

Abstract
In many bacteria glucose is the preferred carbon source and represses the utilization of secondary substrates. In Bacillus subtilis, this carbon catabolite repression (CCR) is achieved by the global transcription regulator CcpA, whose activity is triggered by the availability of its phosphorylated cofactors, HPr(Ser46-P) and Crh(Ser46-P). Phosphorylation of these proteins is catalyzed by the metabolite-controlled kinase HPrK/P. Recent studies have focused on glucose as a repressing substrate. Here, we show that many carbohydrates cause CCR. The substrates form a hierarchy in their ability to exert repression via the CcpA-mediated CCR pathway. Of the two cofactors, HPr is sufficient for complete CCR. In contrast, Crh cannot substitute for HPr on substrates that cause a strong repression. Determination of the phosphorylation state of HPr in vivo revealed a correlation between the strength of repression and the degree of phosphorylation of HPr at Ser46. Sugars transported by the phosphotransferase system (PTS) cause the strongest repression. However, the phosphorylation state of HPr at its His15 residue and PTS transport activity have no impact on the global CCR mechanism, which is a major difference compared to the mechanism operative in Escherichia coli. Our data suggest that the hierarchy in CCR exerted by the different substrates is exclusively determined by the activity of HPrK/P.
As for any organism, nutrient supply is of prime importance for bacteria. In their natural habitats, bacteria often encounter a mixture of different carbon sources that can potentially be used. Therefore, mechanisms have evolved in many bacteria that enable the selective uptake and metabolism of carbon sources that allow the most rapid growth and that promise the best success in the competition with other bacteria or fungi. For many heterotrophic bacteria, glucose is the preferred source of carbon. In the presence of glucose, the genes required for the utilization of secondary carbon sources are not expressed and preexisting enzymes are often inactivated to prevent the waste of resources. This phenomenon is referred to as carbon catabolite repression (CCR; for reviews, see references 6 and 15).
CCR has been extensively studied in the model bacteria Bacillus subtilis and Escherichia coli. Although the physiological outcome is very similar, the global mechanisms underlying CCR are completely different in these bacteria. In E. coli and other enteric bacteria, the EIIAGlc domain of the glucose transporter is the central processing unit in CCR. This protein is part of the phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS), which is responsible for the uptake and concomitant phosphorylation of numerous carbohydrates in many bacteria (7). In this system, the two general phosphotransferases, enzyme I (EI) and histidine protein (HPr), transfer phosphoryl groups from PEP to the various sugar transporters, named enzymes II (EIIs). In the absence of glucose, the EIIAGlc domain is preferentially phosphorylated. In this form, EIIAGlc activates the adenylate cyclase, which leads to an increase in the intracellular cAMP concentration. Binding of cyclic AMP (cAMP) activates the transcription activator CRP (cAMP receptor protein), which is in turn required for the expression of numerous secondary catabolic genes. In the presence of glucose, EIIAGlc is predominantly unphosphorylated and therefore unable to activate adenylate cyclase. In addition, unphosphorylated EIIAGlc inhibits transporters of secondary carbon sources by direct interaction. This operon-specific mechanism, which has been termed inducer exclusion, contributes to the repression of catabolic genes in the presence of glucose. In some cases, e.g., the E. coli lac operon, inducer exclusion might even be the decisive mechanism for CCR (15).
A different mechanism of CCR is operative in the gram-positive soil bacterium B. subtilis and other Firmicutes. Here, the global mechanism of CCR is mediated by the pleiotropic transcription factor CcpA (for reviews, see references 10 and 50). In the presence of glucose, CcpA represses several hundred catabolic genes and activates the transcription of some genes of overflow metabolism (3, 28, 34, 51). The ability of CcpA to bind its target sites, the catabolite responsive elements (cre), is in turn controlled by the presence of its cofactors, HPr(Ser-P) and Crh(Ser-P) (11, 44, 45). Unlike HPr in E. coli, the B. subtilis homolog contains a regulatory phosphorylation site, Ser46, in addition to His15, which is phosphorylated during phosphate transfer to the transported sugar. Crh is homologous to HPr but it lacks His15 and is therefore unable to participate in sugar transport (12). In the presence of glucose, HPr, and presumably also Crh, is phosphorylated by the bifunctional HPr kinase/phosphorylase (HPrK/P) on Ser46, whereas this site is less phosphorylated in cells growing in the absence of sugars (27, 32). Dephosphorylation of HPr(Ser-P) is catalyzed by the phosphorylase activity of HPrK/P (31) and, to some extent, by the protein phosphatase PrpC (48). In vitro experiments suggested that the two antagonistic activities of HPrK/P are regulated by metabolites. High fructose 1,6-bisphosphate (FBP) and ATP or PPi concentrations stimulate the kinase activity, whereas the phosphorylase activity prevails when the Pi concentration is high (20, 37, 39). Only HPr(Ser-P), but none of the other HPr forms, is able to productively interact with CcpA and to exert CCR (38, 44). In vitro experiments suggested that in addition to its role in the activation of HPrK kinase, FBP enhances DNA binding of the CcpA/HPr(Ser-P) complex by directly binding to CcpA (46, 47). The role of Crh in CCR is still unclear.
Traditionally, studies dealing with CCR have been focused on the repression exerted by glucose. Therefore, the terms carbon catabolite repression and glucose repression are often used synonymously. However, for the gram-negative bacterium E. coli it was shown that a large number of carbohydrates in addition to glucose exert CCR (2, 18). Much less is known in B. subtilis. Thus far, CCR exerted by carbon sources different from glucose has not been systematically studied. In the present study, we analyzed CcpA-mediated CCR exerted by a variety of carbon sources in B. subtilis. As a model for the present study, we chose the xynPB operon, which encodes a β-xyloside transporter and β-xylosidase. These functions allow the uptake and degradation of β-xylosides to xylose (Fig. 1) (11, 26). Xylose is subsequently converted to xylulose-5-P by enzymes encoded in the xylAB operon located downstream of the xynPB operon (Fig. 1). Both operons are repressed by the XylR protein, which binds to operator sites present in the respective promoter regions and prevents transcription initiation (4, 11). Binding of xylose releases XylR from its operator sites. B. subtilis is not able to grow on xylose due to the lack of a xylose-specific permease (26, 42, 43). However, xylose can be slowly taken up by the AraE protein, which is sufficient for induction of xynPB and xylAB expression (22). In the presence of glucose, both operons are strongly repressed. Downstream of their promoters, cre sites are present, which are bound by the HPr-Ser46-P/CcpA complex, thereby preventing transcription initiation (11, 21).
FIG. 1.
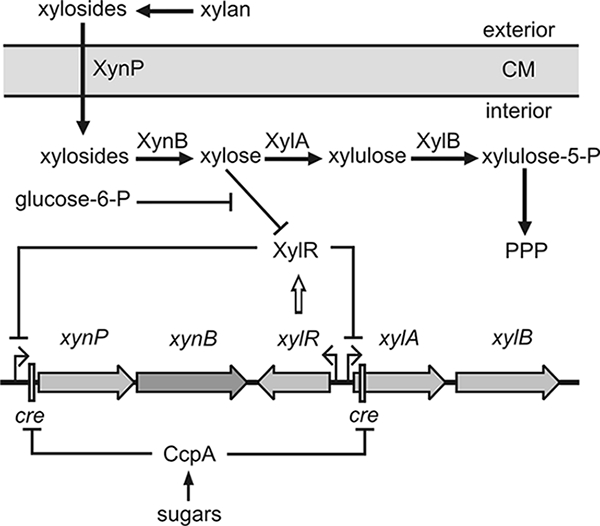
Utilization of β-xylan in B. subtilis. Xylan can be degraded by an extracellular xylanase to β-xylosides, which are subsequently taken up by the β-xyloside transporter XynP encoded in the bicistronic xynPB operon. The β-xylosidase XynB converts β-xylosides to xylose, which is further converted to xylulose-5-P by the functions encoded in the adjacent xylAB operon. Xylulose-5-P finally enters the pentose phosphate pathway (PPP). The xynPB and xylAB operons are repressed by binding of the Xyl repressor (XylR) to operator sites in the absence of the inducer xylose. Both operons are subject to global CCR, which is mediated by binding of CcpA to cre sites downstream of the promoters. In addition, glucose-6-phosphate contributes to CCR by acting as an anti-inducer for XylR.
We show here that a variety of carbohydrates represses xynPB transcription via the CcpA-mediated CCR pathway, but each to a different degree. In general, most substrates transported by the PTS caused a strong repression, whereas CCR by non-PTS substrates was weaker. The Crh protein was completely dispensable for repression exerted by all of these substrates, whereas HPr was essential for the strong CCR caused by sugars such as glucose, fructose, and mannitol. Analysis of the HPr phosphorylation state in vivo revealed that the strength of repression exerted by a particular substrate correlates well with the amount of HPr(Ser-P) in the cell. However, in contrast to E. coli, transport activity of the PTS has no direct role for the global CCR mechanism. Our data suggest that in B. subtilis the strength of CcpA-mediated CCR is determined exclusively by the metabolite-controlled activity of HPrK/P.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The B. subtilis strains used in the present study are listed in Table 1. The presence of the ptsH1 mutation was verified by sequencing of chromosomal DNA of the relevant strains. E. coli DH5α (41) was used for plasmid propagation. B. subtilis was grown in CSE minimal medium supplemented with auxotrophic requirements (at 50 mg liter−1) (30). If necessary, xylose was added at a concentration of 0.2% (wt/vol). Potentially repressing carbon sources were used at a concentration of 0.5% (wt/vol). E. coli was grown in LB medium, and transformants were selected on plates containing ampicillin (100 μg ml−1). LB and SP plates were prepared by the addition of 17 g of Bacto agar (Difco) liter−1 to the medium. For the determination of growth rates, precultures were grown overnight in CE minimal medium supplemented with auxotrophic requirements (at 50 mg liter−1) and a single carbon source at a concentration of 0.5% (wt/vol). The cells were inoculated in the same medium to an optical density at 600 nm (OD600) of 0.1, the bacteria were incubated at 37°C at 200 rpm, and the turbidity at 600 nm was recorded periodically. The growth rate (μ) was determined from the exponential phases of the growth curves using the formula μ = (logb − logB)/(log2 × Δt), where Δt is the time interval in min, b is the OD600 of the culture at the end, and B is the OD600 at the beginning of this time interval.
TABLE 1.
B. subtilis strains used in this study
| Strain | Genotype | Source or referencea |
|---|---|---|
| 168 | trpC2 | Laboratory stock |
| GP270 | trpC2 xylR::ermC | pIW11xylR→168 |
| GP278 | trpC2 xylR::ermC amyE::(xynP-lacZ cat) | 48 |
| GP279 | trpC2 xylR::ermC crh::spc amyE::(xynP-lacZ cat) | QB7097→GP278 |
| GP284 | trpC2 xylR::ermC ptsH1 | pIW11xylR→QB5223 |
| GP287 | trpC2 xylR::ermC ptsH1 crh::spc | pIW11xylR→QB7101 |
| GP289 | trpC2 xylR::ermC hprK::aphA3 | QB7160→GP270 |
| GP297 | trpC2 xylR::ermC crh::spc | QB7097→GP270 |
| GP853 | trpC2 xylR::ermC ccpA::spc | QB5407→GP270 |
| GP858 | trpC2 ΔhprK::aphA3 | QB7160→168 |
| GP864 | trpC2 ΔptsI::ermC | pGP811→168 |
| QB5223 | trpC2 ptsH1 | 30 |
| QB5407 | trpC2 ccpA::spc | 9 |
| QB7097 | trpC2 crh::spc | I. Martin-Verstraete |
| QB7101 | trpC2 ptsH1 crh::spc | I. Martin-Verstraete |
| QB7144 | trpC2 amyE::(xynP-lacZ cat) | 11 |
| QB7160 | trpC2 amyE::(levD-lacZ cat) hprK::aphA3 | 29 |
Arrows indicate construction by transformation.
DNA manipulation.
Transformation of E. coli and plasmid DNA extraction were performed according to standard procedures (41). Restriction enzymes and DNA polymerases were used as recommended by the manufacturers. Chromosomal DNA of B. subtilis was isolated by using a DNeasy tissue kit (Qiagen) according to the supplier's protocol. Plasmid pIW11xylR, used for inactivation of the xylR gene, has been described previously (21). Plasmid pGP811, used for the disruption of ptsI, is a derivative of plasmid pHT181 (25) carrying the 591-bp EcoRI fragment of ptsI inserted in its unique EcoRI site. Plasmid pGP650 carries the hprK(G158A) allele under the control of the constitutive degQ36 promoter. The hprK allele was amplified by using the primers SK11 (GGCGGATCCGTGGCAAAGGTTCGCACAAAAGA) and KS12 (AAAAAGCTTGGTTCTATCGCTTCATTCATTTAACGC) and plasmid pGP407 (16) as a template. Subsequently, the PCR fragment was digested with BamHI and HindIII and inserted between the same sites of plasmid pGP380 (17).
Transformation and enzyme assays.
B. subtilis was transformed with plasmids and chromosomal DNA according to the two-step protocol (23). Transformants were selected on SP plates containing spectinomycin (100 μg ml−1), kanamycin (5 μg ml−1), or erythromycin and lincomycin (2 and 25 μg ml−1, respectively). For enzyme assays, cells were harvested in the exponential growth phase at an OD600 of 0.6 to 0.8. β-Galactosidase and β-xylosidase activities were determined in cell extracts by using o-nitrophenyl galactopyranoside and p-nitrophenyl xyloside as substrates, respectively (26, 41).
Western blot analysis.
For Western blot analyses, crude cell extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane (PVDF; Bio-Rad) by electroblotting. The proteins were detected with rabbit polyclonal antisera raised against CcpA, HPr, or HPrK/P of Bacillus megaterium or B. subtilis (16, 24, 32). The antibodies were visualized by using anti-rabbit immunoglobulin G-AP secondary antibodies (Chemikon International, Temecula, CA) and the CDP* detection system (Roche Diagnostics).
Analysis of the phosphorylation state of HPr in vivo.
HPr phosphorylation was assayed in vivo by Western blot analysis as follows. Bacteria were grown in CSE in the presence of the indicated carbon sources to an OD600 of 0.6. Cells were disrupted by using a French press, and crude extracts were prepared as described previously (27). Proteins (2-μg portions) were separated on nondenaturing 12% polyacrylamide gels. On these gels, phosphorylated HPr migrates faster than the nonphosphorylated protein. HPr(His-P) was dephosphorylated by incubation of the crude extract for 10 min at 70°C. After electrophoresis, the proteins were blotted onto a PVDF membrane. The different forms of HPr were detected by using antibodies directed against B. subtilis HPr (32).
Determination of fructose 1,6-bisphosphate concentrations.
Protein-free cell extracts for the determination of FBP concentrations in B. subtilis were prepared as described previously with few modifications (31). Briefly, cells of the B. subtilis wild-type strain 168 were grown in 50 ml of CSE medium in the presence of the indicated carbon sources. For each growth condition at least three independent experiments were carried out. Cultures were harvested by centrifugation at room temperature for 5 min at 10,000 × g, and pellets were subsequently frozen in liquid nitrogen. The pellets were resuspended in 0.6 M of cold perchloric acid and subsequently incubated on ice for 20 min. The precipitated proteins and cell debris were removed by centrifugation (4°C, 5 min, 13,000 rpm). The pH in the supernatant was adjusted to 7.4 with a solution of cold 0.6 M KOH in 100 mM Tris-HCl (pH 7.4). The precipitated KClO4 was removed by centrifugation. The FBP concentrations were determined in the supernatants as described previously (31).
RESULTS
CCR of β-xylosidase exerted by different carbon sources.
CCR has mainly been studied using glucose as repressing carbon source. To explore whether other carbon sources also exert CCR, we chose the xynPB operon as a model system. For this purpose, we measured the activity of the β-xylosidase XynB as a convenient reporter for regulatory events taking place at the xynPB promoter (see below). First, we tested the wild-type strain 168 in CSE minimal medium containing succinate as a carbon source. In the absence of xylose, only 14 U of β-xylosidase activity was detectable, whereas in the presence of xylose a high β-xylosidase activity of 945 U was measured (Table 2), reflecting the repression of the xyn operon by the XylR protein. Next, we added other carbon sources in addition to succinate and repeated the measurements. Xylose as an inducer for XylR was also included. In all cases, the XynB activities were reduced in comparison to the culture grown on succinate (Table 2). Succinate is known to cause no CCR in B. subtilis (3, 11). Glucose caused the strongest reduction of XynB activity (135-fold), a finding which is in agreement with previous data (11). The sugars salicin, glycerol, mannitol, and fructose also caused repression, but to a weaker extent (12- to 18-fold). Gluconate, sucrose, and sorbitol repressed XynB activity eightfold. Ribose, arabinose, and maltose had only a twofold effect. In conclusion, the substrates formed a hierarchy in their ability to exert CCR.
TABLE 2.
Catabolite repression of β-xylosidase by different carbon sources in various mutants
| Carbon sourcea | Enzyme activityb in U/mg of protein (SD) for strain:
|
||||||
|---|---|---|---|---|---|---|---|
| 168c (wild type) | GP270 (ΔxylR) | GP297 (ΔxylR Δcrh) | GP284 (ΔxylR ptsH1) | GP287 (ΔxylR Δcrh ptsH1) | GP289 (ΔxylR ΔhprK) | GP853 (ΔxylR ΔccpA) | |
| None (CSE) | 945 (281) | 1,585 (560) | 2,748 (540) | 2,590 (304) | 2,397 (214) | 2,287 (218) | 2,142 (384) |
| Ribose | 497 (138) | 939 (164) | 1,360 (169) | 971 (263) | 1,346 (151) | NG | 1,780 (454) |
| Arabinose | 414 (136) | 600 (153) | 876 (35) | 713 (100) | 1,498 (323) | 1,257 (70) | 1,437 (228) |
| Maltose | 437 (127) | 489 (32) | 488 (105) | 710 (81) | 2,226 (206) | 2,023 (408) | 2,078 (632) |
| Gluconate | 116 (11) | 201 (31) | 378 (64) | 244 (46) | 1,220 (179) | 1,163 (170) | 1,628 (554) |
| Sucrose | 126 (20) | 205 (12) | 203 (13) | 309 (88) | 2,770 (136) | 2,271 (292) | 2,858 (123) |
| Salicin | 54 (6) | 175 (19) | 167 (4) | 202 (37) | 2,850 (180) | 2,743 (667) | 2,049 (83) |
| Sorbitol | 114 (20) | 121 (29) | 130 (52) | 113 (19) | 732 (66) | 748 (61) | 734 (51) |
| Glycerol | 82 (15) | 96 (20) | 87 (6) | 135 (17) | 2,164 (110) | 1,273 (160) | 1,138 (127) |
| Mannitol | 72 (13) | 79 (21) | 78 (18) | 606 (190) | 2,080 (395) | 1,689 (269) | 1,265 (454) |
| Fructose | 60 (9) | 63 (5) | 63 (7) | 231 (75) | 2,045 (206) | 1,638 (32) | 1,889 (147) |
| Glucose | 7 (3) | 44 (10) | 60 (21) | 173 (56) | 2,077 (500) | 1,679 (421) | 1,570 (153) |
That is, added to the CSE medium.
The values are the average of at least three independent experiments. NG, no growth.
Xylose was added to induce xynPB expression.
To rule out that enzyme activity rather than synthesis of XynB was affected by the various carbon sources in these and the following experiments, we also performed similar experiments with isogenic strains carrying a transcriptional xynP′-lacZ fusion on the chromosome. The β-galactosidase activities in these strains correlated well with the corresponding β-xylosidase activities (Fig. 2 and data not shown). Somewhat lower repression factors were obtained in the LacZ assays compared to the XynB measurements, making the latter the more sensitive tool to measure CCR. In conclusion, the observed differences of XynB activities in the present study reflect regulatory events taking place at the xynPB promoter.
FIG. 2.
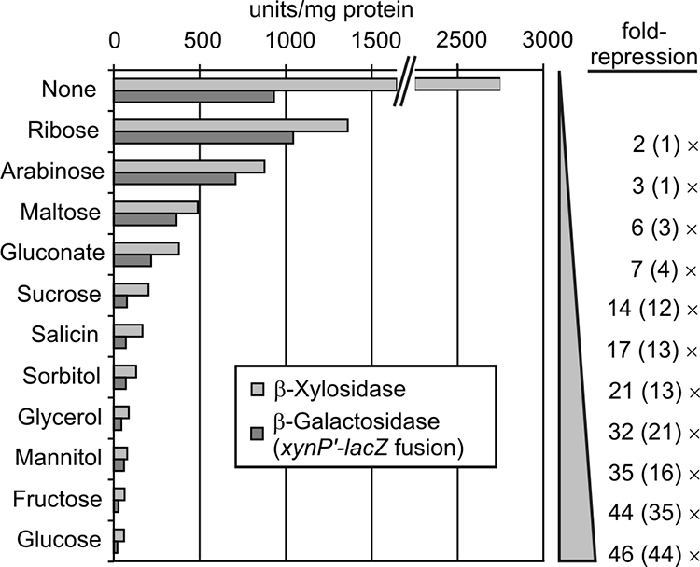
Various carbohydrates repress the xynPB operon via the global CCR pathway. Shown are the β-xylosidase activities in the ΔxylR Δcrh mutant GP297 (light gray bars), in which CCR exclusively relies on the activities of HPrK/P, CcpA, and HPr. To demonstrate that the β-xylosidase activities reflect transcription from the xynPB promoter, the isogenic strain GP279 was used, which carries in addition a xynP′-lacZ fusion on the chromosome. The β-galactosidase activities produced in this strain are shown for comparison (dark gray bars). The repression factors relative to the activities determined in pure CSE medium (None) are depicted at the right.
The XylR repressor protein contributes to CCR of xynPB expression exclusively in the presence of glucose.
As for many other systems, the xynPB operon underlies an operon-specific mechanism of CCR in addition to the general CCR pathway via CcpA. The XylR repressor also contributes to glucose repression of its target genes (5, 21). Uptake of glucose generates glucose-6-phosphate, which binds to XylR and counteracts binding of the inducer xylose (Fig. 1). In principle, the repression of XynB activity by the other carbohydrates may also result from the combined activities of both mechanisms of CCR. To analyze this possible interference, we repeated our experiments with a ΔxylR mutant. As expected, the xylR mutation caused complete derepression of xynPB expression, i.e., high XynB activities were already detectable in the absence of the inducer xylose (Table 2). With respect to the available carbon sources, the enzyme activities followed a similar order as in the xylose-induced wild-type strain (Table 2, compare columns 1 and 2). In most cases, the activities were slightly higher in the ΔxylR mutant, which can be explained by incomplete derepression in the wild-type strain. In contrast, there was a sixfold relief from repression when the cells grew on glucose, which reflects the extra repression exerted by glucose-6-P. In conclusion, XylR only contributes to CCR of xynPB expression when glucose is utilized and is not relevant for CCR elicited by the other carbon sources.
In the ΔxylR mutant, CCR of β-xylosidase completely depends on the general pathway of CCR.
In the ΔxylR mutant, the substrates still formed a hierarchical order in their ability to cause repression. We wanted to determine whether this order is caused exclusively by the global CCR mechanism or whether additional mechanisms are still involved. If only global CCR is involved, an interruption of the general CCR pathway is expected to completely relieve repression. To test this possibility, we used ΔxylR strains, which also lacked HPrK/P or CcpA or its cofactors Crh and HPr. In the latter case, a Δcrh ptsH1 mutant was used, which encodes an HPr mutant with a nonphosphorylatable alanine rather than a serine at position 46. The HPr-Ser46Ala protein is unable to participate in CcpA-mediated CCR but retains its function in PTS sugar uptake (8). All strains produced similar high β-xylosidase activities regardless of the carbon source, with the notable exception of sorbitol (Table 2, compare strains GP287, GP289, and GP853). On sorbitol a threefold repression was still detectable. On all other carbon sources, the elimination of the global regulators of CCR caused a complete relief from CCR. Hence, global CCR is responsible for the hierarchical order of repression caused by the different substrates in the ΔxylR mutant.
The amounts of HPrK, CcpA, and HPr are not affected by the carbon source.
It is possible that the differences in repression exerted by the different substrates are caused by various amounts of HPrK or CcpA or its cofactors in the cell. To address this possibility, we determined the amounts of these proteins in cells grown on the various carbon sources. Protein extracts were separated by SDS-PAGE and subjected to Western blot analyses with specific antisera. Only small differences in the amounts of the three proteins were detectable (Fig. 3). We also attempted to detect the Crh protein. However, although our antiserum readily detected purified Crh protein, no signals were obtained in cell extracts (data not shown). This observation is in agreement with our previous data suggesting that Crh is present in the cell in much lower amounts than HPr (14). In conclusion, the cellular amounts of the global CCR regulators HPrK/P, CcpA, and HPr are not affected by the carbon source.
FIG. 3.

The amounts of HPrK/P, CcpA and HPr are not affected by the utilized carbon source. Wild-type strain 168 was grown to exponential phase in CSE medium containing the indicated carbon sources, and crude extracts were prepared. Of these protein extracts, 10-, 5-, and 2-μg portions were loaded onto 12.5% SDS-PAA gels for the detection of HPrK/P, CcpA, and HPr, respectively. After gel electrophoresis the proteins were blotted onto a PVDF membrane, and the proteins of interest were detected using specific antisera.
HPr and Crh are not interchangeable in CcpA-mediated CCR.
In the ΔxylR mutant, only global CCR is operative in xynPB repression. Hence, the degree of repression exerted by the individual substrates should be reflected by the total number of HPr(Ser-P)/CcpA and Crh(Ser-P)/CcpA complexes competent in DNA binding. Thus far, it is unclear to which extent each of these complexes actually contributes to CCR via CcpA under a certain condition. Hence, it is possible that the cell selectively prefers or relies on one of these complexes depending on the carbon source. To address this question, we compared ΔxylR Δcrh and ΔxylR ptsH1 mutants in which either one of the cofactors for CcpA is missing. The XynB activities in the ΔxylR Δcrh mutant were very similar to those detected in the ΔxylR mutant (Table 2, compare findings for strains GP270 and GP297; Fig. 2). Therefore, Crh is dispensable for CCR exerted by the different carbon sources. Obviously, HPr can completely compensate for the loss of Crh. A different result was obtained using the ΔxylR ptsH1 mutant. In this case, a three- to fourfold relief from repression was observed when the cells grew on glucose or fructose. On mannitol, the β-xylosidase activities were even eightfold higher (Table 2, compare strain GP284 with strains GP270 and GP297). These sugars exert the strongest repression in the wild type and in the ΔxylR mutant. In contrast, repression by all other substrates was not significantly affected by the ptsH1 mutation. These data indicate that Crh is unable to substitute for HPr in the presence of substrates that exert a strong CCR. Hence, HPr is essential for CCR.
The extent to which a substrate causes repression correlates with the amount of HPr(Ser-P) in the cell.
The phosphorylation of HPr by the HPr kinase is a key step in the signaling pathway of CCR in B. subtilis. It is well established that significant amounts of HPr(Ser-P) are present during growth on glucose, whereas little HPr(Ser-P) is detectable in the absence of a sugar, i.e., in CSE minimal medium or LB (27, 32). However, the phosphorylation state of HPr has thus far not been determined during growth in the presence of sugars other than glucose. In order to explore whether the strength of CCR exerted by a given substrate correlates with the amount of HPr(Ser-P) in the cell, we determined the HPr phosphorylation state in vivo. To this end, we prepared extracts from cells grown on the various carbohydrates and subjected them to nondenaturing PAGE, which allows the separation of the nonphosphorylated and phosphorylated forms of HPr. HPr was subsequently detected by Western blot analysis (Fig. 4).
FIG. 4.

Determination of the phosphorylation state of HPr in the presence of different carbon sources. Wild-type strain 168 was grown on CSE medium supplemented with the indicated carbohydrates. Protein extracts were prepared and separated on native 12% PAA gels (top panels). HPr was subsequently detected by Western blotting (odd-numbered lanes). To discriminate between HPr(Ser-P) and HPr(His-P), an aliquot of each cell extract was heated (70°C, 10 min) prior to loading (even-numbered lanes). This causes loss of the phosphohistidine bonds. The lysis buffer used affected the reliability of the Bradford assay for determination of protein concentrations. To account for the differences in the protein estimation and sample loading, 2 μg of each protein extract (according to Bradford-assay) was separated in parallel by SDS-PAGE, and total HPr was detected by Western blot analysis (bottom panels).
As reported previously (27), HPr was present in three different forms when the cells grew in CSE medium: nonphosphorylated, single phosphorylated, and doubly phosphorylated (Fig. 4, lane 1). HPr can be phosphorylated by the EI of the PTS on His15 and by HPrK/P on Ser46. To discriminate between both forms, aliquots of the extracts were incubated at 70°C before material was loaded onto the gel. Heating causes loss of phosphohistidine but not of phosphoserine bonds. As a result, the doubly phosphorylated HPr is converted to HPr(Ser-P) and HPr(His-P) is converted to nonphosphorylated HPr (Fig. 4, even-numbered lanes). From the quantification of the band intensities in both lanes and their comparison, it was possible to calculate the relative amount of each form of HPr (Table 3). In CSE medium, only 13% of the total HPr was phosphorylated at Ser46 and 5% was doubly phosphorylated. The majority of HPr molecules were nonphosphorylated or phosphorylated at His15. Very similar results were obtained when the cells grew on the weakly repressing sugars ribose, gluconate, arabinose, and maltose. The level of HPr(Ser-P) slightly increased up to 32% on gluconate, which is the most strongly repressing substrate among these carbohydrates (Fig. 4, top panel; Table 3). In all cases, HPr(His∼P) and doubly phosphorylated HPr were the predominant forms.
TABLE 3.
Comparison of catabolite repression by different substrates, the relative amounts of the different HPr forms, and the intracellular FBP concentrationa
| Carbon sourceb | Fold repressionc | Relative amt (%)d of HPr form
|
Concn of fructose-1,6-bis-P (mM) | Growth rate (μ) h−1 | |||
|---|---|---|---|---|---|---|---|
| HPr-Ser∼P | HPr-His∼P | HPr | HPr-Ser∼P-His∼P | ||||
| None (CSE) | 13 (3) | 40 (14) | 42 (19) | 5 (2) | 1.8 (0.5) | 0.29 (0.03) | |
| Ribose | 2 | 27 (1) | 37 (6) | 25 (9) | 11 (4) | 6.5 (0.1) | 0.76 (0.07) |
| Arabinose | 3 | 21 (1) | 29 (9) | 13 (10) | 37 (1) | 8.1 (0.5) | 0.98 (0.06) |
| Maltose | 6 | 13 (1) | 47 (4) | 25 (6) | 15 (1) | 10.7 (0.8) | 0.85 (0.01) |
| Gluconate | 7 | 32 (10) | 29 (4) | 9 (2) | 30 (9) | 12.3 (3.6) | 0.94 (0.09) |
| Sucrose | 14 | 66 | 0 | 33 | 1 | 11.5 (1.9) | 0.85 (0.02) |
| Salicin | 17 | 50 (9) | 0 | 50 (9) | 0 | 9.4 (2.1) | 0.91 (0.04) |
| Sorbitol | 21 | 66 (6) | 0 | 32 (7) | 2 (1) | 4.4 (2.1) | 0.83 (0.03) |
| Glycerol | 32 | 46 (3) | 12 (6) | 0 | 42 (4) | 4.3 (1.6) | 0.94 (0.11) |
| Mannitol | 35 | 70 (6) | 0 | 30 (6) | 0 | 4.4 (0.6) | 0.86 (0.03) |
| Fructose | 44 | 60 (2) | 0 | 40 (3) | 0 | 13.3 (3.0) | 0.94 (0.07) |
| Glucose | 46 | 58 (6) | 0 | 35 (10) | 7 (7) | 14.1 (1.3) | 0.95 (0.07) |
Standard deviations are given in parentheses.
That is, added to the CSE medium. For growth rate determinations, CE medium plus a single carbon source was used.
In the ΔxylR Δcrh mutant.
Mean values of at least two independent experiments are presented except for sucrose, for which only one experiment was performed.
A very different result was obtained when the cells grew on sugars that caused a stronger CCR, i.e., sorbitol, sucrose, salicin, mannitol, fructose, and glucose. In these cases, almost no doubly phosphorylated HPr was detectable, and heating of the extracts had almost no effect on the phosphorylation pattern (Fig. 4, lanes 11, 12, and 15 to 24). Hence, there was no detectable HPr(His-P) present in the cells. In contrast, 50 to 70% of all HPr molecules were phosphorylated at Ser46 (Fig. 4, bottom panel; Table 3, sucrose, salicin, sorbitol, glycerol, mannitol, fructose, and glucose). The only other detectable form was nonphosphorylated HPr. As an exception, glycerol behaved different from all other substrates: In contrast to the other strongly repressing substrates, it generated a somewhat lower level of HPr(Ser-P), i.e., 46%. Moreover, no unphosphorylated HPr could be detected. In contrast, doubly phosphorylated HPr was a predominant form in this case (Fig. 4, lanes 13 and 14).
In conclusion, sugars that exert strong (i.e., at least 10-fold; Fig. 2) repression of β-xylosidase activity also generate a high level of HPr(Ser-P) in the cell, whereas in the presence of weakly repressing substrates the amount of HPr(Ser-P) is significantly lower. In addition, the data suggest that the absence of histidine-phosphorylated HPr is a common feature of substrates that generate a strong CCR.
His15-dependent phosphorylation of HPr has no impact on CcpA-mediated CCR.
Interestingly, most of the carbohydrates that generated a strong CCR are substrates of the PTS. Glucose, fructose, mannitol, salicin, and sucrose are all taken up by specific EIIs, which rely on HPr(His-P)-dependent phosphorylation for this function. Hence, utilization of all of these substrates drains away the phosphoryl groups bound to His15 of HPr. Accordingly, no HPr(His-P) can be detected when the cells grow on these substrates (Fig. 4 and Table 3). On the weakly repressing substrates ribose, arabinose, and gluconate, which are non-PTS substrates, a considerable fraction of HPr is phosphorylated at its His15 residue by EI (Table 3). Hence, it is conceivable that in these cases EI and HPrK compete for the phosphorylation of HPr and that CCR is weak, because EI-dependent phosphorylation limits the HPr(Ser-P) amount in the cell and thereby CCR.
To test this possibility, we used a ΔptsI mutant in which HPr cannot get phosphorylated at His15. If histidine phosphorylation of HPr limits CCR, one would expect an increased CCR by non-PTS substrates in this mutant. However, there was virtually no difference in CCR by non-PTS substrates between the ΔptsI mutant and the wild-type strain (Fig. 5). In contrast, on PTS-sugars (and glycerol) an almost complete relief from CCR was observed in the ΔptsI mutant. This was expected, because uptake and metabolism of these substrates are not possible in the ΔptsI mutant. Hence, the cells actually use succinate for growth, which exerts no CCR. To support these data, we also investigated a mutant strain coding for an HPr-His15Ala variant. This strain exhibited β-xylosidase activities almost identical to those determined in the ΔptsI mutant (our unpublished data). In conclusion, the phosphorylation state of HPr at its histidine residue has no direct impact on CcpA-mediated CCR. Therefore, solely different HPrK activities appear to account for the different CCR levels exerted by the various substrates.
FIG. 5.
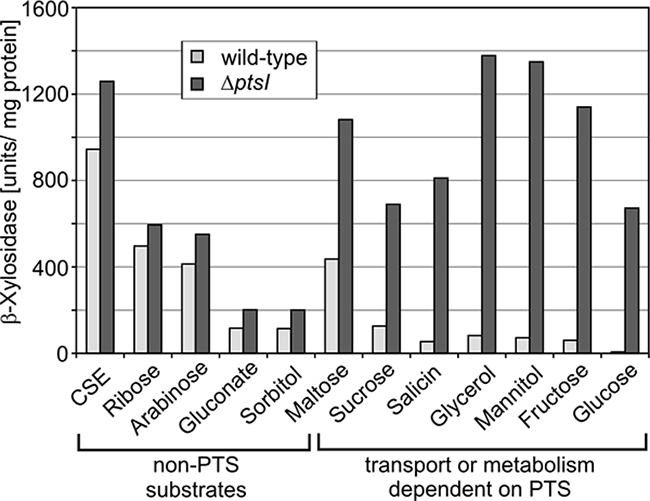
Catabolite repression of β-xylosidase in a mutant lacking EI of the PTS. Strain GP864 (ΔptsI) was grown in CSE medium supplemented with the indicated carbohydrates, and the β-xylosidase activities were determined (dark gray columns). The corresponding activities in the wild-type strain 168 are shown for comparison (light gray columns).
Activity of HPrK/P determines the level of CcpA-exerted CCR in B. subtilis.
Finally, we wanted to confirm that a low HPr kinase activity is responsible for the weak CCR exerted by substrates such as ribose, arabinose, gluconate, or maltose. To this end, we used a strain, which expresses the mutant hprK(G158A) allele rather than wild-type hprK. The G158A exchange is located in the nucleotide binding Walker A motif of HPrK and abolishes phosphorylase activity in vitro (16). As a result, HprK G158A behaves as a constitutive kinase, triggering the slow phosphorylation of HPr. In order to see whether this is indeed the case in vivo, we introduced a plasmid carrying hprK(G158A) into a mutant strain lacking the wild-type gene. A transformant carrying the empty expression plasmid served as a control. These transformants, as well as the wild-type strain, were grown in CSE and CSE plus glucose, and the phosphorylation state of HPr was determined (Fig. 6). As expected, no HPr(Ser-P) was detectable in the ΔhprK mutant (Fig. 6A, lanes 3, 4, 9, and 10). In the strain expressing the hprK(G158A) allele, the fraction of HPr(Ser-P) increased to 38% when the cells grew in CSE, whereas only 13% HPr(Ser-P) were detectable in the wild type (Fig. 6, compare lanes 1 and 2 and lanes 5 and 6). In the presence of glucose 43% HPr(Ser-P) were detectable in the mutant expressing the hprK(G158A) allele (Fig. 6, lanes 11 and 12). The wild-type strain produced 58% HPr(Ser-P) under these conditions (Fig. 6, lanes 7 and 8). In conclusion, the HPrK G158A mutant triggers the phosphorylation of HPr at its Ser residue even in the absence of a repressing sugar. To see whether this increased fraction of HPr(Ser-P) also correlates with a stronger CCR, we determined the β-xylosidase activities in these strains (Table 4). In the ΔhprK mutant carrying the empty expression plasmid, very high β-xylosidase activities were detectable, i.e., xynPB expression was relieved from CCR. In contrast, the strain expressing the hprK(G158A) allele produced only low activities in the range of 60 to 170 U. As an exception, repression by glucose was somewhat stronger (21 U), which can be ascribed to the extra repression by glucose-6-P via XylR. In conclusion, the data show that the HPrK G158A variant phosphorylates HPr under all conditions and thereby triggers strong CCR on all substrates. This confirms that the different levels of CCR exerted by the various carbon sources (Fig. 2) solely result from different activities of HPrK/P.
FIG. 6.

The mutant HprK/P G158A protein phosphorylates HPr at Ser46 even in the absence of a sugar. Strain GP858 (ΔhprK) carrying either plasmid pGP650 encoding the hprK(G158A) allele (lanes 5, 6, 11, and 12) or the empty plasmid (lanes 3, 4, 9, and 10) was grown in CSE or CSE plus glucose. Crude extracts of these strains were subjected to nondenaturating PAGE, and the phosphorylation state of HPr was determined as described for Fig. 4. For comparison, the wild-type strain 168 is also shown (lanes 1, 2, 7, and 8).
TABLE 4.
Catabolite repression by the mutant HPrK/P-G158A protein lacking phosphorylase activity
| Carbon sourcea | Enzyme activityb in U/mg of protein (SD) in strain:
|
||
|---|---|---|---|
| 168c (wild type) | GP858(pGP380) (ΔhprK) | GP858(pGP650) [ΔhprK hprK(G158A)] | |
| None | 945 (281) | 2,077 (323) | 178 (10) |
| Ribose | 497 (138) | NG | 115 (30) |
| Arabinose | 414 (136) | 1,175 (50) | 138 (27) |
| Maltose | 437 (127) | 1,265 (82) | 142 (17) |
| Sucrose | 126 (20) | 1,232 (145) | 124 (66) |
| Gluconate | 116 (11) | 822 (97) | 63 (6) |
| Sorbitol | 114 (20) | 990 (52) | 90 (22) |
| Glycerol | 82 (15) | NG | 70 (15) |
| Mannitol | 72 (13) | 1,592 (460) | 60 (8) |
| Fructose | 60 (9) | 1,364 (355) | 64 (8) |
| Salicin | 54 (6) | 981 (168) | 46 (27) |
| Glucose | 7 (3) | 870 (11) | 21 (4) |
That is, added to the CSE medium.
The values are the averages of at least two independent experiments. NG, no growth.
Xylose was added to induce xynPB expression.
The intracellular FBP concentration and the strength of CCR exerted by a given carbohydrate do not strictly correlate.
FBP has been identified as a key metabolite modulating the activity of HPrK. To determine whether there is a correlation between the intracellular FBP concentration and the level of CCR exerted by a given substrate, we determined the FBP concentrations (Table 3). In cells grown in pure CSE medium, only 1.8 mM FBP was detectable, whereas the FBP concentration increased to 14.1 mM in the presence of glucose. These results are in perfect agreement with a previous study (31). High FBP concentrations in the range of 9.4 to 13.3 mM FBP were also detectable in cells grown on the strongly repressing sugars fructose, salicin, and sucrose. However, there was no strict correlation for the remaining substrates, e.g., the utilization of ribose, arabinose, and maltose generated rather high FBP concentrations (6.5 to 10.7 mM FBP), whereas CCR exerted by these substrates was weak. In contrast, the FBP concentrations were lower (4.3 to 4.4 mM) on mannitol, glycerol, and sorbitol, which all exert a strong CCR. In conclusion, the different levels of repression exerted by the different carbohydrates cannot be explained by the different intracellular FBP levels alone.
DISCUSSION
In this study, we show that in addition to glucose many other carbohydrates cause carbon catabolite repression in B. subtilis. These substrates form a hierarchical order in their capacity to exert repression, suggesting that they trigger the formation of active CcpA complexes to different degrees (Table 2 and Fig. 2). Our data show that the different carbon sources modulate the activities rather than the amounts of the proteins responsible for CCR. In fact, the data suggest a correlation between the ability of a sugar to cause repression and the phosphorylation state of HPr at its Ser46 residue (Fig. 4). We could not observe any interference of the phosphorylation state of HPr at its His15 residue with CcpA-mediated CCR of the xynPB operon, which served as a model system in the present study. Our data suggest that at least in this case HPrK/P activity is the sole factor that accounts for the differences in repression by the various substrates. However, it should be emphasized that in other gene systems additional CcpA-independent mechanisms of CCR exist, which rely on HPr(His-P)-dependent phosphorylation (for a review, see reference 13).
The substrates form a hierarchy in their capacity to exert repression. The general CCR pathway determines this hierarchy, since the absence of either HPrK/P or CcpA or its cofactors resulted in complete derepression on all substrates. Intriguingly, there is a good correlation between the hierarchy in repression (the present study) and the hierarchy of carbon source utilization reported by Monod based on diauxic growth experiments (33). Monod classified the carbon sources utilized by B. subtilis in two groups: A and B. When present in a mixture, the bacteria first utilize the substrates of group A, which include glucose, fructose, mannitol, and sucrose. Subsequently, the cells make use of the group B carbohydrates, e.g., sorbitol, arabinose, or maltose. Based on our observation that group A sugars exert strong CCR, whereas repression by group B sugars is weak, it appears reasonable that the diauxic growth behavior observed by Monod is caused by CcpA-mediated CCR. However, as an inconsistency we observed a strong repression of xynPB by sorbitol, which was assigned by Monod to group B. Interestingly, disruption of the CcpA-mediated CCR did not completely relieve xynPB from repression when the cells grew on sorbitol (Table 2). In contrast, other CcpA-controlled catabolic genes such as rocG were completely derepressed in ccpA and hprK mutants grown on sorbitol (data not shown). Therefore, strong repression by sorbitol appears to be specific for the xynPB operon, which might explain the discrepancy versus Monod's observations.
The Crh protein was completely dispensable for CCR exerted by the different substrates. A different result was obtained for a ptsH1 mutant, in which the Crh protein is the only potential effector for CcpA. In this case, there was a significant relief from CCR exerted by glucose, fructose, and mannitol, but not from CCR exerted by the other sugars. In a previous study it was shown that in a ptsH1 mutant the synthesis of gluconate kinase and glucitol dehydrogenase is relieved from repression by glucose and mannitol but not by glycerol (8). Collectively, these observations suggest that Crh cannot substitute for HPr when the cells grow on substrates that cause a very strong CCR via CcpA. This might be reflected by the up to 100-fold lower synthesis rate of Crh and its 10-fold lower affinity for CcpA compared to HPr (14, 47). Alternatively, Crh might play a more specific role in CCR, when the cells use weaker repressing substrates. Recent work suggested that Crh might be more important for CCR during the transition to stationary phase (19). In conclusion, HPr rather than Crh is the relevant effector for CcpA in vivo, at least during the exponential growth phase.
According to the current model of the global CCR mechanism in B. subtilis, repression is brought about by the HPrK-catalyzed phosphorylation of HPr at its Ser46 residue. Indeed, during growth on weakly repressing carbon sources only a minor fraction of HPr was phosphorylated at Ser46, whereas the majority of HPr molecules were phosphorylated at this site on strongly repressing substrates. This clearly shows that the cell modulates the strength of CCR by dynamically triggering the HPrK/P-dependent (de)phosphorylation of HPr. Interestingly, large amounts of HPr(His-P) and also doubly phosphorylated HPr were formed, when the cells grew on weakly repressing substrates. The formation of significant amounts of doubly phosphorylated HPr in vivo is surprising. Previous in vitro studies suggested that HPr(His-P) is a poor substrate for HPr kinase (39). Moreover, a Ser46Asp exchange in HPr, which mimics phosphorylation at this site, was shown to block EI-dependent phosphorylation in vitro (40). These effects were explained by a diminished affinity for the second phosphoryl group delivering protein. Obviously, the in vitro data do not adequately reflect the situation in vivo.
Most of the strongly repressing substrates are transported by the PTS (glucose, fructose, mannitol, salicin, and sucrose) and are therefore expected to dephosphorylate HPr at its His15 residue, which is in agreement with our finding that no HPr(His-P) was detectable in these cases. On the other hand, these observations raised the possibility that CCR is strong because dephosphorylation of HPr at His15 makes the protein susceptible to HPr kinase-catalyzed phosphorylation, i.e., that EI and HPrK compete for phosphorylation of HPr. However, our experiments with mutants lacking EI did not support this scenario (Fig. 5). Hence, the phosphorylation state of HPr at its His15 residue is irrelevant for CcpA-mediated CCR in B. subtilis. This is very different from E. coli, where the absence of EI leads to permanent repression of secondary catabolic genes (36). In E. coli, many substrates, which are taken up by the PTS cause CCR, because their transport dephosphorylates not only the general PTS proteins EI and HPr but also the EIIAGlc protein (36). In contrast, PTS transport activity has no direct effect on CCR in B. subtilis. Our data using the constitutive HPrK G158A allele (Fig. 6 and Table 3) demonstrate that low HPr kinase activity limits CCR by weakly repressing carbon sources. Hence, different HPrK/P activities account for the different repression levels exerted by the various substrates.
What are the molecular mechanisms that adjust the activity of HPrK/P to the available carbon source? The cells exhibited comparable growth rates on the different carbon sources except for succinate and ribose, on which growth was significantly slower (Table 3, last column). Therefore, it appears unlikely that the growth rate has a direct effect on HPrK/P activity and therefore CCR. It is well known that the two antagonistic activities of HPrK/P are regulated by the concentrations of FBP, ATP, and Pi. The central role of FBP for activity of the HPr kinase has been unequivocally proven in vivo and in vitro. In vivo, any mutation that prevents the formation of FBP results in a complete relief from CCR via CcpA, e.g., there is no CCR by glucose in mutants lacking the glycolytic enzymes glucose-6-phosphate isomerase or phosphofructokinase (35). In vitro, HPr kinase activity is barely detectable below 1 mM FBP. With higher FBP concentrations HPr kinase activity sharply increases and reaches a plateau at about 5 mM FBP (20, 39). In our experiments we detected a low FBP concentration of 1.8 mM in the absence of a sugar (i.e., in CSE medium), whereas in the presence of the various sugars, FBP concentrations in the range of 4.3 to 14.1 mM were detected. Therefore, all sugars generated FBP levels, which are theoretically sufficient for a high HPr kinase activity. This suggests that the different levels of repression exerted by the different carbohydrates cannot be explained by the different intracellular FBP concentrations alone. Therefore, in addition to FBP other metabolites might account for the substrate-dependent differences of HPrK/P activity. Indeed, for Streptococcus bovis, an inverse correlation between HPr(Ser-P) formation and the Pi concentration was observed in vivo (1). In addition, other metabolites, such as ATP, acetyl-phosphate, and glyceraldehyde 3-phosphate, were shown to modulate the activity of B. subtilis HPrK/P in vitro (37).
Hierarchical regulation is a widespread phenomenon if bacteria have the choice between different substrates. If only a few substrates can be used, sophisticated regulatory networks with multiple transcription factors allow the consecutive expression of the respective enzymes. This was observed for the choice of the electron acceptors for E. coli respiration (49). However, individual regulators for the repression of genes that are lower in the hierarchy cannot be used if the bacteria have to choose between a plethora of substrates. Accordingly, in E. coli and B. subtilis, the hierarchy of carbon sources in catabolite repression is established by a single signal, i.e., the control of the phosphorylation states of EIIAGlc and HPr, respectively (18; the present study).
Acknowledgments
We are grateful to Isabelle Martin-Verstraete for the gift of strains. We thank Sabine Lentes for excellent technical assistance.
K.D.S. was supported by a Lichtenberg stipend of the state Niedersachsen. This study was supported by grants of the Fonds der Chemischen Industrie and the Federal Ministry of Education and Research SYSMO network (PtJ-BIO/0313978D) to J.S.
Footnotes
Published ahead of print on 29 August 2008.
REFERENCES
- 1.Asanuma, N., and T. Hino. 2003. Molecular characterization of HPr and related enzymes, and regulation of HPr phosphorylation in the ruminal bacterium Streptococcus bovis. Arch. Microbiol. 179205-213. [DOI] [PubMed] [Google Scholar]
- 2.Bettenbrock, K., T. Sauter, K. Jahreis, A. Kremling, J. W. Lengeler, and E. D. Gilles. 2007. Correlation between growth rates, EIIACrr phosphorylation, and intracellular cyclic AMP levels in Escherichia coli K-12. J. Bacteriol. 1896891-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blencke, H. M., G. Homuth, H. Ludwig, U. Mäder, M. Hecker, and J. Stülke. 2003. Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metab. Eng. 5133-149. [DOI] [PubMed] [Google Scholar]
- 4.Dahl, M. K., J. Degenkolb, and W. Hillen. 1994. Transcription of the xyl operon is controlled in Bacillus subtilis by tandem overlapping operators spaced by four base-pairs. J. Mol. Biol. 243413-424. [DOI] [PubMed] [Google Scholar]
- 5.Dahl, M. K., D. Schmiedel, and W. Hillen. 1995. Glucose and glucose-6-phosphate interaction with Xyl repressor proteins from Bacillus spp. may contribute to regulation of xylose utilization. J. Bacteriol. 1775467-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deutscher, J. 2008. The mechanisms of carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 1187-93. [DOI] [PubMed] [Google Scholar]
- 7.Deutscher, J., C. Francke, and P. W. Postma. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70939-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deutscher, J., J. Reizer, C. Fischer, A. Galinier, M. H. Saier, Jr., and M. Steinmetz. 1994. Loss of protein kinase-catalyzed phosphorylation of HPr, a phosphocarrier protein of the phosphotransferase system, by mutation of the ptsH gene confers catabolite repression resistance to several catabolic genes of Bacillus subtilis. J. Bacteriol. 1763336-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faires, N., S. Tobisch, S. Bachem, I. Martin-Verstraete, M. Hecker, and J. Stülke. 1999. The catabolite control protein CcpA controls ammonium assimilation in Bacillus subtilis. J. Mol. Microbiol. Biotechnol. 1141-148. [PubMed] [Google Scholar]
- 10.Fujita, Y., Y. Miwa, S. Tojo, and K. Hirooka. 2007. Carbon catabolite control and metabolic networks mediated by the CcpA protein in Bacillus subtilis, p. 91-110. In Y. Fujita (ed.), Global regulatory networks in Bacillus subtilis. Transworld Research Network, Trivandrum, India.
- 11.Galinier, A., J. Deutscher, and I. Martin-Verstraete. 1999. Phosphorylation of either Crh or HPr mediates binding of CcpA to the Bacillus subtilis xyn cre and catabolite repression of the xyn operon. J. Mol. Biol. 286307-314. [DOI] [PubMed] [Google Scholar]
- 12.Galinier, A., J. Haiech, M. C. Kilhoffer, M. Jaquinod, J. Stülke, J. Deutscher, and I. Martin-Verstraete. 1997. The Bacillus subtilis crh gene encodes a HPr-like protein involved in carbon catabolite repression. Proc. Natl. Acad. Sci. USA 948439-8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Görke, B., and J. Deutscher. 2007. The regulatory functions of histidyl-phosphorylated HPr in bacilli, p. 1-37. In Y. Fujita (ed.), Global regulatory networks in Bacillus subtilis. Transworld Research Network, Trivandrum, India.
- 14.Görke, B., L. Fraysse, and A. Galinier. 2004. Drastic differences in Crh and HPr synthesis levels reflect their different impacts on catabolite repression in Bacillus subtilis. J. Bacteriol. 1862992-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Görke, B., and J. Stülke. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6613-624. [DOI] [PubMed] [Google Scholar]
- 16.Hanson, K. G., K. Steinhauer, J. Reizer, W. Hillen, and J. Stülke. 2002. HPr kinase/phosphatase of Bacillus subtilis: expression of the gene and effects of mutations on enzyme activity, growth and carbon catabolite repression. Microbiology 1481805-1811. [DOI] [PubMed] [Google Scholar]
- 17.Herzberg, C., L. A. Weidinger, B. Dörrbecker, S. Hübner, J. Stülke, and F. M. Commichau. 2007. SPINE: a method for the rapid detection and analysis of protein-protein interactions in vivo. Proteomics 74032-4035. [DOI] [PubMed] [Google Scholar]
- 18.Hogema, B. M., J. C. Arents, R. Bader, K. Eijkemans, H. Yoshida, H. Takahashi, H. Aiba, and P. W. Postma. 1998. Inducer exclusion in Escherichia coli by non-PTS substrates: the role of the PEP to pyruvate ratio in determining the phosphorylation state of enzyme IIAGlc. Mol. Microbiol. 30487-498. [DOI] [PubMed] [Google Scholar]
- 19.Inacio, J. M., and I. de Sa-Nogueira. 2007. trans-Acting factors and cis elements involved in glucose repression of arabinan degradation in Bacillus subtilis. J. Bacteriol. 1898371-8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jault, J. M., S. Fieulaine, S. Nessler, P. Gonzalo, A. Di Pietro, J. Deutscher, and A. Galinier. 2000. The HPr kinase from Bacillus subtilis is a homo-oligomeric enzyme which exhibits strong positive cooperativity for nucleotide and fructose 1,6-bisphosphate binding. J. Biol. Chem. 2751773-1780. [DOI] [PubMed] [Google Scholar]
- 21.Kraus, A., C. Hueck, D. Gärtner, and W. Hillen. 1994. Catabolite repression of the Bacillus subtilis xyl operon involves a cis element functional in the context of an unrelated sequence, and glucose exerts additional xylR-dependent repression. J. Bacteriol. 1761738-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krispin, O., and R. Allmansberger. 1998. The Bacillus subtilis AraE protein displays a broad substrate specificity for several different sugars. J. Bacteriol. 1803250-3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kunst, F., and G. Rapoport. 1995. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J. Bacteriol. 1772403-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Küster, E., E. J. Luesink, W. M. de Vos, and W. Hillen. 1996. Immunological crossreactivity to the catabolite control protein CcpA from Bacillus megaterium is found in many gram-positive bacteria. FEMS Microbiol. Lett. 139109-115. [DOI] [PubMed] [Google Scholar]
- 25.Lereclus, D., and O. Arantes. 1992. spbA locus ensures the segregational stability of pTH1030, a novel type of gram-positive replicon. Mol. Microbiol. 635-46. [DOI] [PubMed] [Google Scholar]
- 26.Lindner, C., J. Stülke, and M. Hecker. 1994. Regulation of xylanolytic enzymes in Bacillus subtilis. Microbiology 140753-757. [DOI] [PubMed] [Google Scholar]
- 27.Ludwig, H., N. Rebhan, H. M. Blencke, M. Merzbacher, and J. Stülke. 2002. Control of the glycolytic gapA operon by the catabolite control protein A in Bacillus subtilis: a novel mechanism of CcpA-mediated regulation. Mol. Microbiol. 45543-553. [DOI] [PubMed] [Google Scholar]
- 28.Lulko, A. T., G. Buist, J. Kok, and O. P. Kuipers. 2007. Transcriptome analysis of temporal regulation of carbon metabolism by CcpA in Bacillus subtilis reveals additional target genes. J. Mol. Microbiol. Biotechnol. 1282-95. [DOI] [PubMed] [Google Scholar]
- 29.Martin-Verstraete, I., J. Deutscher, and A. Galinier. 1999. Phosphorylation of HPr and Crh by HprK, early steps in the catabolite repression signalling pathway for the Bacillus subtilis levanase operon. J. Bacteriol. 1812966-2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin-Verstraete, I., J. Stülke, A. Klier, and G. Rapoport. 1995. Two different mechanisms mediate catabolite repression of the Bacillus subtilis levanase operon. J. Bacteriol. 1776919-6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mijakovic, I., S. Poncet, A. Galinier, V. Monedero, S. Fieulaine, J. Janin, S. Nessler, J. A. Marquez, K. Scheffzek, S. Hasenbein, W. Hengstenberg, and J. Deutscher. 2002. Pyrophosphate-producing protein dephosphorylation by HPr kinase/phosphorylase: a relic of early life? Proc. Natl. Acad. Sci. USA 9913442-13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monedero, V., S. Poncet, I. Mijakovic, S. Fieulaine, V. Dossonnet, I. Martin-Verstraete, S. Nessler, and J. Deutscher. 2001. Mutations lowering the phosphatase activity of HPr kinase/phosphatase switch off carbon metabolism. EMBO J. 203928-3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monod, J. 1942. Recherches sur la croissance des cultures bacteriennes. Thesis. Hermann et Cie, Paris, France.
- 34.Moreno, M. S., B. L. Schneider, R. R. Maile, W. Weyler, and M. H. Saier, Jr. 2001. Catabolite repression mediated by the CcpA protein in Bacillus subtilis: novel modes of regulation revealed by whole-genome analyses. Mol. Microbiol. 391366-1381. [DOI] [PubMed] [Google Scholar]
- 35.Nihashi, J., and Y. Fujita. 1984. Catabolite repression of inositol dehydrogenase and gluconate kinase syntheses in Bacillus subtilis. Biochim. Biophys. Acta 79888-95. [DOI] [PubMed] [Google Scholar]
- 36.Postma, P. W., J. W. Lengeler, and G. R. Jacobson. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57543-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramström, H., S. Sanglier, E. Leize-Wagner, C. Philippe, A. Van Dorsselaer, and J. Haiech. 2003. Properties and regulation of the bifunctional enzyme HPr kinase/phosphatase in Bacillus subtilis. J. Biol. Chem. 2781174-1185. [DOI] [PubMed] [Google Scholar]
- 38.Reizer, J., U. Bergstedt, A. Galinier, E. Kuster, M. H. Saier, Jr., W. Hillen, M. Steinmetz, and J. Deutscher. 1996. Catabolite repression resistance of gnt operon expression in Bacillus subtilis conferred by mutation of His-15, the site of phosphoenolpyruvate-dependent phosphorylation of the phosphocarrier protein HPr. J. Bacteriol. 1785480-5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reizer, J., C. Hoischen, F. Titgemeyer, C. Rivolta, R. Rabus, J. Stülke, D. Karamata, M. H. Saier, Jr., and W. Hillen. 1998. A novel protein kinase that controls carbon catabolite repression in bacteria. Mol. Microbiol. 271157-1169. [DOI] [PubMed] [Google Scholar]
- 40.Reizer, J., S. L. Sutrina, M. H. Saier, G. C. Stewart, A. Peterkofsky, and P. Reddy. 1989. Mechanistic and physiological consequences of HPr(ser) phosphorylation on the activities of the phosphoenolpyruvate:sugar phosphotransferase system in gram-positive bacteria: studies with site-specific mutants of HPr. EMBO J. 82111-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sambrook, J., and D. Russell. 2001. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 42.Schmiedel, D., and W. Hillen. 1996. A Bacillus subtilis 168 mutant with increased xylose uptake can utilize xylose as sole carbon source. FEMS Microbiol. Lett. 135175-178. [Google Scholar]
- 43.Schmiedel, D., and W. Hillen. 1996. Contributions of XylR, CcpA, and cre to diauxic growth of Bacillus megaterium and to xylose isomerase expression in the presence of glucose and xylose. Mol. Gen. Genet. 250259-266. [DOI] [PubMed] [Google Scholar]
- 44.Schumacher, M. A., G. S. Allen, M. Diel, G. Seidel, W. Hillen, and R. G. Brennan. 2004. Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell 118731-741. [DOI] [PubMed] [Google Scholar]
- 45.Schumacher, M. A., G. Seidel, W. Hillen, and R. G. Brennan. 2006. Phosphoprotein Crh-Ser46-P displays altered binding to CcpA to effect carbon catabolite regulation. J. Biol. Chem. 2816793-6800. [DOI] [PubMed] [Google Scholar]
- 46.Schumacher, M. A., G. Seidel, W. Hillen, and R. G. Brennan. 2007. Structural mechanism for the fine-tuning of CcpA function by the small molecule effectors glucose 6-phosphate and fructose 1,6-bisphosphate. J. Mol. Biol. 3681042-1050. [DOI] [PubMed] [Google Scholar]
- 47.Seidel, G., M. Diel, N. Fuchsbauer, and W. Hillen. 2005. Quantitative interdependence of coeffectors, CcpA and cre in carbon catabolite regulation of Bacillus subtilis. FEBS J. 2722566-2577. [DOI] [PubMed] [Google Scholar]
- 48.Singh, K. D., S. Halbedel, B. Görke, and J. Stülke. 2007. Control of the phosphorylation state of the HPr protein of the phosphotransferase system in Bacillus subtilis: implication of the protein phosphatase PrpC. J. Mol. Microbiol. Biotechnol. 13165-171. [DOI] [PubMed] [Google Scholar]
- 49.Unden, G., and J. Bongaerts. 1997. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1320217-234. [DOI] [PubMed] [Google Scholar]
- 50.Warner, J. B., and J. S. Lolkema. 2003. CcpA-dependent carbon catabolite repression in bacteria. Microbiol. Mol. Biol. Rev. 67475-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshida, K., K. Kobayashi, Y. Miwa, C. M. Kang, M. Matsunaga, H. Yamaguchi, S. Tojo, M. Yamamoto, R. Nishi, N. Ogasawara, T. Nakayama, and Y. Fujita. 2001. Combined transcriptome and proteome analysis as a powerful approach to study genes under glucose repression in Bacillus subtilis. Nucleic Acids Res. 29683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]