

Abstract
Current evidence indicates that methylation of cytosine in mammalian DNA is restricted to both strands of the symmetrical sequence CpG, although there have been sporadic reports that sequences other than CpG may also be methylated. We have used a dual-labeling nearest neighbor technique and bisulphite genomic sequencing methods to investigate the nearest neighbors of 5-methylcytosine residues in mammalian DNA. We find that embryonic stem cells, but not somatic tissues, have significant cytosine-5 methylation at CpA and, to a lesser extent, at CpT. As the expression of the de novo methyltransferase Dnmt3a correlates well with the presence of non-CpG methylation, we asked whether Dnmt3a might be responsible for this modification. Analysis of genomic methylation in transgenic Drosophila expressing Dnmt3a reveals that Dnmt3a is predominantly a CpG methylase but also is able to induce methylation at CpA and at CpT.
The existence of non-CpG methylation in mammalian DNA has been a contentious issue. Conventional wisdom has it that mammalian methylation occurs predominantly (or even exclusively) at CpG dinucleotides (1, 2). However, some studies analyzing the nearest neighbors of 5-methylcytosine (5mC) on a genome-wide level have indicated that non-CpG methylation is present in significant quantities (3–6). Indeed, one report (6) has indicated that 55% of all methylation in human spleen DNA could be at dinucleotides other than CpG. It is likely that these wide variations in the reported frequency of non-CpG methylation result from artifacts intrinsic in the conventional nearest neighbor technique, but until the advent of specialized sequencing strategies, there was no way of confirming or refuting these studies.
In fact, specialized sequencing strategies have offered little support for the existence of significant quantities of non-CpG methylation. One study reported non-CpG methylation in the promoter region of integrated adenovirus DNA, suggesting that it might be part of a de novo methylator response to invading viral genomes (7). Other work using mammalian cell lines and bisulphite sequencing (which enables the positive identification of all 5mC residues) reported non-CpG methylation within a stably integrated segment of plasmid DNA (8). The host cell lines appeared to have the capacity to maintain methylation at symmetric CpNpG sites, but some de novo CpNpG methylation at other sites within the integrated segment was also noted. Two reports indicating that non-CpG methylation might also be present in endogenous DNA elements, notably in mammalian replication origins (9, 10), have subsequently been disputed (11). The findings may have been attributable to failure of cytosine deamination in the bisulphite sequencing protocol. However, Woodcock et al. have observed nonsymmetric CpA and CpT methylation in an L1 element from human embryonic fibroblasts (12).
Despite the relative lack of supporting evidence from sequencing, it could still be argued that non-CpG methylation is present but is confined to selective areas of the genome. If this were the case, then only a global strategy would reliably detect it. We have developed an improved nearest neighbor technique to investigate this possibility and have also sequenced random fragments of bisulphite-modified DNA. Our results indicate that non-CpG methylation is easily detectable in embryonic stem (ES) cells but not in the soma, and that it is likely to arise as a result of a de novo cytosine-5 methyltransferase that is highly expressed in the embryo.
Materials and Methods
ES Cell Culture.
ES cells were grown in the presence of wild-type irradiated feeder cells. To eliminate possible contamination of the ES cells by these feeder cells, the cells were passaged twice in medium containing 1000 units of leukemia inhibitory factor before being harvested for DNA extraction.
DNA Preparation.
DNAs were extracted as previously described (13). RNA was removed by treatment with RNaseA and RNaseT1 followed by ethanol precipitation. DNAs were extracted from a number of mouse ES cell lines that differed according to their expression of DNA methyltransferase 1 (Dnmt1). These differences were engineered by targeted deletion of Dnmt1 and by stable integration of Dnmt1 minigenes (see Table 1 for genotypes and references). DNA was also extracted from liver, kidney, spleen, and lung, and from the mouse erythroleukemia cell line.
Table 1.
ES cells used for nearest neighbor analysis
| Genotype | Exp. | Description | Ref. |
|---|---|---|---|
| Dnmt1c/c | − | Homozygous mutant ES | 14 |
| Dnmt1s/s | − | Homozygous mutant ES | 14 |
| Dnmt1n/n | −* | Hypomorphic mutant ES | 15 |
| Dnmt1s/chip | +− | cDNA homologously integrated into Dnmt1s/s ES | 16 |
| Dnmt1s/s;cDNA | + | cDNA minigene randomly integrated into Dnmt1s/s ES | 17 |
| Dnmt1c/c;cDNA | + | cDNA minigene randomly integrated into Dnmt1c/c ES | 17 |
| Dnmt1+/+ | ++ | Wild-type ES | 15 |
| Dnmt1c/c;BAC | +++ | Dnmt1 BAC integrated into Dnmt1c/c ES | † |
| Dnmt1+/+;BAC | ++++ | Dnmt1 BAC integrated into wild-type ES | † |
*With residual activity.
†D.B. and R.J. (unpublished results).
Expression of Dnmt3a in Drosophila melanogaster.
The methods concerning Dnmt3a-expressing flies and extraction of their genomic DNA have previously been described (18).
An Overview of the Nearest Neighbor Analysis (NNA) Technique.
The technique presented here differs in two main respects from conventional NNA by nick labeling (2). First, the DNA is cut with a restriction enzyme and labeled by “fill-in.” This step improves the reproducibility of the results. Second, in the “dual-label” NNA procedure, the DNA is labeled with two different isotopes of phosphorous, which enables the frequency of non-CpG methylation (indicated by the 32P isotope) to be measured directly against the frequency of CpG methylation (indicated by the 33P isotope), and allows the elimination of confounding background radioactivity from the measurements.
Single-Label NNA.
The percentage of CpG methylated at FokI cut sites was determined in all DNA samples. DNA (10 μg) was digested with 30 units of FokI (GGATGN9–13, New England Biolabs) for 2 h at 37°C in a 150-μl reaction volume. The enzyme was heat-inactivated (60°C for 20 min), and the restriction fragments were precipitated in ethanol. After resuspension in 16 μl of TE buffer (10 mM Tris/1 mM EDTA, pH 8), the sample was split into four aliquots. One aliquot was used for single-labeling with [α-32P]dGTP, and the other aliquots were dual-labeled (see Dual-Label NNA). In addition, the percentage of CpA and CpT methylated at MvaI cut sites (CC/WGG) was also determined in DNA extracted from Drosophila flies expressing Dnmt3a. In this case, 10 μg of DNA was digested with 50 units of MvaI (MBI Fermentas, Vilnius, Lithuania; 5-h digest) and labeled with [α-32P]dATP and [α-32P]dTTP. In each labeling reaction, ≈2 μg (4 μl) of DNA was labeled in a 12-μl reaction volume with 20 μCi of [α-32P]dNTP (10 μCi/μl and 3000 Ci/mmol; Amersham Pharmacia; 1 Ci = 37 GBq) by using 2.5 units of Klenow (Amersham Pharmacia) and the manufacturer's fill-in buffer. After labeling for 15 min at 15°C, the reaction was stopped by addition of 2 μl of 0.2 M EDTA. Unincorporated nucleotides were removed by using Sephadex G50 spin columns (Boehringer Mannheim). The DNA was digested to deoxyribonucleotide 3′-monophosphates, separated by two-dimensional TLC, and quantified by scintillation as previously described (2).
Dual-Label NNA.
Three mixtures of labeling nucleotides were made, containing 1 vol of [α-33P]dGTP (≈2000 Ci/mmol, 10 μCi/μl; DuPont) with 1 vol of [α-32P]dATP, [α-32P]dTTP, or [α-32P]dCTP (each 3000 Ci/mmol, 10 μCi/μl; Amersham Pharmacia). Aliquots (2 μl) of these mixtures were used for labeling. The labeling, subsequent removal of unincorporated nucleotides, and digestion of the labeled DNA to deoxyribonucleotide 3′-monophosphates were identical to the procedure already described for the single-label analysis.
Separation of the Dual-Labeled Deoxyribonucleotide 3′-Monophosphates by TLC.
A 2.4-μl sample of the digest was spotted in 0.6-μl aliquots (with intermittent drying to prevent dispersion) onto a 20 × 20 cm glass-backed cellulose TLC plate (Merck) 1.5 cm from the bottom right corner. The digest first was separated by two-dimensional TLC using an isobutyric acid-based solvent [66 vol isobutyric acid (Sigma):18 vol water:3 vol 30% ammonia solution) in both dimensions. The labeled 5mCp nucleotide was detected by autoradiography and scraped off the TLC plate, and the radioactivity was eluted in 100 μl of water at 50°C for 30 min with intermittent vortexing. The sample was pelleted by centrifugation, and the supernatant was recovered and dried down in a DNA speed vacuum. After resuspending the sample in 2 μl of water, any contaminating background radioactivity (not related to 5mCp) was removed by another two-dimensional TLC in the isobutyric-based solvent (first dimension) followed by a solution of saturated ammonium sulfate (80 vol saturated ammonium sulfate:18 vol 1 M sodium acetate:2 vol isopropanol; second dimension). The purified 5mCp nucleotide was detected by autoradiography, scraped off the TLC plate, and eluted in water as described above. The 33P and 32P isotope activities within each sample were measured immediately after addition to 6 ml of scintillant with a Beckman LS 6000IC counter and a dual-label quench curve. The 5mCp samples were counted for 1 h to incur counting errors of <2% for the low-abundance isotope (32P).
Dual-Label Quench Curve.
The quench curve for simultaneous measurement of 33P and 32P activities was set up according to the procedure recommended by Beckman in the LS 6000IC operating manual and the Advanced Technology Guide (19). The counting windows used were 0–257.5 KeV (lower energy window) and 257.5–1280 KeV (higher energy window) (1 eV = 1.602 × 10−19 J). 32P activity was assumed to be detected with 99% efficiency at zero quench, and 33P activity was assumed to be counted with 95% efficiency at zero quench. To test the accuracy of the quench curve, 33P and 32P isotopes were mixed in varying proportion and the ratio of isotope activities predicted was plotted against the ratio of isotope activities measured. The relationship was found to be perfectly linear over a range of 33P:32P dpm/dpm activities from 0.1 to 150.
Quantification of Non-CpG Methylation.
Analysis of the DNA sequence in a typical 100-kb mouse DNA segment indicated that the frequencies of NpG, NpC, NpT, and NpA at FokI cut sites are equal. The relative specific activities of the labeling nucleotides (specific activity of the 33P nucleotide ÷ specific activity of the 32P nucleotide) therefore were taken to be equal to the 33P ÷ 32P dpm activity ratio of the whole-labeled DNA (DNAdpm RATIO) after separation of the unincorporated labeling nucleotides but before TLC.
The molar ratio of 33P:32P isotopes in 5mCp was found by dividing the 5mCp activity ratio (5mCdpm RATIO) by the relative specific activity. The 5mCMOLAR RATIO represents the frequencies of mCpG ÷ mCpA, mCpT, or mCpC (depending on the labeling mixture). Absolute levels of a methylated non-CpG dinucleotide were then determined by finding the reciprocal of 5mCMOLAR RATIO (e.g., the number of mCpA per mCpG) and by correcting for the extent of CpG methylation. Values were expressed per 100 (total) CpG at FokI cut sites (see Eq. 1).
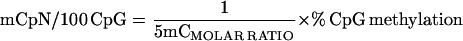 |
1 |
Bisulphite Modification, PCR, Cloning, and Sequencing of RsaI Fragments.
One microgram of DNA was digested for 6 h with 10 units of RsaI (Promega) in a 20-μl reaction volume. The restriction fragments were blunt-end ligated with phosphorylated EcoRI linkers (Promega), using T4 DNA ligase (Promega). The linked DNA fragments were modified with the bisulphite modification kit from Oncor, according to the manufacturer's instructions. The deaminated restriction fragments were PCR amplified with primers directed at the deaminated sequence of the EcoRI linkers, the remaining half of the RsaI site (5′-AATTTTGTTGTTGTTGAT-3′ and 5′-TTAATTCCATTACTATCAAC-3′). After 40 cycles of amplification (94°C denaturation for 1 min, 55°C annealing for 1 min, and 72°C extension for 1 min) with Taq polymerase, the PCR products were size-fractionated on a 1% agarose gel in TAE buffer (0.04 M Tris acetate, pH 8.3/1 mM EDTA), and a gel slab corresponding to 200–300 bp was cut out. The PCR products were recovered from the gel by using the QIAquick gel extraction kit (Qiagen, Chatsworth, CA), cloned into the pGEM T-easy cloning vector (Promega), and sequenced by using T7 and Sp6 sequencing primers and an ABI prism 373 automated sequencer (Applied Biosystems).
Two forms of sequence are revealed by using this technique: (i) a C-depleted, T-rich sequence corresponding to the modified strand, and (ii) a G-depleted, A-rich sequence corresponding to the complement of the modified strand. Therefore, any C in the modified strand or G in the complement of the modified strand represents a 5mC residue in the original sequence, and any G or A in the modified strand represents a G or A in the original sequence. However, any T in the modified strand is either a C or a T in the original sequence (as C deaminates to U and sequences as T). Hence, the occurrences of all mCpG, mCpA, and mCpmC can be positively identified, but it is not possible to distinguish between mCpT and mCpC in the original sequence.
PCR Amplification of the Deaminated DNA Fragment J1–22.
This fragment was amplified on three separate occasions from bisulphite-modified DNA by using the following primers: 5′-AAAATATGTAATTTTTATATATAGGTTT-3′ and 5′-CTTTTTAAACAAAATAATTAAACAATTA-3′. PCR conditions were as described for the linker PCR above, except that a 52°C annealing temperature was used. Cloning and sequencing were carried out as for the random PCR-amplified RsaI fragments.
Results
NNA Indicates That Non-CpG Methylation Is More Prevalent in Wild-Type ES Cells Than in Tissues Despite Similar Levels of CpG Methylation.
In ES cells with wild-type DNA methyltransferase 1 alleles (Dnmt1+/+), the average percentage methylation at CpG was 62.2% (Fig. 1A, black bars). In the somatic DNAs, the percentage CpG methylation levels were marginally higher, varying from 66% in liver to 81.6% in lung (Fig. 1A, clear bars). However, quantification of the amount of non-CpG methylation indicated that the levels of all methylated non-CpG dinucleotides were higher in ES cells than in tissues (Fig. 1 B–D). The mean level (±2 SD) of mCpA methylation in the Dnmt1+/+ ES cells was 15.8 ± 3.1 mCpA per 100 CpG at FokI cut sites, whereas in the somatic DNAs (mouse erythroleukemia cell line and tissue DNAs), the mean level was 3.4 ± 2.6 mCpA per 100 CpG (Fig. 1B). The difference was significant at P = 0.0013 (parametric two-sample t test). The levels of mCpT were severalfold lower than those of mCpA. In the Dnmt1+/+ ES cell samples, the mean level was 3.1 ± 0.7 mCpT per 100 CpG, but this was still 2 to 3 times higher than that observed in the somatic DNAs, which had a mean level of 1.25 ± 0.8 mCpT per 100 CpG (Fig. 1C). This difference was also significant (P = 0.0008). The least frequently methylated dinucleotide was mCpC. The mean level of this dinucleotide in the Dnmt1+/+ ES cells was 1.16 ± 0.38 mCpC per 100 CpG, but this was only marginally higher than the mean level of 0.83 ± 0.24 mCpC per 100 CpG found in the somatic DNAs (Fig. 1D). This difference was not significant at 95% confidence with the parametric two-sample t test (P = 0.11), but was significant when using a nonparametric Mann–Whitney test (P = 0.0369). As the severalfold difference in mCpA and mCpT between Dnmt1+/+ ES cells and tissues occurred on a background of similar levels of CpG methylation, erroneous labeling of mCpG sites (as mCpA, mCpT, or mCpC) could not have accounted for the differences in non-CpG methylation observed. The amount of any such error would have been similar in each of the DNAs analyzed. We conclude that mCpC methylation, if present, is at too low a level to be reliably detected in our assay.
Figure 1.

mCpA and mCpT are severalfold more prevalent in wild-type ES cells than in tissues, even in the absence of Dnmt1. Bar charts to illustrate levels of mCpG (A), mCpA (B), mCpT (C), and mCpC levels (D) as measured by NNA. Black bars indicate wild-type ES cells; clear bars indicate Dnmt1-null ES cells; light-gray bars indicate somatic DNAs; and checkered bars indicate Dnmt1 partial mutant, Dnmt1 rescued, and Dnmt1 overexpressing ES cells (see Table 1 for explanation of genotypes and references).
Levels of Dnmt1 Affect CpG Methylation but Not Non-CpG Methylation.
As expected, percentage CpG methylation levels were much lower in the Dnmt1s/s and Dnmt1c/c ES cells (Fig. 1A, light gray bars) than in the wild-type ES cells (18% vs. 62.2%, respectively). Both of these mutant ES cell lines are Dnmt1-null (14). However, levels of mCpA and mCpT were largely unaffected by null mutations of Dnmt1. The average levels found were 10.7 ± 3.3 CpA per 100 CpG and 2.8 ± 0.8 CpT per 100 CpG in the Dnmt1-null ES cells (Fig. 1 B and C, light gray bars). Therefore, on average, the mutant ES cells retained 68% and 91% of the wild-type mCpA and mCpT levels, respectively. This result indicates that non-CpG methylation is not caused by Dnmt1. The mCpA and mCpT levels in the Dnmt1-null ES cells also were significantly higher than those observed in the somatic DNAs (P < 0.01; Fig. 1 B and C).
Additional ES cell lines with varying levels of active Dnmt1 also were analyzed. These lines included the partial loss-of-function Dnmt1 mutant cell line Dnmt1n/n, “rescued” Dnmt1 mutant cell lines, and wild-type ES cell lines overexpressing Dnmt1 (Fig. 1, checkered bars; see Table 1 for details). The level of CpG methylation was seen to vary appreciably depending on the Dnmt1 genotype, with the Dnmt1BAC-expressing ES cells tending to have the highest levels of CpG methylation and the Dnmt1n/n ES cells having a level of CpG methylation that was intermediate between the Dnmt1c/c ES cells and the Dnmt1+/+ ES cells. High levels of non-CpG methylation were prevalent in all of these additional ES cell lines, irrespective of the level of functional Dnmt1 (and the level of CpG methylation), again confirming that non-CpG methylation must be the result of a separate methyltransferase activity.
The Contribution of Non-CpG Methylation to Total Methylation.
As the quantification of all methylated dinucleotides was made relative to the total number of CpG (methylated and unmethylated) at FokI cut sites, it also was possible to calculate the relative contribution of each dinucleotide to total cytosine methylation at FokI cut sites. In wild-type ES cells, 75.5 ± 6% (±2 SD) of the total methylation was at mCpG, 19.3 ± 6% at mCpA, 3.8 ± 0.5% at mCpT, and 1.4 ± 0.4% at mCpC. In Dnmt1-null ES cells (Dnmt1s/s and Dnmt1c/c), the contribution of non-CpG methylation to total methylation was much greater, with 54.7 ± 7.6% of the residual methylation at mCpG, 33.5 ± 5.2% at mCpA, 8.9 ± 1.4% at mCpT and 2.9 ± 2.2% at mCpC. In the tissue DNAs, on average, 93 ± 3.8% of the total methylation was at mCpG, 4.2 ± 3% at mCpA, 1.6 ± 1% at mCpT, and 1 ± 0.3% at mCpC.
Bisulphite Sequencing Confirms That Non-CpG Methylation Is Significant in ES Cell DNA but Minimal in Spleen DNA.
DNA from wild-type ES cells, Dnmt1c/c ES cells, and spleen were analyzed (see Table 2). Generally, only one strand of the PCR product was sequenced, but all occurrences of non-CpG methylation were confirmed by sequencing the complementary strand. Clones in which there appeared to have been complete failure of deamination (4 of 272 clones) were discarded from the analysis. As predicted by the NNA, mCpA was the most frequently methylated non-CpG dinucleotide in ES cells. In 14.5 kb of DNA sequence from wild-type ES cells, 8 kb of mCpA were detected, whereas in 21.3 kb of DNA sequence from Dnmt1-null ES cells, 10 kb of mCpA were detected. No mCpA was detected in spleen despite sequencing 18.8 kb of genomic DNA. This lack of any detectable mCpA in spleen indicates that the mCpA detected in wild-type and Dnmt1c/c ES cells is very unlikely to be attributable to an artifact such as failure of complete cytosine deamination or PCR error. All DNA samples were processed in parallel with the same reagents. Low levels of mCpT/C were detected in all DNAs, and no mCpmC moieties were detected in any of the sequences. Total non-CpG methylation was found to be 15% of the total methylation in wild-type ES cells, 37% of the total in Dnmt1c/c ES cells, and 2% of the total in spleen, the latter being entirely caused by mCpT/C. Generally, these figures agree well with those derived from NNA (Table 3).
Table 2.
Quantification of CpG and non-CpG methylation detected by random sequencing
| DNA | Total no. of clones | Cumulative sequence length, bp | No. with non-CpG methylation | No. of mCpG | No. of mCpA | No. of mCpT/C | No. of mCpmC |
|---|---|---|---|---|---|---|---|
| Dnmt1+/+ | 85 | 14,499 | 7 | 61 | 8 | 3 | 0 |
| Dnmt1c/c | 92 | 21,330 | 11 | 18 | 10 | 1 | 0 |
| Spleen | 95 | 18,881 | 2 | 95 | 0 | 2 | 0 |
Table 3.
The % contributions of CpG and non-CpG methylation to total methylation
| DNA | Dinucleotide | % Contribution
|
|
|---|---|---|---|
| NNA | Sequencing | ||
| Dnmt1+/+ ES | mCpG | 75.5 | 85 |
| mCpA, T, and C | 24.5 | 15 | |
| Dnmt1c/c ES | mCpG | 57.9 | 62 |
| mCpA, T, and C | 42.1 | 38 | |
| Spleen | CpG | 95 | 98 |
| mCpA, T, and C | 5 | 2 | |
Non-CpG Methylation Occurs at Both Symmetric and Asymmetric Sites.
The deaminated sequences surrounding the non-CpG methylated sites were examined to ascertain whether non-CpG methylation occurred within a particular consensus sequence. Determining a consensus sequence is hampered by the fact that the deaminated sequence contains redundancy in respect of T; any Ts that are present in the modified strand may have been either T or C in the original sequence. However, the locations of A and G in the modified strand are not affected by modification, so whether non-CpG methylation occurs at symmetric or asymmetric sites can be deduced. Non-CpG methylation was not restricted to symmetric sites such as Cp(N)npG. There also was no clear flanking consensus sequence for non-CpG methylation. Similarly, there was no consistent flanking consensus sequence for CpG methylation in the Dnmt1 mutants (data not shown).
Some mCpA Sites Are Preferentially Methylated.
To determine whether certain non-CpG methylated sites were preferentially methylated, we repeatedly sequenced DNA strands that were found to be methylated at mCpA in the random screen of RsaI fragments. One fragment from wild-type ES cells (J1–22) was found to have three sites of mCpA methylation in the initial screen (Fig. 2). By using the modified sequence, internal primers were designed specifically to amplify this sequence from each of the original samples of bisulphite-modified DNA. The PCR was performed on three separate occasions. The PCR products from each reaction were cloned. A total of 60–70 clones (20 from each reaction) were picked, and the plasmid DNAs were prepared for sequencing. One of the mCpA sites (nucleotide 122 in Fig. 2) had an increased frequency of methylation. In Dnmt1+/+ ES cells, 12 of 67 clones (18%) were methylated at this site, and in the Dnmt1c/c mutants, 6 of the 62 clones (10%) were methylated. In spleen, only 1 of 60 sequences was methylated at this site. Because the global amount of methylation at CpA is equivalent to 1–2% of CpA being methylated in ES cells, the frequency of methylation at this site is approximately 10 times higher than would be expected by chance in wild-type ES cells and 5 times higher than would be expected by chance in Dnmt1-null ES cells.
Figure 2.

Bisulphite genomic sequencing confirms the presence of non-CpG methylation in ES cell DNA. Sequence of J1–22, a PCR product derived from a bisulphite-modified RsaI fragment from wild-type ES cells (Dnmt1+/+) by using primers directed toward the linker DNA. The small arrows indicate the positions of nonsymmetrically methylated cytosine residues. The large solid arrows indicate the modified EcoRI linker DNA sequences. The dashed arrows indicate the sequences to which primers were designed for specific amplification of this DNA segment from bisulphite-modified but unlinked DNA.
Dnmt3a Methylates at CpG and to a Lesser Extent at CpA and CpT in Vivo.
To directly analyze the in vivo sequence specificity of Dnmt3a, we expressed Dnmt3a in Drosophila (18). Drosophila has no endogenous DNA methylation, and therefore even small amounts of non-CpG methylation could be analyzed. Single-label NNA was performed on FokI-digested genomic DNA from Dnmt3a-transgenic flies labeled with all four [α-32P]dNTPs, as well as on MvaI-digested DNA (CC/WGG) labeled with [α-32P]dATP and with [α-32P]dTTP. DNA from wild-type flies also was labeled as a control. The results of the [α-32P]dGTP and [α-32P]dATP labeling are presented in Fig. 3. In the DNA from transgenic flies, 10% of CpG, 1% of CpA, and 0.3% of CpT were found to be methylated at FokI cut sites. There was no convincing CpC methylation. At MvaI cut sites, 0.5% of CpA were methylated, but methylation at CpT was negligible (<0.1% of CpT). No methylation of any kind was detected in the wild-type controls. These data indicate that Dnmt3a has a relaxed substrate specificity, but has approximately a 10-fold preference for methylating CpG over CpA in Drosophila. This finding is consistent with the 2:1 ratio of mCpG:mCpA found in Dnmt1-null mouse ES cells, because the frequency of CpG is suppressed 4- to 5-fold in mouse DNA, and it is therefore reasonable to expect that the ratio of CpG:CpA methylation would be lower. The fact that some mCpT was clearly detectable at FokI cut sites but not at MvaI cut sites in the transgenic flies also indicates that Dnmt3a may be less prone to methylating CpT dinucleotides within certain sequence contexts.
Figure 3.

Dnmt3a induces CpG, CpA, and CpT methylation in Drosophila. NNA of genomic DNA extracted from wild-type (A, C, and E) and Dnmt3a-transgenic (B, D, and F) Drosophila. (A and B) FokI-digested DNAs labeled with [α-32P]dGTP. (C and D) FokI-digested DNA labeled with [α-32P]dATP. (E and F) MvaI-digested DNAs labeled with [α-32P]dATP. In the MvaI experiments (E and F), a small amount of labeling Ap, Tp, and Gp can also be seen. This finding is attributable to background labeling at nicks in the DNA. The identities of the labeled nucleotides are shown in B and F.
Discussion
This study demonstrates that, in contrast with somatic tissues, which have negligible non-CpG methylation, 15–20% of total cytosine methylation in ES cells is at sequences other than CpG. NNA and sequencing experiments also indicate that non-CpG methylation is not caused by Dnmt1 but is caused by another methyltransferase, most likely by the activity of the de novo methyltransferase Dnmt3a. Dnmt3a is highly expressed in ES cells and poorly expressed in tissues. This finding correlates well with the occurrence of non-CpG methylation in ES cells but not in tissues. In addition, this enzyme was shown to induce CpG as well as non-CpG methylation in Drosophila, supporting the notion that it catalyses non-CpG methylation in ES cells. Methylation at CpA was the most common form of non-CpG methylation found in both the NNA and the bisulphite sequencing experiments and also was the predominant form seen with the Dnmt3a transgene in Drosophila. As it could be argued that the absence of some crucial factor in Drosophila causes reduced specificity of methylation by Dnmt3a, analysis of non-CpG methylation in Dnmt3a−/− and Dnmt3b−/− ES cells will be the definitive test of the potential of these enzymes to induce non-CpG methylation. Dnmt3b was not tested in this study, but its high sequence homology with Dnmt3a (20) indicates that it too may methylate at non-CpG dinucleotides in addition to CpG.
NNA also predicted that mCpT sites, although 3–4 times less frequent, would be more prevalent in ES cells than in spleen. Also, mCpC sites would have sequenced as CpT if present. However, the random sequencing experiment did not show a difference between mCpT/C levels in ES cells and spleen. The most likely explanation for this is that because mCpT and mCpC are present in much lower frequency than mCpA, the background level of PCR mutations, which would be predicted to occur with approximately the same frequency as mCpT [1 to 2 × 10−4 (21)], tends to artifactually elevate the apparent level of CpT in the PCR products. This event would obscure any genuine difference that might otherwise be found between ES cells and spleen. Our failure to identify any mCpC methylation by NNA in Drosophila indicates either that its level is too low to be detected by any of these techniques, or that it is catalyzed by a separate methyltransferase.
It is clear that NNA by nick translation is prone to error, which may account for variable levels of non-CpG methylation reported by other authors using this technique (2, 4, 6). The fill-in method presented in this paper is far less prone to artifact than are previous techniques, but certain features indicate that the modifications introduced in our technique may not have abolished all artifactual labeling. In particular, the amount of mCpA methylation detected in the Dnmt1-null ES cells was a little less than that detected in the wild-type ES cells (10.7 vs. 15.8 mCpA per 100 CpG). This finding could indicate that DNAs that are more methylated at CpG also may be more prone to labeling as mCpA, possibly because of erroneous detection of some genuine mCpG sites as mCpA. The degree of this error clearly would depend on the level of mCpG. As the tissue DNAs and wild-type ES cell DNAs are highly methylated at CpG, erroneous labeling of this kind might account for most of the low-level non-CpG methylation detected in the somatic tissue DNAs, as well as for a fraction of the larger amount of non-CpG methylation detected in the wild-type ES cells. This explanation is supported by the tendency of the NNA technique to detect larger amounts of mCpA than the sequencing technique in DNAs with high levels of CpG methylation. NNA showed 4.2% and 19.3% of the total methylation at mCpA in spleen and wild-type ES cells, respectively, whereas sequencing showed 0% and 11% of the total at mCpA. Correspondingly, in cells with a low level of mCpG (Dnmt1c/c), mCpA contributes 33.5% of the total methylation as assessed by NNA and 34.5% as assessed by sequencing.
In the original publication describing the cloning of Dnmt3a and Dnmt3b, no non-CpG methylation was observed when baculovirus-expressed proteins were used for the in vitro methylation of synthetic oligonucleotides (20). Two possibilities might account for this discrepancy. First, the activities of Dnmt3a and Dnmt3b in vitro are low when compared with Dnmt1. Failure to detect non-CpG methylation simply may have been because of the very low levels that would have been present. Second, the rather low activities of the enzymes in vitro may be attributable to their having particular substrate requirements that are not met by the synthetic oligonucleotides. Our data indicate that non-CpG methylation may occur preferentially at certain sequences.
The functional significance of non-CpG methylation in early development is still uncertain and will be difficult to determine with any certainty, because non-CpG and CpG methylation are catalyzed by the same enzyme. The data in this paper and in the publication by Woodcock et al. (12) indicate that certain non-CpG sites may be preferentially methylated, leading to the possibility of maintenance methylation at certain sites by reiterative de novo methylation. Consistent methylation at certain sites may point to a functional role for this kind of methylation. However, the recent knockout studies of Dnmt3a and Dnmt3b in mice indicate that, as is the case for CpG methylation, their absence has no obvious consequence for ES cells (22). The effect on development is more difficult to determine. Dnmt3a−/−/Dnmt3b−/− mice die shortly after gastrulation, but Dnmt3a−/− mice survive until birth but die shortly thereafter.
In light of the findings of this study and the evidence that the Dnmt3 enzymes, and not Dnmt1, are responsible for de novo methylation (18, 22), we propose the following mechanism for the establishment of DNA methylation in mammalian development (Fig. 4). In early preimplantation embryos, the genome becomes demethylated (23), probably as a result of active and passive demethylation (24, 25). After implantation of the blastocyst, DNA methylation is restored. The de novo activity of Dnmt3 and its expression in early development indicate that it is the major enzyme responsible for reestablishing methylation patterns. However, the Dnmt3 enzymes are inefficient methylators of the DNA on their own. HPLC analysis indicates that in Dnmt1-null ES cells, only 1% of cytosines are methylated as compared with 4% in wild-type ES cells (data not shown). Dnmt3a also has a relaxed sequence specificity, methylating some non-CpG dinucleotides (predominantly CpA) as well as CpG. In early postimplantation development, Dnmt1 supports methylation by Dnmt3 by completing the methylation of those CpG sites that have been hemimethylated by Dnmt3 and by maintaining methylation of all reciprocally methylated CpG. When Dnmt3 is down-regulated in later development, no further de novo methylation occurs, and non-CpG methylation is concomitantly lost. Consistent with this model, non-CpG methylation is found in ES cells when Dnmt3 is active but disappears in tissues when Dnmt3 is expressed at low levels. The CpG specificity of methylation in the soma reflects the mCpG-specific maintenance methyltransferase activity of Dnmt1.
Figure 4.

Model for the reestablishment of DNA methylation after implantation. (A) In early preimplantation development, the genome becomes substantially demethylated. Dnmt1 is transcribed and is active but is not targeted to the nucleus (26). Nothing is known about the activities of Dnmt3a and Dnmt3b in this early stage of development (hence the dotted lines), the earliest stage at which transcripts have been documented being embryonic day 6.5 (20). (B) Northern blot analysis indicates that Dnmt3a activity is particularly high at embryonic day 10.5 (20), and it is reasonable to assume that this finding correlates with Dnmt3a activity. Hence, at this stage, de novo methylation at CpG, CpA, and CpT (square symbols) will be occurring as well as maintenance of mCpG sites by Dnmt1 (round symbols). (C) In late development, Dnmt3 activity is low. Therefore, maintenance methylation predominates, and non-CpG methylation is diminished.
Acknowledgments
This work was supported by the Medical Research Council of the U.K. (Clinical Training Fellowship awarded to B.H.R.) and by the Welsh Bone Marrow Transplant Fund. F.L. received a research fellowship from the Deutsche Forschungsgemeinschaft.
Abbreviations
- 5mC
5-methylcytosine
- ES cells
embryonic stem cells
- Dnmt
DNA methyltransferase
- NNA
nearest neighbor analysis
References
- 1.Sinsheimer R L. J Biol Chem. 1955;215:579–583. [PubMed] [Google Scholar]
- 2.Gruenbaum Y, Stein R, Cedar H, Razin A. FEBS Lett. 1981;124:67–71. doi: 10.1016/0014-5793(81)80055-5. [DOI] [PubMed] [Google Scholar]
- 3.Salomon R, Kaye A. Biochim Biophys Acta. 1970;204:340–351. [PubMed] [Google Scholar]
- 4.Grafstrom R H, Yuan R, Hamilton D L. Nucleic Acids Res. 1985;13:2827–2842. doi: 10.1093/nar/13.8.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nyce J, Liu L, Jones P A. Nucleic Acids Res. 1986;14:4353–4367. doi: 10.1093/nar/14.10.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodcock D M, Crowther P J, Diver W P. Biochem Biophys Res Commun. 1987;145:888–894. doi: 10.1016/0006-291x(87)91048-5. [DOI] [PubMed] [Google Scholar]
- 7.Toth M, Mueller U, Doerfler W. J Mol Biol. 1990;214:673–683. doi: 10.1016/0022-2836(90)90285-T. [DOI] [PubMed] [Google Scholar]
- 8.Clark S J, Harrison J, Frommer M. Nat Genet. 1995;10:20–27. doi: 10.1038/ng0595-20. [DOI] [PubMed] [Google Scholar]
- 9.Tasheva E S, Roufa D J. Somatic Cell Mol Genet. 1995;21:369–383. doi: 10.1007/BF02310205. [DOI] [PubMed] [Google Scholar]
- 10.Tasheva E S, Roufa D J. Mol Cell Biol. 1994;14:5636–5644. doi: 10.1128/mcb.14.9.5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rein T, Zorbas H, DePamphilis M L. Mol Cell Biol. 1997;17:416–426. doi: 10.1128/mcb.17.1.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodcock D M, Lawler C B, Linsenmeyer M E, Doherty J P, Warren W D. J Biol Chem. 1997;272:7810–7816. doi: 10.1074/jbc.272.12.7810. [DOI] [PubMed] [Google Scholar]
- 13.Laird P W, Zijderveld A, Linders K, Rudnicki M A, Jaenisch R, Berns A. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lei H, Oh S P, Okano M, Juttermann R, Goss K A, Jaenisch R, Li E. Development (Cambridge, UK) 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- 15.Li E, Bestor T H, Jaenisch R. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 16.Tucker K L, Beard C, Dausmann J, Jackson-Grusby L, Laird P W, Lei H, Li E, Jaenisch R. Genes Dev. 1996;10:1008–1020. doi: 10.1101/gad.10.8.1008. [DOI] [PubMed] [Google Scholar]
- 17.Tucker K L, Talbot D, Lee M A, Leonhardt H, Jaenisch R. Proc Natl Acad Sci USA. 1996;93:12920–12925. doi: 10.1073/pnas.93.23.12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyko F, Ramsahoye B H, Kashevsky H, Tudor M, Mastrangelo M, Orr-Weaver T L, Jaenisch R. Nat Genet. 1999;23:363–366. doi: 10.1038/15551. [DOI] [PubMed] [Google Scholar]
- 19.Steiner R. Advanced Technology Guide for LS 6500 Series Scintillation Counters (BR-7885B) Fullerton, CA: Beckman; 1996. [Google Scholar]
- 20.Okano M, Xie S, Li E. Nat Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 21.Tindall H R, Kunkel T A. Biochemistry. 1988;27:6008–6013. doi: 10.1021/bi00416a027. [DOI] [PubMed] [Google Scholar]
- 22.Okano M, Bell D W, Haber D A, Li E. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 23.Monk M, Boubelik M, Lehnert S. Development (Cambridge, UK) 1987;99:371–382. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 24.Shemer R, Kafri T, O'Connell A, Eisenberg S, Breslow J L, Razin A. Proc Natl Acad Sci USA. 1991;88:11300–11304. doi: 10.1073/pnas.88.24.11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rougier N, Bourc'his D, Gomes D M, Niveleau A, Plachot M, Paldi A, Viegas-Pequignot E. Genes Dev. 1998;12:2108–2113. doi: 10.1101/gad.12.14.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlson L L, Page A W, Bestor T H. Genes Dev. 1992;6:2536–2541. doi: 10.1101/gad.6.12b.2536. [DOI] [PubMed] [Google Scholar]