D. Barford. 2005. Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. EMBO J. 241-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zenk, S. F., D. Stabat, J. L. Hodgkinson, A. K. Veenendaal, S. Johnson, and A. J. Blocker. 2007. Identification of minor inner-membrane components of the Shigella type III secretion system “needle complex.” Microbiology 1532405-2415. [DOI] [PubMed] [Google Scholar]
- 71.Zhang, Y., and D. C. Chan. 2007. Structural basis for recruitment of mitochondrial fission complexes by Fis1. Proc. Natl. Acad. Sci. USA 10418526-18530. [DOI] [PMC free article] [PubMed] [Google Scholar]