

Abstract
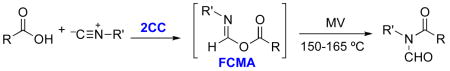
Microwave induced two component coupling (2CC) reaction of carboxylic acids with isonitriles gives rise to various N-formylamides. The formimidate carboxylate mixed anhydride (FCMA) is proposed as the reactive intermediate, which undergoes 1,3-O → N acyl transfer to give the observed product. The formation and survival of the labile FCMA system has been evaluated.
Recently we reported on the microwave induced coupling of carboxylic acids with isonitriles, giving rise to various N-formylamides (cf. 4, Scheme 1).1 We suggested the term two component coupling (2CC) to differentiate this work from earlier studies.2 As we discussed previously, one likely mechanistic interpretation of the 2CC reaction is that 4 arises from a 1,3-O→N acyl transfer within 3.3,4 The latter comes about from a protonation-addition sequence in the joining of 1 and 2. To the best of our knowledge, no structure corresponding to a formimidate-carboxylate mixed anhydride 3 (hereafter referred to in this paper as a FCMA), had been documented in a convincing way, let alone fully characterized.5–7 Our thoughts and experiences in this area led us to suppose that a generic FCMA, 3, would be a highly reactive acyl donor. Accordingly, a recent report5a to the effect that FCMA 7 is produced at room temperature as a crystalline product from the reaction of acid 5 and isonitrile 6 in water, provoked our curiosity. Moreover we noted that the spectroscopic properties of the alleged 7 (particularly its reported IR spectrum),8 do not correspond to what would be expected from such a structure.9
Scheme 1.

In our hands, reaction of 5 and 6 in water did indeed produce, as reported by the authors,5a a crystalline product, mp 69–71 ºC. Surprisingly at the time, microwave heating of this solid in chloroform failed to produce any discernible amounts of what would have been the expected product, 8, given the claimed structure 7.1 Adding to the puzzle, it was found that the crystalline product could not be retrieved after it had been dissolved in chloroform, even without thermolysis (i.e. at room temperature). Instead, evaporation of the solvent leaves a residue which does not have the properties of its precursor, allegedly 7. The residue from chloroform could be separated into components 5 and 9 by exploiting their differing acidic and neutral solubility properties, respectively.
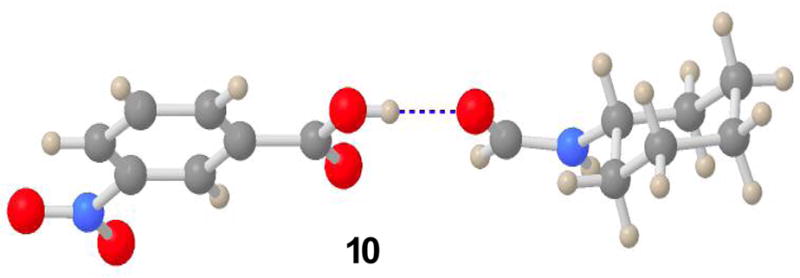
Fortunately, it proved possible to obtain some diffraction-worthy crystals from the product of the reaction of 5 and 6. Crystallographic analysis of the sample revealed the structure to be 10, a stable complex (fascinating in its own right!) between N-formylcyclohexylamine 9 and m-nitrobenzoic acid 5 (see Figure 1).10 Apparently, the fragile molecular association between 5 and 9 unravels upon dissolution in chloroform. Thus, the claim that the reaction of 5 and 6 produces FCMA 7 is not correct
Figure 1.
Also noteworthy was a report, in the same paper, describing a high yielding formation of amides (cf. 11, Scheme 2) from reactions of various benzoic acids (cf. 5) and isonitriles (cf. 6) conducted in methanol at room temperature.5a Our previous work,1 admittedly conducted in chloroform, showed virtually no reaction between acids and isonitriles at room temperature. Moreover we suspected that if a FCMA (cf. 3) intermediate were produced, it would have suffered conversion to the corresponding methyl ester. Accordingly, we repeated the reaction under the authors’ conditions, i.e. methanol as the solvent, at room temperature. As before, we had no difficulty in duplicating the published gross observations but we were not in agreement on the assignments. However, the assignments of simple amidic structures to the resultant crystalline products are not correct. First, several of the alleged amides had actually been previously reported in the literature.11 In each case that could be checked, there was a large discrepancy in melting points between the alleged “amides” reported from the isonitrile based coupling reactions and those previously reported. In each case the melting points of the purported amides reported5a were much higher than those previously reported for the authentic amides. Furthermore, in several cases, we prepared authentic amide samples ourselves by standard (cf. DCC) coupling methods. The NMR spectra of the authentic amides were very different from those of the amides claimed as arising from the isonitrile method.5a
Scheme 2.

In pursuing the matter, it became clear that the product of the reaction of 5 + 6 in methanol is not the amide 11 but rather the salt 13, arising from the neutralization of the acid 5 and cyclohexyl amine 12. Indeed, the same material as that synthesized by the authors (cf. 13) was generated by simply mixing equivalent amounts of 5 and 12. It is likely that 12 arises from a well precedented, though mechanistically unclear, methanol-mediated conversion of isonitriles to amines.12 Neutralization of the amine 12 provides the actual product, i.e. salt 13.
Another earlier paper by Gloede et. al. on the reaction of isonitriles and carboxylic acids (Scheme 3) provoked skepticism on our part.5b,c It was reported that the reaction of p-nitrobenzoic acid 14 and cyclohexylisonitrile 6, when conducted in methanol under reflux, gave rise to FCMA 15, m.p. 174–176 °C. Again, for obvious reasons,13 we wondered whether such a FCMA could have persisted in methanol. Accordingly, we repeated the experiment and obtained, exactly as reported, a crystalline compound, m.p. 173–175 °C (in addition to varying quantities of methyl ester 16). However, it was soon found that the high melting product is actually 17; i.e. the p-nitrobenzoic acid salt of 1,3-dicyclohexylamidine 18.14 This structure was confirmed by spectroscopic analysis of the product formed from mixing two equivalents 14 and one equivalent 18.15 Furthermore, removal of p-nitrobenzoic acid by basic workup afforded amidine 18 as the product. While the definition of a specific pathway for formation of 17 from among several obvious possibilities is not available from our data, qualitatively, it must involve, in some form, the methanolytic progression of 6 toward cyclohexylamine 12 as discussed above. The formation of the amidine 18 may well reflect an addition reaction of cyclohexylamine with 616 or an acid–mediated condensation between N-cyclohexylformamide 9 (or its equivalent) and cyclohexylamine 12 (or its functional equivalent).17 While this uncertainty remains to be sorted out, it is clear that the published assertions which claimed the formation and survival of labile FCMA systems in the presence of putative acyl acceptors (for instance, methanol or water as solvents) are not correct.18 In addition to the cases studied above, there may well be other instances where such claims warrant reexamination.5e
Scheme 3.

Motivated by the results described above, we asked whether a relatively weak nucleophile, such as methanol, could compete with 1,3-O→N acyl transfer in the context of a microwave mediated 2CC experiment (Scheme 4). Under these near stoichiometric conditions, substantial methanol induced conversion of isonitrile to amine12 would hopefully be attenuated. We started by studying the reaction of ca. 1:1:1 equivalents of acid 19, isonitrile 6 and methanol under the usual microwave mediated thermolysis. In the event, there was obtained ca. 38% yield of methyl ester 20, the expected two-component coupling product 21 (30%) and traces of the amide 22 (4%). Separately, it was demonstrated that amide 22 does not arise from methanolytic deformylation of 21 under closely simulated methanol conditions. It is likely that 22 comes about from small amounts of cyclohexylamine (12) or its equivalent arising from 6. Thus, acylation of 12 by FCMA 23 could lead to 22.
Scheme 4.

Finally, it was of interest to study the possibility of a 2CC reaction between phthaloyl glycine 24 and serine isonitrile benzyl ester 25 as a model for interdiction by an intramolecular hydroxyl group (Scheme 5). Remarkably, even at room temperature, the 2CC reaction does occur, giving rise to 26, although in only ca. 25% yield. In an important control experiment, it was shown that hydroxyl protected serine isonitrile derivative 27,19 seemingly does not react with 24 at all at room temperature.
Scheme 5.

Our data do not allow us to distinguish between several obvious variations of the general scheme suggested below (Scheme 6). Globally, the teaching seems to be that an otherwise unfavorable formation of a FCMA can be driven to product 26 through neighboring hydroxyl participation to enable the 1,3-O→N acyl transfer at room temperature.18
Scheme 6.

The formation of 26 points to an eventual approach to serine ligation.20 In the succeeding paper, we probe subtle but important mechanistic issues as well as new directions for the 2CC reaction.
Supplementary Material
Detailed experimental procedures, copies of all spectral data, full characterization (PDF), and a cif file of X-ray for compound 10. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
Support for this research was provided by the National Institutes of Health (CA28824 to SJD). This investigation was supported by a “Research Centers in Minority Institutions” award, RR-03037 (to LJT), from the National Center for Research Resources, National Institutes of Health. Special thanks go to Rebecca Wilson for editorial consultation and Dana Ryan for assistance with the preparation of the manuscript. We thank Dr. Jianglong Zhu and Dr. Brendan Crowley for their helpful discussions. We thank Dr. George Sukenick for NMR spectroscopic assistance, and Ms. Hui Fang and Ms. Sylvi Rusli for mass spectrometric assistance. We also thank Prof. Shaabani for supplying experimental details regarding ref. 5a and Dr. Isaka for a personal communication regarding ref. 5e.
References
- 1.(a) Li X, Danishefsky SJ. J Am Chem Soc. 2008;130:5446. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jones GO, Li X, Hayden AD, Houk KN, Danishefsky SJ. Org Lett. 2008 doi: 10.1021/ol8016287. ASAP. [DOI] [PubMed] [Google Scholar]
- 2.Passerini M. Gazz Chim Ital. 1921;51:181.Ugi I, Meyr R, Fetzer U, Steinbrückner C. Angew Chem. 1959;71:386.For reviews, see Banfi L, Riva R. Org React. 2005;65:1.Dömling A, Ugi I. Angew Chem Int Ed. 2000;39:3168. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u.Dömling A. Chem Rev. 2006;106:17. doi: 10.1021/cr0505728.
- 3.The reaction described here is a special instance of a Mumm rearrangement which involves a 1,3-O→N acyl transfer not in the formimidate series. However, the formimidate context reported here renders this reaction unique. For literature access to the Mumm reaction, see: Mumm O, Hesse H, Volquartz H. Ber. 1915;48:379.Curtin DY, Miller LL. J Am Chem Soc. 1967;89:637.Sheehan JC, Corey EJ. J Am Chem Soc. 1952;74:4555.Schwarz JSP. J Org Chem. 1972;37:2906.Darbeau RW, White EH, Nunez N, Coit B, Daigle M. J Org Chem. 2000;65:1115. doi: 10.1021/jo991600n.
- 4.In our first paper in this series (reference 1), we described a direct pathway to 4, via a formal cycloaddition pathway, which does not pass through 3. In this paper, we focus on the presumed pathway from 3 to 4. However, involvement of the direct cycloaddition pathway to reach 4 is not ruled out.
-
5.Shaabani A, Soleimani E, Rezayan AH. Tetrahedron Lett. 2007;48:6137.Gloede J, Gross H. Zeitschrift fuer Chemie. 1968;8:219.Gloede J. J fuer Praktische Chemie. 1982;324:667.Shono T, Kimura M, Ito Y, Nishida K, Oda R. Bull Chem Soc Japan. 1964;37:635. The structure was presumed on the basis of its infrared spectrum. Isaka M, Boonkhao B, Rachtawee P, Auncharoen P. J Nat Prod. 2007;70:656. doi: 10.1021/np060509t. Actually, it is not clear whether the FCMA structure claimed in the paper is correct (see figure below). Personal communication from Dr. M. Isaka.
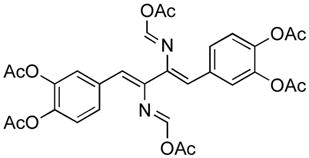 For cyclic structures of formimidate-carboxylate mixed anhydride, see: Black DSC, Boscacci AB. Aus J Chem. 1977;30:1109.Kobayashi S, Tsukamoto Y, Saegusa T. Macromolecules. 1990;23:2608.Reed PE, Katzenellenbogen JA. J Med Chem. 1991;34:1162. doi: 10.1021/jm00107a041.Gomory A, Somogyi A, Tamas J, Stajer G, Bernath G, Komaromi I. Inter J Mass Spec & Ion Proc. 1991;107:225.
For cyclic structures of formimidate-carboxylate mixed anhydride, see: Black DSC, Boscacci AB. Aus J Chem. 1977;30:1109.Kobayashi S, Tsukamoto Y, Saegusa T. Macromolecules. 1990;23:2608.Reed PE, Katzenellenbogen JA. J Med Chem. 1991;34:1162. doi: 10.1021/jm00107a041.Gomory A, Somogyi A, Tamas J, Stajer G, Bernath G, Komaromi I. Inter J Mass Spec & Ion Proc. 1991;107:225.
- 6.In some instances, spectroscopic suggestion of such an intermediate has been described. cf. Hou J-L, Ajami D, Rebek J., Jr J Am Chem Soc. 2008;130:7810. doi: 10.1021/ja802288k.Restorp P, Rebek J., Jr J Am Soc Chem. 2008 doi: 10.1021/ja803854r. ASAP.
- 7.Greater progress in characterization has been achieved with related mixed anhydrides which are not in the formimidate series. cf. inter alia Kawasaki K, Tsumura S, Katsuki T. Synlett. 1995;12:1245.Schwarz JSP. J Org Chem. 1972;37:2906.Cambie RC, Hayward RC, Roberts JL, Rutledge PS. J Chem Soc Perkin Trans 1. 1974;15:1858.
- 8.The authors reported absorptions at 1687 and 1598 cm−1 for the alleged mixed anhydride 7.
- 9.Typical IR absorptions reported for a non-formimidate mixed anhydride of the type 7 are ca. 1710–1660 cm−1; see reference 7.
- 10.The NMR spectrum of the diffraction worthy crystal is the same as that of the bulk material. For a crystal structure based on a hydrogen bonded complex between two “small molecules”, see: Leiserowitz L, Nader F. Acta Cryst. 1977;B33:2719.
- 11.(a) Attaur-Rahman, Basha A, Waheed N. Tetrahedron Lett. 1976;3:219. [Google Scholar]; (b) Cooley JH, Stone DM, Oguri H. J Org Chem. 1977;42:3096. [Google Scholar]; (c) Hendrickson JB, Hussoin MS. J Org Chem. 1989;54:1144. [Google Scholar]; (d) Rigby JH, Laurent S. J Org Chem. 1998;63:6742. [Google Scholar]; (e) Fernholz H, Schmidt HJ. Angew Chem Int Ed. 1969;8:521. [Google Scholar]; (f) Callens E, Burton AJ, Barrett AGM. Tetrahedron Lett. 2006;47:8699. [Google Scholar]; (g) Katritzky AR, He HY, Suzuki K. J Org Chem. 2000;65:8210. doi: 10.1021/jo000792f. [DOI] [PubMed] [Google Scholar]; (h) Shrestha-Dawadi PB, Jochims JC. Synthesis. 1993;4:426. [Google Scholar]; (i) Siebenmann C, Schnitzer RJ. J Am Chem Soc. 1943;65:2126. [Google Scholar]
- 12.(a) Gassman PG, Haberman LM. Tetrahedron Lett. 1985;26:4971. [Google Scholar]; (b) Kotha S, Brahmachary E. Bioorg Med Chem. 2002;10:2291. doi: 10.1016/s0968-0896(02)00039-1. [DOI] [PubMed] [Google Scholar]; (c) Priestley ES, Decicco CP. Org Lett. 2000;2:3095. doi: 10.1021/ol006284+. [DOI] [PubMed] [Google Scholar]; (d) Kotha S, Screenivasachary N, Mohanraja K, Durani S. Bioorg Med Chem Lett. 2001;11:1421. doi: 10.1016/s0960-894x(01)00227-x. [DOI] [PubMed] [Google Scholar]
- 13.Microwave heating of Gloede’s product (purported 15) in chloroform again failed to produce the desired N-formylamide.
- 14.It should be noted that the elemental analysis, “%N 10.45,” (reference 3b) of Gloede’s product 15 (calcd. %N 10.14) fits better for compound 17 (calcd. %N 10.33) than that concluded by Gloede. As reported by the authors, the claimed 15 does not react with methanol under reflux conditions.
- 15.Authentic 1,3-dicyclohexylamidine (18) was prepared according to a known procedure: Taylor EC, Ehrhart WA. J Org Chem. 1963;28:1108.
- 16.Simon JR. Synthesis. 2001;13:2011. [Google Scholar]
- 17.(a) Price CC, Roberts RM. J Am Chem Soc. 1946;68:1255. doi: 10.1021/ja01211a035. [DOI] [PubMed] [Google Scholar]; (b) Olguín LF, Jiménez-Estrada M, Bárzana E, Navarro-Ocaña A. Synlett. 2005;2:340. [Google Scholar]
- 18.This concept was suggested to account for the small amount of amide formation at room temperature; see reference 1. For cyclization examples that do not follow Baldwin’s rule, see: Astudillo MEA, Chokotho NCJ, Jarvis TC, Johnson CD, Lewis CC, McDonnel PD. Tetrahedron. 1985;41:5919.
- 19.Both isonitriles 25 and 27 were prepared in racemic form.
- 20.Okamoto R, Kajihara Y. Angew Chem Int Ed. 2008;47:5402. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures, copies of all spectral data, full characterization (PDF), and a cif file of X-ray for compound 10. This material is available free of charge via the Internet at http://pubs.acs.org