

Abstract
During the late phase of retroviral replication, newly synthesized Gag proteins are targeted to the plasma membrane (PM), where they assemble and bud to form immature virus particles. Membrane targeting by human immunodeficiency virus type 1 (HIV-1) Gag is mediated by the PM marker molecule phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], which is capable of binding to the matrix (MA) domain of Gag in an extended lipid conformation and of triggering myristate exposure. Here, we show that, as observed previously for HIV-1 MA, the myristyl group of HIV-2 MA is partially sequestered within a narrow hydrophobic tunnel formed by side chains of helices 1, 2, 3, and 5. However, the myristate of HIV-2 MA is more tightly sequestered than that of the HIV-1 protein and does not exhibit concentration-dependent exposure. Soluble PI(4,5)P2 analogs containing truncated acyl chains bind HIV-2 MA and induce minor long-range structural changes but do not trigger myristate exposure. Despite these differences, the site of HIV-2 assembly in vivo can be manipulated by enzymes that regulate PI(4,5)P2 localization. Our findings indicate that HIV-1 and HIV-2 are both targeted to the PM for assembly via a PI(4,5)P2-dependent mechanism, despite differences in the sensitivity of the MA myristyl switch, and suggest a potential mechanism that may contribute to the poor replication kinetics of HIV-2.
Keywords: HIV-1, HIV-2; Gag; myristyl, myr; matrix, MA; phosphatidylinositol-4,5-bisphosphate, PI(4,5)P2
Introduction
Human immunodeficiency virus types 1 and 2 (HIV-1 and HIV-2) are closely related primate lentiviruses derived by zoonosis from simians. HIV-1 was transmitted from chimpanzees and is genetically similar to simian immunodeficiency virus (SIV) from chimpanzees, whereas HIV-2 is similar to the sooty mangabee monkey strain of SIV (SIVSMM).1 HIV-1 and HIV-2 both infect CD4+ T cells and macrophages and are capable of causing AIDS. However, HIV-2 is considerably less pathogenic than HIV-1, and most HIV-2-infected individuals live relatively normal life spans even in the absence of antiretroviral therapy.2–4 For this reason, HIV-2 has received considerably less attention compared with HIV-1.
Individuals infected with HIV-2 typically have low plasma viral loads. Variations in HIV-1 viral load correlate with clinical status, with each threefold increase in plasma viral RNA correlating with a 50% decrease in 10-year survival.5 The viral loads in HIV-2-infected individuals have been found to be significantly lower than those in most HIV-1-infected individuals and similar to the loads observed in HIV-1 long-term nonprogressors.6,7 Although HIV-2-infected individuals mount a stronger immune response, it is not clear if or how these phenotypes are coupled.8 HIV-2 strains replicate much more slowly than do HIV-1 in vitro;9 this does not appear to be due to enhanced sensitivity to chemokines10 and may instead be due to the inherent properties of the virus. Understanding the molecular basis for differences in viral replication rates could facilitate the development of vaccines or improved therapeutics for the treatment of HIV-1-infected individuals.
Recent studies showed that HIV-2 is unable to assemble and bud from transfected yeast spheroplasts, despite the fact that HIV-1 is able to produce virus-like particles in this system.11 This defect was attributed to a reduced ability of HIV-2 Gag proteins to remain stably associated with the plasma membrane (PM) at virus assembly sites.11 Gag is the major structural protein of retroviruses,12,13 and membrane binding is mediated by its N-terminal matrix (MA) domain.14–31 MA contains an N-terminal myristyl (myr) group that can adopt sequestered and exposed conformations,32 consistent with a myr switch mechanism for regulating membrane binding.33–36 Exposure of the HIV-1 myr group can be increased by factors that promote protein self-association, such as inclusion of the proximal capsid domain of Gag.32,37–40 Although these findings explain why Gag and Gag/RNA assemblies bind membranes more tightly than the isolated MA protein in vitro, they do not provide insights into the mechanism of specific membrane targeting in vivo.
More recent studies25 revealed that membrane targeting by HIV-1 Gag is regulated by phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2], a cellular factor associated predominantly with the inner leaflet of the PM.41–43 PI(4,5)P2 is considered a major landmark for cellular proteins that need to be concentrated at the PM41–43 and is linked to a number of cellular functions.43 Depletion of PI(4,5) P2 inhibits HIV-1 assembly and leads to accumulation of Gag at the membranes of late endosomes and multivesicular bodies,25 and induction of PI(4,5)P2-enriched endosomes retargets Gag assembly and budding to these endosomes.25 Soluble PI(4,5)P2 analogs with truncated acyl chains are capable of binding to HIV-1 MA and of triggering exposure of the myr group.29 It thus appears that HIV-1 hijacks the PI(4,5)P2 trafficking system to target Gag to assembly sites on the PM.
Since it has yet to be determined if other retro-viruses utilize a similar PI(4,5)P2-dependent mechanism and in view of the different clinical, cellular, and biochemical properties of HIV-1 and HIV-2, we initiated structural and PI(4,5)P2 binding studies of the HIV-2 MA protein in vitro and assessed the influence of PI(4,5)P2 on HIV-2 assembly in cells. Our findings indicate that, like HIV-1, membrane targeting by HIV-2 Gag is mediated by PI(4,5)P2. However, the myr switch of HIV-2 MA is significantly less sensitive than that of the HIV-1 protein, suggesting a potential mechanism for inhibited membrane binding in yeast and reduced replication rates in vitro.
Results
NMR analysis of the HIV-2 unmyristylated and myristylated MA proteins
Both unmyristylated [myr(−)] and myristylated [myr(+)] forms of HIV-2 MA were co-expressed in Escherichia coli and purified by column chromatography, and purity and myristylation efficiency were verified by mass spectrometry. Two-dimensional (2D) 1H–15N heteronuclear single quantum coherence (HSQC) spectra obtained for HIV-2 myr(−)MA and myr(+)MA were very similar, except for signals associated with residues Leu31, Gly49, and the stretch of residues near the N-terminus (Gly2–Glu17) (Fig. 1). For both proteins, the 1H and 15N NMR signals were insensitive to concentration over the range of 50 μM to ∼1 mM. These findings contrast with those obtained previously for HIV-1 myr(+)MA, in which a subset of signals was shown to shift progressively toward the frequencies observed for myr(−)MA upon increasing the protein concentration. These changes observed for the HIV-1 protein were attributed to a concentration-dependent shift in a monomer–trimer equilibrium that occurs with concomitant exposure of the myr group.32 The absence of concentration-dependent shifts for HIV-2 myr(+)MA indicates that the protein exists in a unique conformation under these conditions. Sedimentation equilibrium data obtained for both myr(−)MA and myr(+)MA are similar in appearance and confirm that both proteins remain monomeric at concentrations as high as 110 μM (Fig. 1). The protein precipitates from solution at concentrations above 1 mM, which could indicate the formation of an insoluble myr-exposed species.
Fig. 1.
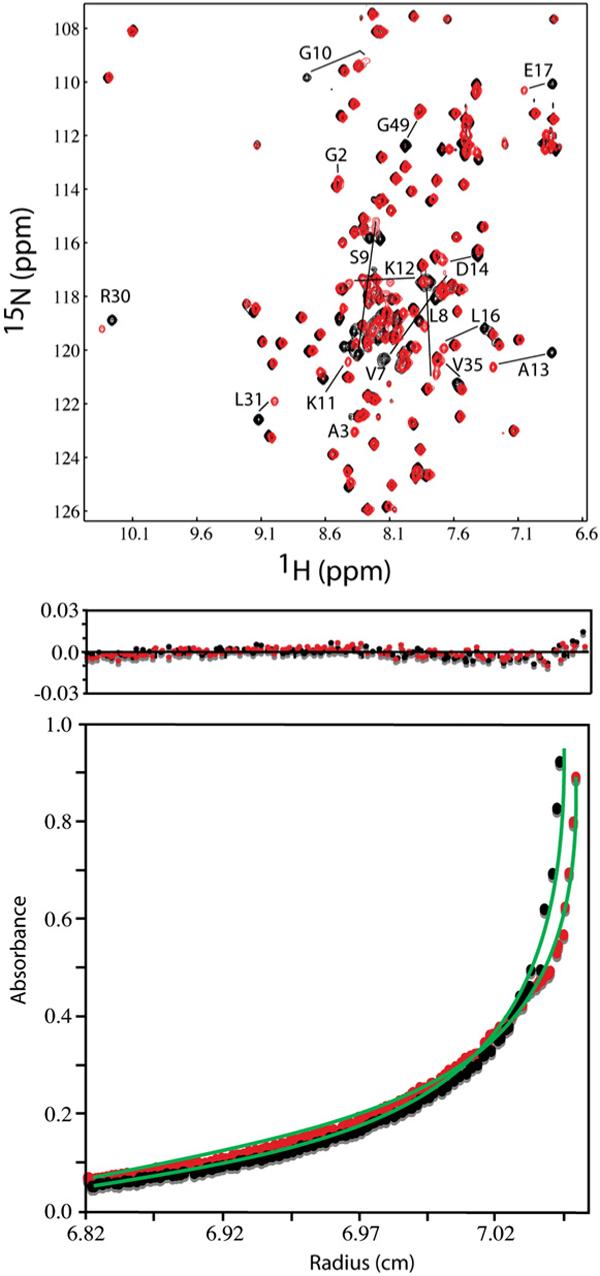
(a) Overlay of 2D 1H–15N HSQC spectra collected for HIV-2 myr(−)MA (black) and myr(+)MA (red) proteins (500 μM, 35 °C). Significant chemical shift changes occur for residues that are affected by the positioning of the myr group. (b) Representative sedimentation profiles obtained for myr(−)MA (black) and myr(+)MA (red) proteins (26,000 rpm, 20 °C, 110 μM). For both proteins, sedimentation profiles fit best to monomeric species.
A combination of 2D [1H–1H nuclear Overhauser enhancement spectroscopy (NOESY), 1H–15N HSQC, and 1H–13C heteronuclear multiple quantum coherence] and 3D as well as 4D [15N-, 13C-, 15N/13C-, 13C/13C-, and 13C-edited/12C-double-half-filtered NOE] data was collected for myr(−) MA and myr(+)MA proteins. The main spectral differences between myr(−)MA and myr(+)MA were associated with the first 17 residues (Gly2–Glu17) of the protein. For myr(−)MA, Val7 exhibited numerous backbone and side-chain NOEs with residues Ile34, Leu51, Glu52, Ile85, and His89, whereas Leu8 exhibited NOEs with residues Ala13, Leu16, Ile34, Ile85, and His89. These NOEs are either weak or absent in the spectra collected for myr(+) MA. The 2D NOESY, 3D 13C-edited NOE, and 3D 13C-edited/12C-double-half-filtered NOE data obtained for myr(+)MA show numerous unambiguous NOEs between the myristate group and the side chains of Val7, Leu8, Leu16, Ile34, Ala38, Glu48, Leu51, Glu52, and Ile85, and representative portions of the 3D 13C-edited/12C-double-half-filtered NOE data collected for 13C-labeled myr(+)MA with the unlabeled myristate group are shown in Fig. 2. These data indicate that the myristate group is buried within the core of the protein and makes contacts with the side chains of Ile34, Glu52, and Ile85. In addition, strong NOE cross-peaks were observed between the terminal methyl group (myr-C14H3; ∼0.65 ppm) and the side chains of Leu16 and Ile85, indicating a close packing of the myr group against these hydrophobic residues. In all spectra obtained, no new intraprotein NOE that would be indicative of a significantly different protein conformation was detected. Together, the NMR data indicate that myristylation does not dramatically alter conformation of the protein and that the relatively small spectral changes observed for residues Gly2–Glu17 reflect minor local adjustment to allow insertion of the myr group.
Fig. 2.
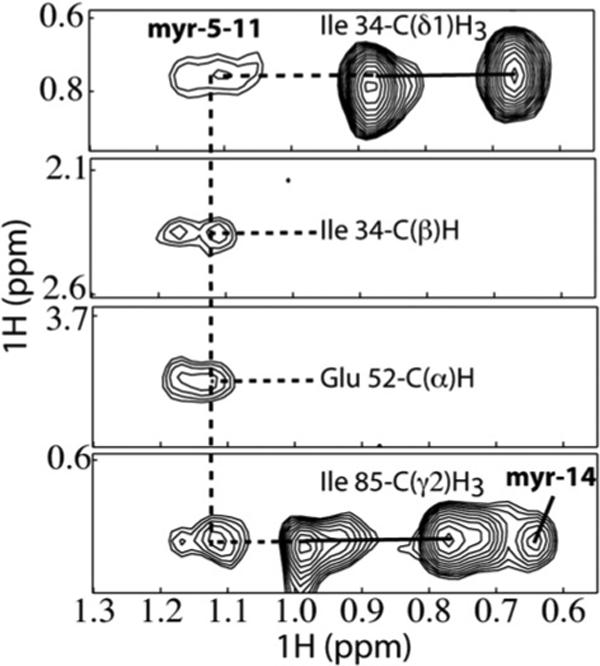
Three-dimensional 13C-edited/12C-double-half-filtered NOE data obtained for HIV-2 myr(+)MA showing unambiguously assigned intermolecular NOEs between the myristate group and key residues of a 13C-labeled protein sample (myristate group is not 13C labeled). Continuous and dashed lines denote 1H–12C breakthrough doublets and NOE peaks, respectively.
Structures of HIV-2 myr(−)MA and myr(+)MA
The myristylated and unmyristylated forms of the HIV-2 myr(+)MA protein consist of five α-helices and a 310-helix capped by a three-stranded β-sheet. In both proteins, helices 1−4 pack against a long central helix (helix 5). The observed folding is similar to that observed in the X-ray structure of the trimeric unmyristylated SIV myr(−)MA protein,44 except that our data indicate that both forms of the HIV-2 protein are monomeric in solution, and C-terminal residues that form a short β-hairpin in SIV myr(−)MA are disordered in HIV-2 myr(+)MA and myr(−)MA. Myristylation does not induce significant structural changes in the overall shape of the HIV-2 myr(+)MA protein. Superpositions of the 20 lowest-penalty structures calculated for HIV-2 myr(−)MA and myr(+)MA are shown in Fig. 3 (see also Table 1). The 25 C-terminal residues appear to be disordered, based on the absence of significant amide-to-amide (i to i + 1 and i to i − 1) and amide–side-chain NOEs, as well as near-zero residual dipolar couplings (RDCs). For myr(+)MA, the myr group adopts an extended conformation, with the myr-C14H3 group packing in close proximity to the side chains of core residues Leu16, Ile34, and Leu85 and other myristate methylene groups interacting with the side chains of Val7, Leu8, Ile34, Ala38, Val35, Glu48, Leu51, and Glu52 (Fig. 4).
Fig. 3.
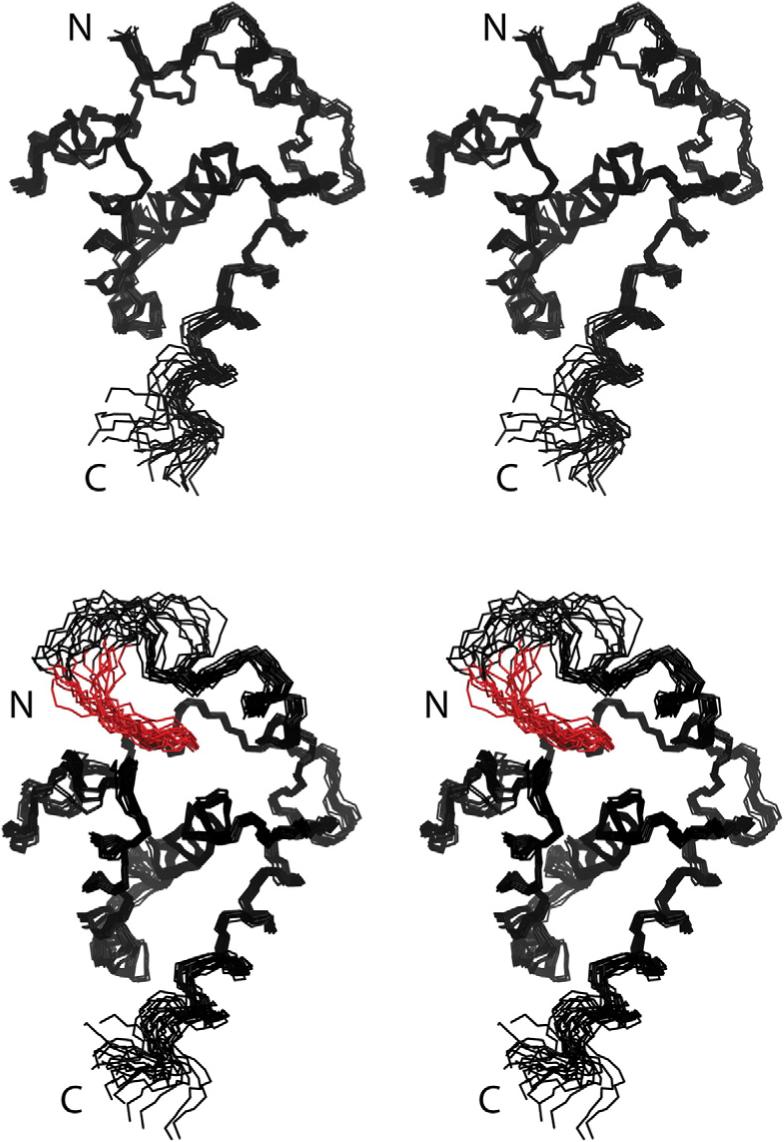
Stereoviews showing the best-fit backbone superpositions of the 20 refined structures calculated for HIV-2 myr(−)MA (top) and myr(+)MA (bottom) proteins. The myristate group is shown in red. The last 15 residues of the protein are not shown because they are disordered.
Table 1.
Statistical data
| myr(−) MA | myr(+) MA | myr(+)MA/ di-C4-PI(4,5)P2 | |
|---|---|---|---|
| NMR-derived restraints | |||
| 1H-1H distance restraints | 850 | 851 | 881 |
| Intraresidue | 97 | 97 | 99 |
| Sequential (|i - j| = 1) | 160 | 160 | 160 |
| Medium and long range (|i - j| > 1) | 593 | 583 | 607 |
| Backbone H bonds (4/H bond) | 0 | 0 | 0 |
| Side-chain H bonds (4/H bond) | 24 | 20 | 20 |
| Dipolar coupling restraints | 85 | 86 | 85 |
| Intermolecular | 0 | 0 | 24 |
| Average restraints/refined residue | 15.9 | 15.7 | 15.7 |
| Target function (Å2) | |||
| Mean (SD) | 0.58 (0.05) | 0.48 (0.05) | 0.93 (0.5) |
| Minimum | 0.48 | 0.38 | 0.31 |
| Maximum | 0.65 | 0.62 | 1.60 |
| Restraint violations | |||
| Average maximum upper distributed (Å) | 0.15 | 0.11 | 0.26 |
| ±SD | 0.04 | 0.03 | 0.18 |
| Average maximum van der Waals (Å) | 0.26 | 0.25 | 0.28 |
| ±SD | 0.01 | 0.01 | 0.07 |
| Average maximum RDC (Hz) | 0.03 | 0.02 | 0.02 |
| ±SD | 0.01 | 0.01 | 0.02 |
| PROCHECK statistics (%) | |||
| Residues in most favored regions | 84.6 | 84.4 | 82.9 |
| Residues in additional allowed regions | 14.4 | 14.4 | 15.2 |
| Residues in generously allowed regions | 0.7 | 1.1 | 1.7 |
| Residues in disallowed regions | 0.3 | 0.2 | 0.2 |
| Structure convergence [Å2; mean (SD)] | |||
| Main-chain atoms | 0.38 (0.07) | 0.47 (0.16) | 0.43 (0.19) |
| All heavy atoms | 0.84 (0.06) | 0.92 (0.14) | 1.22 (0.30) |
Fig. 4.

A representative structure of HIV-2 myr(+)MA (slate) and comparison with the HIV-1 myr(+)MA protein (sand). (a) Semitransparent surface representation of MA showing the penetration of the myr group (red sticks) and interactions with the side chains of Val7, Leu8, Leu16, Ile34, and ILe85 (green sticks). (b) Cartoon representation of the HIV-2 and HIV-1 myr(+)MA proteins comparing the sequestration of the myristate group (red) in the hydrophobic cavity formed by residues Val7, Leu8, Leu16, Ile34, and Ile85 (green spheres). (c) Superimposition of representative structures of HIV-2 and HIV-1 myr(+)MA. Myristate groups of HIV-2 and HIV-1 myr(+)MA proteins are packing against Leu16 and Trp16, respectively. NMR data revealed that helix 6 is flexible for both proteins. (d) An expanded view of the protein core of HIV-2 and HIV-1 myr(+)MA showing the myristate packing against side chains of Leu16 and Trp16, respectively.
As observed previously for the corresponding HIV-1 proteins,32 significant NMR spectral and structural differences were observed for residues Gly2–Glu17 of the myristylated and unmyristylated HIV-2 MA proteins. Residues Arg9–Glu17 form a compact helix (e.g., Ala13 packs tightly against Leu16) with a 310 character in myr(−)MA but form a more extended α-helix in the myristylated MA protein. In addition, the side chain of Lys12, which is located near the N-terminus of helix 1, is exposed to solvent and does not make long-range contacts in myr(+)MA but packs tightly against the side chain of Val88 and forms a Lys12--to-His89 Nδ2 hydrogen bond in myr(−)MA. Interestingly, Lys12 is substituted by Glu in HIV-1, and this residue forms a His89 Hδ2 to the Glu12 COO− salt bridge in the HIV-1 myr(−)MA structure (Fig. 5).
Fig. 5.
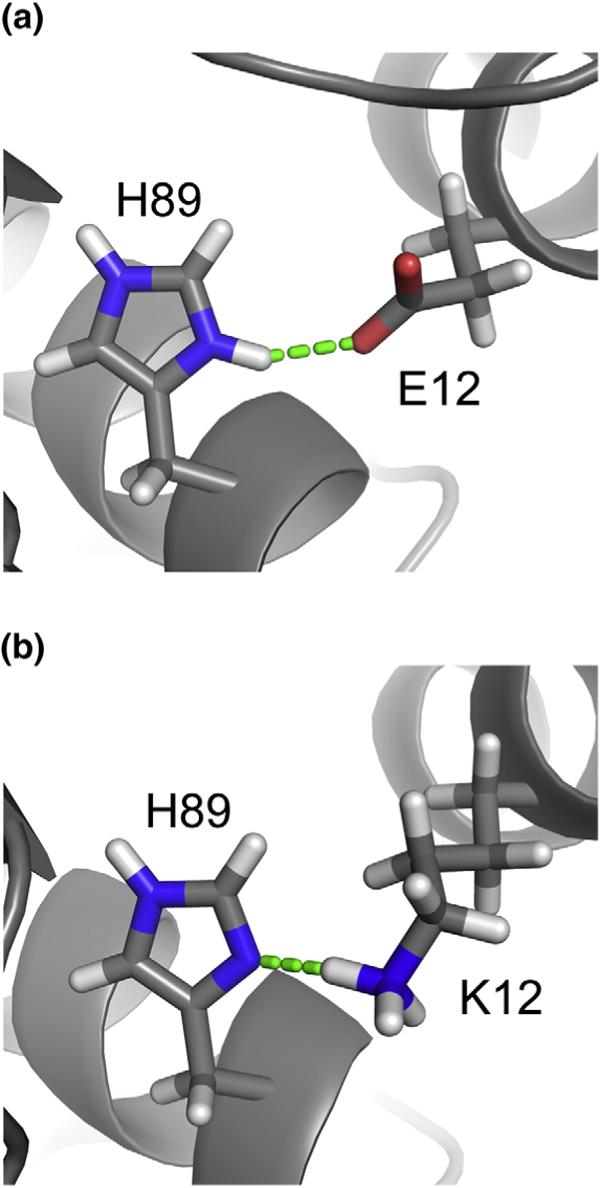
Comparison of the HIV-2 myr(−)MA structure with the myristate-exposed HIV-1 myr(+)MA structure. The packing of helix 1 is stabilized by a buried His(+)— Glu(−) salt bridge in the HIV-1 protein, whereas the packing is stabilized by a buried His−Lys(+) salt bridge in the HIV-2 protein.
PI(4,5)P2 binds to HIV-2 MA
Addition of substoichiometric amounts of native PI(4,5)P2 species to either myr(−)MA or myr(+)MA led to severe broadening in the 1H–15N HSQC NMR spectra. Signals for backbone NH groups were broadened beyond detection at 1:1 PI(4,5)P2/MA stoichiometries. Signal broadening is mainly due to the formation of micelles in aqueous solution, which is promoted by the long 1′- and 2′-acyl chains.45 Previous studies have shown that phosphoinositide derivatives with long 1′- and 2′-acyl chains were solubilized when a combination of detergents was used.46–48 Attempts to study native PI(4,5)P2 binding in the presence of dodecylphosphocholine, 1,2-diheptanoyl-sn-glycero-3-phosphocholine, or a combination of these detergents and Chaps (3-[(3-cholamidopropyl)dimethylammonio]propanesulfonic acid) were unsuccessful due to detergent-induced protein unfolding. Thus, studies were conducted with soluble forms of PI(4,5)P2 containing truncated lipids.
Representative 1H–15N HSQC NMR data obtained upon titration of HIV-2 myr(+)MA with di-C4-PI(4,5)P2 are shown in Fig. 6. Titration of di-C4-PI(4,5)P2 led to significant changes in the backbone 1Hand 15N NMR chemical shifts of residues Arg22, Lys27, Lys28, Arg30, His33, ILe34, Trp36, Ala37, Asn39, Leu41, Leu46, Leu75, Ser77, Leu78, and Thr81 [ΔδHN ((Δδ1-H)2 + (Δδ15N)2)1/2=0.1−0.6 ppm] (Fig. 6). These residues reside on a β-II–V cleft and were previously shown to contribute to the PI(4,5)P2 binding site of HIV-1 myr(+)MA. Nonlinear least-squares fits of the titration data afforded a dissociation constant (Kd) value of 143±16 μM. Similar binding studies conducted on HIV-2 myr(−)MA led to very similar changes in the 2D HSQC spectra and afforded a Kd value of 129±14 μM. These values are in close agreement with those observed for di-C4-PI(4,5)P2 binding to HIV-1 myr(−)MA and myr(+)MA (Kd=240±60 and 150±30 μM, respectively). Titration of di-C8-PI(4,5)P2 into HIV-2 myr(+)MA led to similar chemical shift changes at relatively low di-C8-PI(4,5)P2/MA ratios (<1:1) but also induced sample precipitation. No detectable change was observed in the 2D 1H–15N HSQC spectra of myr(+)MA upon titration with di-C4-PI, di-C4-PI(3)P, di-C4-PI(4)P, di-C4-PI(5)P, di-C4-phosphatidylcholine, and di-C4-PI(3,5)P2.
Fig. 6.
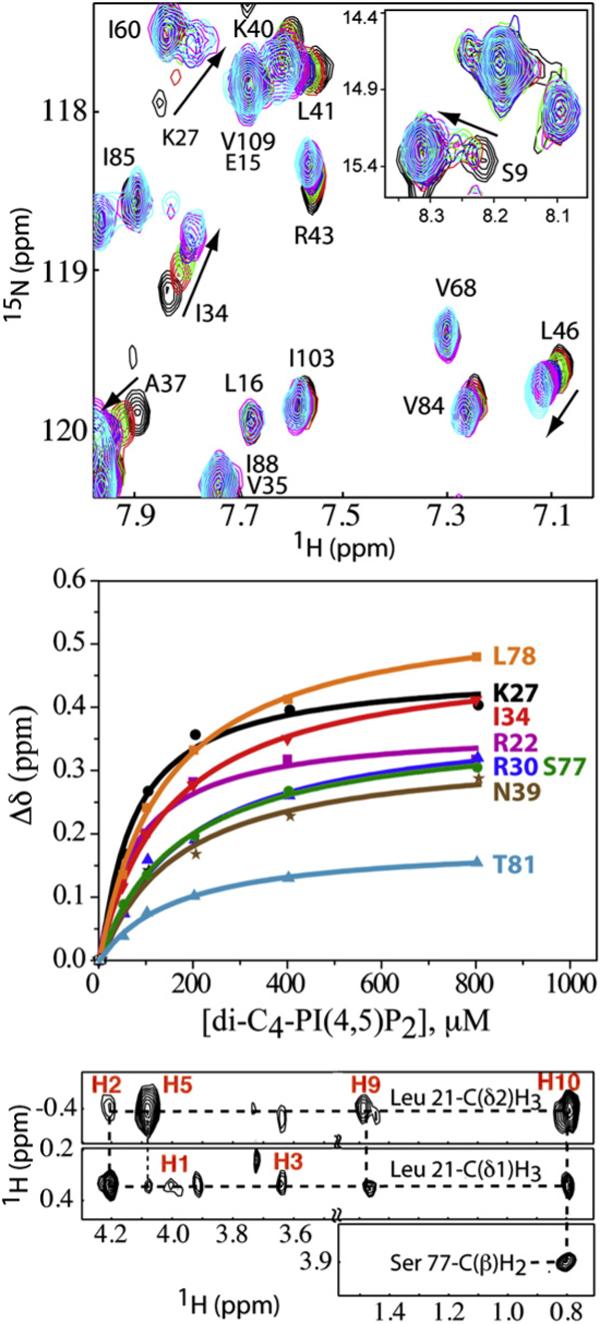
(a) Overlay of 2D 1H–15N HSQC spectra upon titration of HIV-2 myr(+)MA with di-C4-PI(4,5)P2 [60 μM, 35 °C; di-C4-PI(4,5)P2/MA = 0:1 (black), 1:1 (red), 2:1 (green), 4:1 (blue), 8:1 (magenta), and 16:1 (cyan)]. (b) 1H and 15N NMR chemical shift titration data, which fit to 1:1 binding isotherms (Kd = 143±16 μM). (c) Representative 13C-edited/12C-double-half-filtered NOE data showing unambiguously assigned intermolecular NOEs between residues Leu21, Ser77, and di-C4-PI(4,5)P2. Dashed lines denote intermolecular NOE peaks.
Structure of the myristate-sequestered di-C4-PI(4,5)P2/MA complex
The structure of the di-C4-PI(4,5)P2/MA complex was determined using a combination of 15N-, 13C-, 15N/13C-, 13C/13C-, and 13C-edited/12C-double-half-filtered NOE experiments.49,50 Unambiguous intermolecular NOEs were observed between the myristate group and the side chains of Val7, Leu8, Leu16, Ile34, Ala38, Glu48, Leu51, Glu52, and Ile85, indicating that the myr group remains sequestered, at least to a large extent, upon di-C4-PI(4,5)P2 binding. In addition, NOE cross-peaks were observed between the inositol ring protons and the side chains of Leu21 and Lys27, and the H1′,H2′, and H3′ groups of the glycerol moiety show NOE cross-peaks to the side chains of His33 and Trp36 (Fig. 6). No NOE cross-peak was observed between the 1′-acyl chain and protein residues, which suggests that the 1′-acyl chain is exposed to solvent.
The NMR data are consistent with a single binding mode, with di-C4-PI(4,5)P2 binding to a highly basic surface on the myr(+)MA protein (Fig. 7). di-C4-PI (4,5)P2 binds to a β-II–V cleft, with the glycerol moiety packing against the side chains of His33 and Trp36 and the 2′-acyl chain packing within the β-II–V cleft against the side chains of Leu21, Lys27, Tyr29, His33, Trp36, and Ser77. The phosphoinositide head group packs against Leu21 and Lys27, burying the 2′-acyl chain. In addition to the hydrophobic contacts, the 1′-phosphodiester is poised to make favorable electrostatic interactions with the positively charged side chains of His33 and Lys27, and the 4′- and 5′-phosphates are positioned to form salt bridges with the side chains of Arg22 and Lys76, respectively (Fig. 7).
Fig. 7.
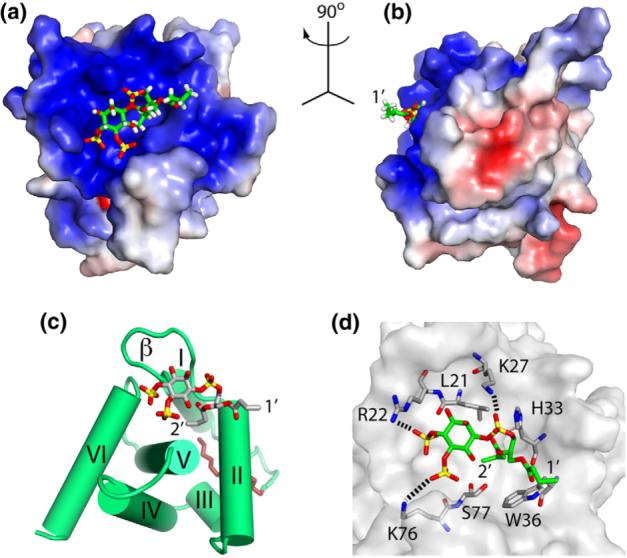
Structure of the HIV-2 myr(+)MA/di-C4-PI(4,5)P2 complex. (a,b) Interactions between di-C4-PI(4,5)P2 (sticks) and MA (colored according to electrostatic surface potential) showing the 2′-fatty acid inserting in a preexisting cleft and the inositol ring packing against a basic patch of the protein. (c) PI(4,5)P2 binding to the β-II–V cleft. (d) A network of interactions implicated in PI(4,5)P2 binding.
di-C4-PI(4,5)P2 binding primes, but does not trigger, the myr switch
The HIV-1 myr(+)MA protein exists as a preexisting equilibrium between myristate-sequestered and myristate-exposed conformations, and titration with di-C4-PI(4,5)P2 leads to a shift in equilibrium toward the myr-exposed conformer.29 This equilibrium shift leads to large changes in the backbone 1H and 15N NMR chemical shifts for most of the N-terminal residues and residues of helix 1.29 Although a few residues near the N-terminus of the HIV-2 myr(+) MA protein exhibited small di-C4-PI(4,5)P2-dependent 1H–15N chemical shifts (for example, see Fig. 6), the NMR signals for most of these residues were not significantly affected by di-C4-PI(4,5)P2 binding. However, the linewidths of nearly all these residues were selectively and significantly increased upon titration with di-C4-PI(4,5)P2, indicating a destabilization of, or change in, the structure of helix 1. Likely explanations are that di-C4-PI(4,5)P2 binding promotes the formation of a very small population of a myristate-exposed conformer and that this conformer undergoes chemical exchange on an approximately millisecond time scale with the myristate-sequestered species. The population of the myristate-exposed species would have to be relatively small because the chemical shifts of most residues are not significantly perturbed.
The subcellular site of HIV-2 Gag assembly is regulated by PI(4,5)P2
A previous study demonstrated that disruption of PM PI(4,5)P2 levels induced a retargeting of HIV-1 Gag to intracellular compartments.25 Overexpression of the PI(4,5)P2 phosphatase 5-phosphatase IV (5ptaseIV) caused HIV-1 Gag to assemble in multi-vesicular bodies, and overexpression of a dominant-active mutant of Arf6 (Arf6/Q67L) induced the formation of PI(4,5)P2-enriched endosomal-like structures to which HIV-1 Gag was targeted.25 To determine whether the localization of HIV-2 Gag assembly would be similarly altered by PI(4,5)P2 disruption, we co-transfected the HIV-1 molecular clone pNL4−3 or the HIV-2 molecular clone pROD10 with plasmids expressing 5ptaseIV or Arf6/Q67L. We also tested the effect of a 5ptaseIV mutant from which the phosphatase domain was deleted (5ptaseIV/Δ1) as a negative control.25 Transfected cells were metabolically radiolabeled with [35S]Met/Cys, and cell and viral lysates were prepared and immunoprecipitated with HIV-immunoglobulin (Ig) (for HIV-1) or an antibody specific for HIV-2 Gag. Cell- and virion-associated Gag levels were quantified, and virus release efficiency was calculated. As reported previously,25 both 5ptaseIVand Arf6/Q67L overexpression markedly reduced the efficiency of HIV-1 particle production (Fig. 8). The effect of 5ptaseIV on particle production was eliminated by the Δ1 deletion. Interestingly, 5ptaseIV and Arf6/Q67L overexpression also significantly reduced levels of released HIV-2 virions, indicating that the production of both HIV-1 and HIV-2 particles is inhibited by PI(4,5)P2 perturbation.
Fig. 8.

Disruption of PI(4,5)P2 inhibits HIV-1 and HIV-2 particle production. HeLa cells were transfected with HIV-1 (pNL4−3) or HIV-2 (pROD10) molecular clones alone (−) or were co-transfected at a 1:2 DNA ratio with vectors expressing 5ptaseIV, the Δ1 deletion mutant of 5ptaseIV, or Arf6/Q67L. Transfected cells were metabolically radiolabeled with [35S]Met/Cys for 2 h. Cell and viral lysates were prepared and immunoprecipitated with HIV-Ig (for HIV-1 samples) or with the anti-p27gag monoclonal antibody R1C7 (for HIV-2 samples). Gag proteins were quantified by phosphorimager analysis. Virus release efficiency was calculated as the amount of virion-associated Gag as a fraction of total (cell plus virion) Gag.
To determine the molecular basis for the reduced HIV-2 particle production observed upon 5ptaseIV and Arf6/Q67L overexpression, we fixed and examined cells transfected as described above by transmission electron microscopy (EM). In the absence of PI(4,5)P2 disruption, both HIV-1 and HIV-2 assemblies were observed predominantly at the PM (Fig. 9a and b). As reported previously,25 overexpression of 5ptaseIV and Arf6/Q67L induced the mistargeting of HIV-1 assembly to intracytoplasmic vesicles (Fig. 9a). This effect is not due to a defect in endocytosis imposed by PI(4,5)P2 disruption.25 Significantly, this altered targeting phenotype was also observed for HIV-2 (Fig. 9b). Quantitative results from the EM analysis showing the effects of 5ptaseIV, 5ptaseIV/Δ1, and Arf6/Q67L on particle production are presented in Table 2.
Fig. 9.

Disruption of PI(4,5)P2 induces a relocalization of HIV-1 and HIV-2 assembly to intracellular vesicles. HeLa cells were transfected with HIV-1 (pNL4−3) or HIV-2 (pROD10) molecular clones alone (−) or were co-transfected with vectors expressing 5ptaseIVor Arf6/Q67L. Transfected cells were fixed and examined by thin-section transmission EM. Scale bars represent 100 nm unless otherwise indicated.
Table 2.
Quantitative assessment of intracellular versus extracellular particle production
| Condition | Total virions scored | Intracellular (%) | PM associated/ Extracellular (%) |
|---|---|---|---|
| HIV-1 | 252 | 23 | 77 |
| HIV-1+ 5ptaseIV | 582 | 60 | 40 |
| HIV-1+ 5ptaseIV/ Δ1 | 283 | 25 | 75 |
| HIV-1+ Arf6/ Q67L | 858 | 54 | 46 |
| HIV-2 | 1448 | 9 | 91 |
| HIV-2 + 5ptaseIV | 2091 | 44 | 56 |
| HIV-2+ 5ptaseIV/ Δ1 | 2030 | 29 | 71 |
| HIV-2+ Arf6/ Q67L | 1080 | 67 | 33 |
Data from analysis of EM images (see the text for details).
Discussion
The overall structure of HIV-2 myr(+)MA is very similar to the structures previously determined for the HIV-129,32,51,52 and SIV44 myr(−)MA proteins. All these proteins fold as a cluster of six α-helices and a three-strand meandering β-sheet. The N-terminal myr group of HIV-2 myr(+)MA is partially sequestered within a narrow hydrophobic tunnel that does not exist in the unmyristylated protein. The most significant differences between the myristylated and unmyristylated HIV-2 MA proteins involve the structure and orientation of helix 1. In the myristylated HIV-2 MA protein, these residues adopt a classic α-helix, with the side chains of Val7, Leu8, Leu16, Ile34, Ala38, Val35, Glu48, Leu51, Glu52, and Leu85 packing against the myr group and the side chain of Lys12 exposed to solvent and disordered. In the unmyristylated HIV-2 myr(−)MA protein, helix 1 exhibits a mixture of α and 310 characters, with the side chain NH+4 group of Lys12 forming a buried salt bridge with His89. In the myristylated HIV-2 MA protein, the side chain of Lys12 is exposed to solvent and does not make long-range contacts. Interestingly, Lys12 is substituted by Glu in HIV-1, and this residue forms a His89 Hδ2to the Glu12 COO− salt bridge in the HIV-1 myr(−)MA structure. A lysine is conserved at position 12 in all reported strains of HIV-2 and SIV, whereas this position is exclusively occupied by glutamate in all reported strains of HIV-1. It is unclear why HIV-1 would evolve to use a buried protonated histidine indole to form a long-range salt bridge with a glutamate COO− side chain, whereas HIV-2 and SIV utilize a neutral His89 indole in a hydrogen bond with a lysine group.
In view of the high degree of structural similarity between the myristate-sequestered forms of HIV-1 and HIV-2 myr(+)MA, we were surprised to find that the proteins exhibit significantly different myr switch behaviors. Increasing the concentration of HIV-1 myr(+)MA from 0.025 to 0.6 mM results in an equilibrium shift from a predominantly myristate-sequestered monomeric state to a predominantly myristate-exposed multimeric state. In contrast, no evidence for trimerization or aggregation was observed for HIV-2 myr(+)MA by analytical ultra-centrifugation. In addition, the 1H–15N HSQC spectra obtained for HIV-2 myr(+)MA were unaffected by changes in concentration over the range 0.05−1.0 mM. These data collectively indicate that HIV-2 myr(+)MA remains monomeric and that the myr group remains sequestered, even at protein concentrations up to ∼1 mM.
We previously showed that soluble PI(4,5)P2 analogs containing truncated acyl chains are capable of binding to a cleft on the surface of HIV-1 MA. Residues that comprise this cleft are highly conserved among all reported strains of HIV-1, HIV-2, and SIV. Of the 454 HIV-1/SIV-chimpanzee Gag strains viewed† (curated alignments), Ser77, Asn80, and Lys/Arg22 are strictly conserved; Leu21, Trp36, and Thr97 are substituted once; and Lys27 is substituted twice. Conservation among the 66 strains of HIV-2/SIVSMM and 78 strains of SIV Gag was also analyzed. In HIV-2/SIVSMM strains, Leu21, Arg22, Lys27, Trp36, Ser77, and Thr97 are all strictly conserved, whereas N80 has one semiconserved substitution. Among the remaining SIV strains, Leu21, Trp36, and Thr97 are strictly conserved, Arg22 is substituted six times, Lys27 is substituted twice, and Ser77 is conservatively substituted in nine strains. These residues are strictly conserved between the HIV-1NL4−3 and HIV-2ROD10 strains studied here, and as such, it is not surprising that di-C4-PI(4,5)P2 binds both proteins with similar affinities and binding modes. In both cases, the 2′-acyl chain is sequestered within the cleft and buried below the inositol ring and the 1′-acyl chain extends away from and does not interact with the protein. Such an “extended lipid” biding mode, which has been predicted in phospholipid–cytochrome c models,53–55 would allow PI(4,5)P2 to serve as a membrane anchor by interacting directly with both the membrane and the MA.29
One of the striking differences between HIV-1 and HIV-2 myr(+)MA is that the HIV-1 protein exists as an equilibrium mixture of rapidly interconverting myristate-sequestered and myristate-exposed species, whereas HIV-2 myr(+)MA exclusively adopts the myristate-sequestered conformation. The binding of di-C4-PI(4,5)P2 or di-C8-PI(4,5)P2 to HIV-1 myr(+)MA induces a shift in the myr switch equilibrium toward the myristate-exposed species. However, we did not observe a similar effect for HIV-2 myr(+)MA. Careful examination of the 1H–15N HSQC titration data revealed that most of the signals for residues of helix 1, especially those near the N-terminus of the helix, undergo selective broadening at increasing concentrations of di-C4-PI(4,5)P2, and some signals exhibit both broadening and small progressive chemical shift changes. These findings indicate that di-C4-PI(4,5)P2 binding induces a long-range allosteric effect that destabilizes helix 1. The data are consistent with a model in which, at high di-C4-PI(4,5)P2 concentrations, a small fraction of HIV-2 myr(+)MA adopts a myristate-exposed conformation that undergoes relatively rapid (millisecond time scale) exchange with the predominant myristate-sequestered species. Addition of di-C8-PI(4,5)P2 also leads to selective broadening of helix 1 residues and induces precipitation of the protein. The longer acyl chains of di-C8-PI(4,5)P2 are known to enhance binding to HIV-1 myr(+)MA [relative to di-C4-PI(4,5)P2] and induce precipitation, and we speculate that the di-C8-PI(4,5)P2 may be functioning similarly upon binding to HIV-2 myr(+)MA.
At this point, it is unclear why the HIV-1 and HIV-2 myr(+)MA proteins exhibit such different myr switch behaviors. Of the amino acids that interact directly with the sequestered myristate in HIV-1 myr (+)MA, only one (Trp16) is substituted in the HIV-2 protein (by Leu16). Of course, other substitutions may also have a significant effect. For example, in both the HIV-1 and HIV-2 myr(−)MA structures, the packing of helix 1 against the core of the protein is mediated by a salt bridge with the partially buried side chain of His89. As indicated above, the nature of the salt bridge is substantially different between these proteins. In HIV-1 myr(−)MA, the indole ring of His89 is protonated and forms a salt bridge with the negatively charged side chain of Glu12. In HIV-2 myr(+)MA, the residue at position 12 is a lysine, and the indole ring of His89 is neutral and accepts a hydrogen bond from the positively charged side-chain group of Lys12. It is striking that His89 is conserved among all known strains of HIV-1, HIV-2, and SIV and that Glu and Lys residues are conserved among all known strains of HIV-1 and HIV-2/SIV MA proteins, respectively. Regardless, the structural differences associated with helix 1 appear to be due to reorganizations needed to fill the void that is normally occupied by the myr group. These structural features and differences are similar to those observed previously for HIV-1 myr(−)MA and myr(+)MA proteins.29,32,51,52 Mutagenesis studies (underway) should help us determine if substitution of Lys12 by Glu in HIV-2 myr(+)MA is sufficient to invoke an HIV-1-like myr switch phenotype.
It is unclear why HIV-1 evolved a more sensitive myr switch compared with HIV-2. Since the myr group is essential for tight membrane binding, one might expect that the propensity of HIV-2 to bind membranes might be reduced somewhat compared with the HIV-1 protein. In this regard, several strains of HIV-2 were recently shown to be unable to assemble and bud from yeast cells, despite the fact that HIV-1 is able to produce virus particles in this context.11 This defect was attributed to a reduced ability of HIV-2 Gag proteins to remain stably associated with the PM at virus assembly sites,11 which could be due to the weaker myr switch. Interestingly, virus assembly could be enhanced by substituting the N-terminal half of HIV-2 MA by the corresponding residues of HIV-1 MA.11 As described above, a number of amino acids in the N-terminal half of HIV-2 MA are known to interact directly with the myr group in the myristate-sequestered form of the protein, and it has been shown for HIV-132 that substitution of selected residues (even single conservative substitutions) can dramatically influence both the behavior of the myr switch in vitro30 and the ability of Gag to bind the PM and assemble in vivo.35,56–58 We thus speculate that differences in the ability of HIV-1 and HIV-2 Gag to assemble and bud from certain cell types may at least partly be due to differences in the efficiency of the myr switch, which should inhibit membrane binding and possibly Gag self-association. A weak myr switch might also explain why HIV-2 replicates much more slowly than HIV-1 in vitro9 and may thus contribute to the lower pathogenicity of HIV-2 relative to HIV-1.2–4
Materials and Methods
Sample preparation
For construction of the His6-tagged myr(−)MA plasmid, its coding sequences were PCR amplified from the ROD10 isolate (Centralized Facility for AIDS Reagents, UK; National Center for Biotechnology Information accession code M15390) and cloned into E. coli expression vector pET16b at its NcoI and XhoI sites in-frame with the C-terminal His6 tag and stop codon of the plasmid. The plasmid that co-expresses the myr(+)MA protein with a C-terminal His6 tag was constructed by ligating the MA gene into a co-expression vector harboring the yeast N-terminal myristyltransferase gene.32 Leu75, which was mutated to serine during the cloning of myr(−)MA and myr(+)MA, was corrected with the use of a QuickChange XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). Plasmids were sequenced at the Yale University W. M. Keck Foundation Biotechnology Resource Laboratory (New Haven, CT).
Protein samples were prepared as follows: Cells were first grown in 4 L of LB medium at 37 °C until the OD600 reached ∼0.6−0.7. Next, cells were spun down and washed with 1× M9 salt before transferring them to 1 L of M9 minimal medium containing 15NH4Cl as the sole nitrogen source to produce 15N-labeled protein and 13C-labeled glucose as the sole carbon source to produce 13C-labeled protein. For myr(+)MA samples, cells were supplemented with myristic acid (10 mg/L; Sigma) and then grown for 1 h before induction with isopropyl-D-thiogalactoside (1 mM). Cells were grown at 37 °C for ∼12−14 h and lysed using a microfluidizer, and the proteins were purified by cobalt affinity chromatography (Clontech, Mountain View, CA) and ion exchange column chromatography (SP column). Molecular weights and efficiency of myristylation were confirmed by electrospray ionization mass spectrometry [Mmeas/Mcalc = 15,694.2:15,694.0 and 15,904.4:15,904.2 for myr(−)MA and myr(+)MA, respectively]. Phosphoinositides PI(4,5)P2, di-C4-PI(4,5)P2, di-C8-PI(4,5)P2, di-C4-PI, di-C4-PI(3)P, di-C4-PI(4)P, di-C4-PI(5)P, di-C4-PI(3,5)P2, di-C8-PI(3,4,5)P2, as well as di-C4-PI (Echelon) and di-C4-phosphatidylcholine (Avanti Polar Lipids) were obtained commercially and used without further purification. Samples for all NMR experiments were prepared in 20 mM sodium phosphate at pH 6.3, 100 mM NaCl, 50 mM glutamate, 50 mM arginine, and 10 mM DTT. Protease inhibitor cocktail (1×, Calbiochem, San Diego, CA) and sodium azide [0.02% (w/v)] were added to the NMR samples to maintain protein stability during the course of the NMR data collection. NaCl was excluded from the NMR buffer for the di-C4-PI(4,5)P2 and di-C4-PI(4,5)P2 titration experiments.
NMR spectroscopy
NMR data were collected with Bruker DRX (800 MHz of 1H) and DMX (600 MHz of 1H) spectrometers equipped with cryoprobes, processed with NMRPipe,59 and analyzed with NMRView.60 A combination of 2D, 3D, and 4D NOESY data was obtained for natural abundance, 15N-labeled protein samples, and 15N-,13C-labeled protein samples (35 °C). Protein backbone signals were assigned using standard triple-resonance methods (HNCA and HNCOCA), and side-chain signals were assigned from 3D and 4D 15N-, 13C-, and 15N/13C-edited NOESY data. Intermolecular 1H–1H NOEs between the 15N-,13C-labeled protein and the unlabeled myristate group were assigned from 2D (1H–1H) and 3D (13C- and 13C-edited/12C-double-half-filtered) NOESY data (see Refs. 49,50,61 and citations therein). 1H–15N RDCs were measured using a modified in-phase/antiphase HSQC experiment62 for both myr(−)MA and myr(+)MA samples aligned in polyacrylamide gel.63 Binding isotherms from 1H–15N NMR HSQC titration experiments were calculated with ORIGIN 7.0 software (MicoCal, Northampton, MA).
Analytical ultracentrifugation
Sedimentation equilibrium measurements were collected on a Beckman XL-I Optima system equipped with a four-hole An-60 rotor (Beckman Coulter). Cells were equipped with double-sectored charcoal-filled epon centerpieces of path length 12 or 3 mm and quartz windows. Protein samples were prepared in 20 mM sodium phosphate buffer at pH 6.3 containing 100 mM NaCl, 50 mM glutamate, 50 mM arginine, and 2 mM Tris (2-carboxy-ethyl)-phosphine–HCl. Loading concentrations varied from 50 to 150 μM for all samples. Data were collected at rotor speeds of 22,000, 26,000, and 30,000 rpm; temperature was 20 °C. Scans were obtained by using an absorption optics system with 0.002-cm step size and four averages per point and were acquired at wavelengths of 280 and 295 nm. Partial specific volumes (v-bar) and molar extinction coefficients were calculated by using the program SEDNTERP, and buffer densities were measured pycnometrically. Data analysis was performed by using NONLIN.64 Equilibrium association constants were determined by global analysis of data acquired from samples prepared at three loading concentrations and centrifuged at three rotor speeds.
Structure calculations
Structures were calculated in torsion angle space with CYANA‡ starting from random initial angles. Upper interproton distance bounds of 2.7, 3.3, and 5.0 Å (with appropriate corrections for pseudoatoms) were employed for NOE cross-peaks of strong intensity, medium intensity, and weak intensity, respectively, which were qualitatively determined following intensity normalization of the different NOE data sets. No backbone hydrogen bond or chemical shift-based torsion angle restraint was employed. Structures of myr(−)MA and myr(+)MA calculated with RDC restraints exhibited improved convergence. RDC data obtained for myr(−)MA and MA were very similar and, consistent with the NOE data, indicate that myr sequestration does not significantly alter the structure protein. Statistical information is provided in Table 1. Structures were evaluated with Procheck∥ and figures were generated with PyMOL§.
Proviral clones, expression plasmids, antibodies, and cells
HIV-165 and HIV-2ROD66 molecular clones have been described previously. The 5ptaseIV expression plasmid pcDNA4TO Myc5ptaseIV was a kind gift from P. Majerus (Washington University School of Medicine, St. Louis, MO).67 The Δ1 5ptaseIV mutant lacking the phosphatase signature domain was constructed as described previously.25 The hemagglutinin-tagged Arf6/Q67L expression plasmid pXS Arf6Q67L-HA was a generous gift from J. Donaldson [National Heart, Lung, and Blood Institute, National Institutes of Health (NIH)].68 HeLa cells were maintained and transfected as previously described.69 Immunoprecipitation of HIV-1 Gag was performed by using HIV-Ig, obtained through the NIH AIDS Research and Reference Reagent Program. HIV-2 Gag was detected with the anti-p27gag monoclonal antibody R1C7, a gift from J. Kappes (University of Alabama at Birmingham, Birmingham, AL).70
Transfections, virus release assays, and EM analysis
Transfections were performed in a six-well plate seeded at 4 × 105 cells/well using the Lipofectamine LTX reagent (Invitrogen). One day after transfection, cells were metabolically labeled for 2 h with [35S]Met/Cys in Met−/Cys− RPMI 1640 medium supplemented with 2% dialyzed fetal bovine serum (Sigma Aldrich). Preparation of cell and viral lysates and immunoprecipitation of viral proteins were performed as previously described.69 Virus release efficiency was calculated as the amount of virion-associated Gag as a fraction of total (cell plus virion) Gag synthesized during the metabolic labeling period. For EM analysis, HeLa cells were plated in a six-well plate at 4 × 105 cells/well. Cells were transfected using the ExGen500 in vivo transfection reagent (Fermentas). One day after transfection, fixation and processing of HeLa cells were carried out as previously described.71 Cells were examined with a Hitachi H7600 electron microscope.
Between ∼250 and ∼2000 virions (present in 10−20 virus-positive cells) under each condition were scored as being either associated with the PM or in an intracellular compartment to quantify the effect of PI(4,5)P2 disruption on the localization of HIV-1 and HIV-2 assembly. From these results, the percentage of virions that were PM associated/extracellular and that of virions that were intracellular were calculated.
Atomic coordinates
The atomic coordinates have been deposited in the Protein Data Bank¶ and are available under accession codes 2K4E, 2K4H, and 2K4I for the HIV-2 myr(−)MA, myr(+)MA, and myr(+)MA/di-C4-PI(4,5)P2 structures, respectively.
Acknowledgements
This work was supported by the NIH through grant AI30917 (M.F.S.); the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH (E.O.F.); the Intramural AIDS Targeted Antiviral Program (E.O.F.); and federal funds from the National Cancer Institute, NIH, under contract N01-CO-12400 (K.N.). We thank Rob Edwards and Chen Yu (University of Maryland Baltimore County Howard Hughes Medical Institute staff), Dr. Dorothy Beckett (University of Maryland, College Park, MD), and David King (University of California, Berkeley, Howard Hughes Medical Institute, Berkley, CA) for technical support; Dr. Bechet, A. M. L. Lever, and the Centralized Facility for AIDS Reagents (UK) for providing the HIV-2 cDNA (pROD10) plasmid; J. Donaldson and P. Majerus for providing Arf6/Q67L and 5ptaseIV expression vectors, respectively; J. Kappes for the anti-p27gag monoclonal antibody R1C7; and the AIDS Research and Reference Reagent Program for HIV-Ig.
Abbreviations used
- 2D
two-dimensional
- 5ptaseIV
5-phosphatase IV
- EM
electron microscopy
- HIV
human immunodeficiency virus
- HIV-2 Gag
myristoylated HIV-2 Gag polyprotein
- HIV-2 MA
HIV-2 matrix protein
- HSQC
heteronuclear single quantum coherence
- Ig
immunoglobulin
- myr
myristyl
- myr(−)
unmyristylated protein
- myr(+)
myristylated protein
- NOESY
nuclear Overhauser enhancement spectroscopy
- PI(4,5)P2
phosphatidylinositol-(4,5)-bisphosphate
- PM
plasma membrane
- RDC
residual dipolar coupling
- SIV
simian immunodeficiency virus
- SIVSMM
SIV from sooty mangabee monkey
Footnotes
References
- 1.Lemey P, Pybus OG, Wang B, Saksena NK, Salemi M, Vandamme A-M. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl Acad. Sci. USA. 2003;100:6588–6592. doi: 10.1073/pnas.0936469100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poulsen AG, Aaby P, Larsen O, Jensen H, Naucler A, Lisse IM, et al. 9-year HIV-2-associated mortality in an urban community in Bissau, West Africa. Lancet. 1997;349:911–914. doi: 10.1016/S0140-6736(96)04402-9. [DOI] [PubMed] [Google Scholar]
- 3.Schim van der Loeff MF, Jaffar S, Aveika AA, Sabally S, Corrah T, Harding E, et al. Mortality of HIV-1, HIV-2 and HIV-1/HIV-2 dually infected patients in a clinic-based cohort in The Gambia. AIDS. 2002;16:1775–1783. doi: 10.1097/00002030-200209060-00010. [DOI] [PubMed] [Google Scholar]
- 4.Ariyoshi K, Jaffar S, Alabi AS, Berry N, Schim van der Loeff MF, Sabally S, et al. Plasma RNA viral load predicts the rate of CD4 T cell decline and death in HIV-2-infected patients in West Africa. AIDS. 2000;14:339–344. doi: 10.1097/00002030-200003100-00006. [DOI] [PubMed] [Google Scholar]
- 5.Mellors JW, Rinaldo CR, Gupta P, Jr, White RM, Todd JA, Kingsley LA. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 6.Popper SJ, Sarr AD, Travers KU, Gueye-Ndiaye A, Mboup S, Essex ME, Kanki PJ. Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. J. Infect. Dis. 1999;180:1116–1121. doi: 10.1086/315010. [DOI] [PubMed] [Google Scholar]
- 7.Vesanen M, Stevens CE, Taylor PE, Rubinstein P, Saksela K. Stability in controlling viral replication identifies long-term nonprogressors as a distinct subgroup among human immunodeficiency virus type 1-infected persons. J. Virol. 1996;70:9035–9040. doi: 10.1128/jvi.70.12.9035-9040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duvall MG, Precopio ML, Ambrozak DA, Jaye A, McMichael AJ, Whittle HC, et al. Polyfunctional T cell responses are a hallmark of HIV-2 infection. Eur. J. Immunol. 2008;38:350–363. doi: 10.1002/eji.200737768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaak H, van der Ende ME, Boers PHM, Schuitemaker H, Osterhaus ADME. In vitro replication capacity of HIV-2 variants from long-term aviremic individuals. Virology. 2006;353:144–154. doi: 10.1016/j.virol.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 10.Blaak H, Boers PHM, van der Ende ME, Schuitemaker H, Osterhaus ADME. CCR5-restricted HIV type 2 variants from long-term aviremic individuals are less sensitive to inhibition by β-chemokines than low pathogenic HIV type 1 variants. AIDS Res. Human Retroviruses. 2008;24:473–484. doi: 10.1089/aid.2007.0001. [DOI] [PubMed] [Google Scholar]
- 11.Morikawa Y, Goto T, Yasuoka D, Momose F, Matano T. Defect of human immunodeficiency virus type 2 Gag assembly in Saccharomyces cerevisiae. J. Virol. 2007;81:9911–9921. doi: 10.1128/JVI.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gheysen D, Jacobs E, de Foresta F, Thiriart C, Francotte M, Thines D, De Wilde M. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell. 1989;59:103–112. doi: 10.1016/0092-8674(89)90873-8. [DOI] [PubMed] [Google Scholar]
- 13.Ganser-Pomillos BK, Yeager M, Sundquist WI. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008;18:203–217. doi: 10.1016/j.sbi.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, et al. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell. 2005;120:663–674. doi: 10.1016/j.cell.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Hermida-Matsumoto L, Resh MD. Localization of human immunodeficiency virus type 1 Gag and Env at the plasma membrane by confocal imagine. J. Virol. 2000;74:8670–8679. doi: 10.1128/jvi.74.18.8670-8679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiernan RE, Ono A, Englund G, Freed EO. Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J. Virol. 1998;72:4116–4126. doi: 10.1128/jvi.72.5.4116-4126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiernan RE, Ono A, Freed EO. Reversion of a human immunodeficiency virus type 1 matrix mutation affecting Gag membrane binding, endogenous reverse transcriptase activity, and virus infectivity. J. Virol. 1999;73:4728–4737. doi: 10.1128/jvi.73.6.4728-4737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ono A, Orenstein JM, Freed EO. Role of the Gag matrix domain in targeting human immuno-deficiency virus type 1 assembly. J. Virol. 2000;74:2855–2866. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scarlata S, Cater C. Role of HIV-1 Gag domains in viral assembly. Biochim. Biophys. Acta. 2003;1614:62–72. doi: 10.1016/s0005-2736(03)00163-9. [DOI] [PubMed] [Google Scholar]
- 20.Ehrlich LS, Fong S, Scarlata S, Zybarth G, Carter C. Partitioning of HIV-1 Gag and Gag-related proteins to membranes. Biochemistry. 1996;35:3933–3943. doi: 10.1021/bi952337x. [DOI] [PubMed] [Google Scholar]
- 21.Ono A, Freed EO. Plasma membrane rafts play a critical role in HIV-1 assembly and release. Proc. Natl. Acad. Sci. USA. 2001;98:13925–13930. doi: 10.1073/pnas.241320298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spearman P, Horton R, Ratner L, Kuli-Zade I. Membrane binding of human immunodeficiency virus type 1 matrix protein in vivo supports a conformational myristyl switch mechanism. J. Virol. 1997;71:6582–6592. doi: 10.1128/jvi.71.9.6582-6592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou W, Parent LJ, Wills JW, Resh MD. Identification of a membrane-binding domain within the amino-terminal region of human immuno-deficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 1994;68:2556–2569. doi: 10.1128/jvi.68.4.2556-2569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouamr F, Scarlata S, Carter CA. Role of myristylation in HIV-1 Gag assembly. Biochemistry. 2003;42:6408–6417. doi: 10.1021/bi020692z. [DOI] [PubMed] [Google Scholar]
- 25.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl Acad. Sci. USA. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jouvenet N, Neil SJD, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demirov D, Freed EO. Retrovirus budding. Virus Res. 2004;106:87–102. doi: 10.1016/j.virusres.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 28.Freed EO. HIV-1 Gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- 29.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl Acad. Sci. USA. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saad JS, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, et al. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J. Mol. Biol. 2007;366:574–585. doi: 10.1016/j.jmb.2006.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adamson CS, Freed EO. Human immunodeficiency virus type 1 assembly, release and maturation. Adv. Pharmacol. 2007;55:347–387. doi: 10.1016/S1054-3589(07)55010-6. [DOI] [PubMed] [Google Scholar]
- 32.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl Acad. Sci. USA. 2004;101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou W, Resh MD. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 1996;70:8540–8548. doi: 10.1128/jvi.70.12.8540-8548.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hermida-Matsumoto L, Resh MD. Human immunodeficiency virus type 1 protease triggers a myristoyl switch that modulates membrane binding fo Pr55gag and p17MA. J. Virol. 1999;73:1902–1908. doi: 10.1128/jvi.73.3.1902-1908.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paillart J-C, Gottlinger HG. Opposing effects of human immunodeficiency virus type 1 matrix mutations support a myristyl switch model of Gag membrane targeting. J. Virol. 1999;73:2604–2612. doi: 10.1128/jvi.73.4.2604-2612.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Resh MD. A myristoyl switch regulates membrane binding of HIV-1 Gag. Proc. Natl Acad. Sci. USA. 2004;101:417–418. doi: 10.1073/pnas.0308043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ebbets-Reed D, Scarlata S, Carter CA. The major homology region of the HIV-1 Gag precursor influences membrane affinity. Biochemistry. 1996;35:14268–14275. doi: 10.1021/bi9606399. [DOI] [PubMed] [Google Scholar]
- 38.Liang C, Hu J, Russell RS, Roldan A, Kleiman L, Wainberg MA. Characterization of a putative α-helix across the capsid-SP1 boundary that is critical for the multimerization of human immunodeficiency virus type 1 Gag. J. Virol. 2002;76:11729–11737. doi: 10.1128/JVI.76.22.11729-11737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Accola MA, Hoglund S, Gottlinger HG. A putative α-helical structure which overlaps the capsid-p2 boundary in the human immunodeficiency virus type-1 Gag precursor is crucial for viral particle assembly. J. Virol. 1998;72:2072–2078. doi: 10.1128/jvi.72.3.2072-2078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ono A, Demirov D, Freed EO. Relationship between human immunodeficiency virus type-1 Gag multimerization and membrane binding. J. Virol. 2000;74:5142–5150. doi: 10.1128/jvi.74.11.5142-5150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin TFJ. PI(4,5)P2 regulation of surface membrane traffic. Curr. Opin. Cell Biol. 2001;13:493–499. doi: 10.1016/s0955-0674(00)00241-6. [DOI] [PubMed] [Google Scholar]
- 42.Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature. 2005;438:597–604. doi: 10.1038/nature04397. [DOI] [PubMed] [Google Scholar]
- 43.McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 44.Rao Z, Belyaev AS, Fry E, Roy P, Jones IM, Stuart DI. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature. 1995;378:743–747. doi: 10.1038/378743a0. [DOI] [PubMed] [Google Scholar]
- 45.Janmey PA, IIda K, Yin HL, Stossel TP. Phosphoinositide micelles and polyphosphoinositide-containing vesicles dissociate endogenous gelsolin–actin complexes and promote actin assembly from the fast-growing end of actin filaments blocked by gelsolin. J. Biol. Chem. 1987;262:12228–12236. [PubMed] [Google Scholar]
- 46.Kutateladze T, Overduin M. Structural mechanism of endosome docking by the FYVE domain. Science. 2001;291:1793–1796. doi: 10.1126/science.291.5509.1793. [DOI] [PubMed] [Google Scholar]
- 47.Kutateladze T. Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim. Biophys. Acta. 2006;1761:868–877. doi: 10.1016/j.bbalip.2006.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee SA, Kovacs J, Stahelin RV, Cheever ML, Overduin M, Setty TG, et al. Molecular mechanism of membrane docking by the Vam7p PX domain. J. Biol. Chem. 2006;281:37091–37101. doi: 10.1074/jbc.M608610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley & Sons; New York, NY: 1986. [Google Scholar]
- 50.Kay LE, Clore GM, Bax A, Gronenborn AM. Four-dimensional heteronuclear triple-resonance NMR spectroscopy of interleukin-1β in solution. Science. 1990;249:411–414. doi: 10.1126/science.2377896. [DOI] [PubMed] [Google Scholar]
- 51.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Crystal structures of the trimeric HIV-1 matrix protein: implications for membrane association. Proc. Natl Acad. Sci. USA. 1996;93:3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 53.Kinnunen PKJ, Koiv A, Lehtonen JYA, Rytomaa M, Mustonen P. Lipid dynamics and peripheral interactions of proteins with membrane surfaces. Chem. Phys. Lipids. 1994;73:181–207. doi: 10.1016/0009-3084(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 54.Rytomaa M, Kinnunen PKJ. Reversibility of the binding of cytochrome c to liposomes. J. Biol. Chem. 1995;270:3197–3202. doi: 10.1074/jbc.270.7.3197. [DOI] [PubMed] [Google Scholar]
- 55.Touminen EKJ, Wallace CJA, Kinnunen PKJ. Phospholipid–cytochrome c interaction. J. Biol. Chem. 2002;277:8822–8826. doi: 10.1074/jbc.M200056200. [DOI] [PubMed] [Google Scholar]
- 56.Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 1994;68:5311–5320. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ono A, Freed EO. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 1999;73:4136–4144. doi: 10.1128/jvi.73.5.4136-4144.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ono A, Huang M, Freed EO. Characterization of human immunodeficiency virus type 1 matrix revertants: effects on virus assembly, Gag processing, and Env incorporation into virions. J. Virol. 1997;71:4409–4418. doi: 10.1128/jvi.71.6.4409-4418.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 60.Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 61.Folkers PJM, Folmer RHA, Konings RNH, Hilbers CW. Overcoming the ambiguity problem encountered in the analysis of nuclear Overhauser magnetic resonance spectra of symmetric dimer proteins. J. Am. Chem. Soc. 1993;115:3798–3799. [Google Scholar]
- 62.Ishii Y, Markus MA, Tycko R. Controlling residual dipolar couplings in high-resolution NMR of proteins by strain induced alignment in a gel. J. Biomol. NMR. 2001;21:141–151. doi: 10.1023/a:1012417721455. [DOI] [PubMed] [Google Scholar]
- 63.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J. Biomol. NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 64.Johnson ML, Faunt LM. Parameter estimation by least-squares methods. Methods Enzymol. 1992;210:1–37. doi: 10.1016/0076-6879(92)10003-v. [DOI] [PubMed] [Google Scholar]
- 65.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clavel F, Guyader M, Guetard D, Salle M, Montagnier L, Alizon M. Molecular cloning and polymorphism of the human immune deficiency virus type 2. Nature. 1986;324:691–695. doi: 10.1038/324691a0. [DOI] [PubMed] [Google Scholar]
- 67.Kisseleva MV, Cao L, Majerus PW. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. J. Biol. Chem. 2002;277:6266–6272. doi: 10.1074/jbc.M105969200. [DOI] [PubMed] [Google Scholar]
- 68.Brown FD, Rozelle AL, Yin HL, Balla T, Donaldson JG. Phosphatidylinositol 4,5-bisphosphate and Arf6-regulated membrane traffic. J. Cell Biol. 2001;154:1007–1017. doi: 10.1083/jcb.200103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Freed EO, Martin MA. Evidence for a functional interaction between the V1/V2 and C4 domains of human immunodeficiency virus type 1 envelope glycoprotein gp120. J. Virol. 1994;68:2503–2512. doi: 10.1128/jvi.68.4.2503-2512.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Minassian AA, Kalyanaraman VS, Gallo RC, Popovic M. Monoclonal antibodies against human immunodeficiency virus (HIV) type 2 core proteins: cross-reactivity with HIV type 1 and simian immunodeficiency virus. Proc. Natl Acad. Sci. USA. 1988;85:6939–6943. doi: 10.1073/pnas.85.18.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonda MA, Aaronson SA, Ellmore N, Zeve VH, Nagashima K. Ultrastructural studies of surface features of human normal and tumor cells in tissue culture by scanning transmission electron microscopy. J. Natl Cancer Inst. 1976;56:245–263. doi: 10.1093/jnci/56.2.245. [DOI] [PubMed] [Google Scholar]