

Abstract
Invasion plasmid antigen C (IpaC) is secreted by the Shigella flexneri type III secretion system (TTSS) as an essential trigger of epithelial cell invasion. At the molecular level, IpaC possesses a distinct functional organization. The IpaC C-terminal region between amino acids 319 and 345 is predicted to form a coiled-coil structure. Such alpha-helical motifs appear to be a recurring structural theme among TTSS components. Together with IpaB, this IpaC region is also required for the formation of translocon pores in target cell membranes. In contrast, mutations within the C-terminal tail of IpaC (defined by residues 345 to 363) have no effect on contact hemolysis (a putative measure of translocon pore formation), but they can contribute significantly to IpaC’s ability to trigger S. flexneri entry into cultured cells. Here we describe the molecular dissection of the IpaC C-terminus and how changes in this region affect selected virulence-related activities. IpaC invasion function requires its immediate C-terminus and this general region may be involved in its ability to trigger actin nucleation. In contrast, IpaC could not be shown to interact directly with Cdc42, a host GTPase closely tied to Shigella invasion.
Keywords: Shigella, type III secretion, invasion, actin, IpaC
1. Introduction
Shigella flexneri is a gram-negative facultative intracellular pathogen that causes bacillary dysentery (shigellosis), a self-limiting gastroenteritis responsible for significant mortality in developing nations [1]. Shigella spp. are also a public health problem in industrialized regions, especially in institutional settings such as daycare centers and anywhere normal public health measures are compromised [2]. After ingestion, S. flexneri travels to the colon where it exploits M cells as a conduit for breaching the colonic epithelium [3]. In the submucosa, Shigella enters macrophages to induce apoptosis [4] and ultimately invades epithelial cells by inducing localized cytoskeletal rearrangements at the site of pathogen contact [5].
Epithelial cell invasion is an essential step in the pathogenesis of S. flexneri and the genes required for this are encoded on a large virulence plasmid [6]. The mxi/spa operon encodes a type III secretion apparatus (TTSA) that secretes translocator and effector proteins into the membrane and cytoplasm of target cells. The ipa operon encodes six proteins, five of which are secreted by the TTSA [7]. Of the secreted proteins, the invasion plasmid antigens IpaB, IpaC and IpaD are essential for Shigella invasion [8]. Upon host cell contact, IpaB and IpaC are inserted into the host cell membrane where they have been described as forming a pore-like transmembrane channel called the translocon [9]. Effector proteins have been proposed to pass through the translocon to gain access to the host cell cytoplasm. TTSA-mediated insertion of translocon pores into the host membrane is a theme proposed for many gram-negative pathogens [10]; however, the IpaB/IpaC pore may be unique because it is implicated in directly subverting the host cell signaling pathways that promote pathogen entry [11, 12] and destabilizing membranes to allow vacuolar escape [12-14].
IpaC is proposed to induce the cytoskeletal rearrangements that allow Shigella invasion of epithelial cells [11, 12, 15]. How IpaC triggers actin recruitment is not firmly established, but a role for the small Rho-family GTPase Cdc42 in this process has been proposed [16]. It was also shown that IpaC can nucleate actin in vitro [17], which is a property first associated with its Salmonella homologue SipC [18, 19]. The specific role of Cdc42 with respect to IpaC-mediated actin nucleation has not been extensively studied at the biochemical level.
IpaC has been shown to possess a distinct functional organization [14, 15, 17, 20-22]. Its N-terminal 20 amino acids provide a signal for type III secretion. Adjacent N-terminal regions (residues 50 to 80), along with C-terminal regions, target IpaC for association with its cytoplasmic chaperone IpgC [20, 22, 23]. IpaC’s central hydrophobic region (residues 100-170) has two putative transmembrane helices that are responsible for its penetration of phospholipid membranes and contribute to overall protein stability in the absence of phospholipids [17, 20]. The IpaC C-terminus possesses a putative coiled-coil domain that may be involved in oligomerization [17, 20, 24], however, the precise structure of the IpaC C-terminus has not yet been determined.
In this study, we more closely examine the functional significance of the IpaC C-terminus using molecular and mutational analyses in conjunction with protein binding studies, Shigella invasion of cultured cells, contact-mediated hemolysis, and in vitro analysis of putative IpaC functions. The acquired data suggest that the C-terminal tail of IpaC is required specifically for Shigella invasiveness, but not for pore formation. IpaC was not found, however, to directly interact with Cdc42 or Rac1 in vitro. Meanwhile some deletion mutations at the IpaC C-terminus appear to interfere with its ability to induce actin nucleation. It thus appears that the IpaC C-terminus may contribute to the effector function of this protein, but this function does not appear to occur from direct interactions with the GTPases generally accepted to be responsible for Shigella entry into host cells.
2. Results
2.1. IpaC does not appear to interact directly with Cdc42 in vitro
It has been shown that Rho-family GTPases are intimately involved in Shigella invasion [5, 25, 26]. Because IpaC is able to direct actin rearrangements in target cells in the absence of other Shigella components [5], it has been suggested that it directly activates Cdc42 to initiate the regulatory cascade needed to promote Shigella entry [5, 25, 26]. An important part of this activity appears to be located at the IpaC C-terminus since antibodies against this region can block its ability to induce filopodia formation [5]. The necessary involvement of IpaC in Cdc42 activation could mean that IpaC interacts directly with Cdc42 as part of its activity. In Salmonella typhimurium the IpaC homolog SipC is also an essential effector of invasion, however, the secreted TTSS effector SopE2 is the protein found to interact with and activate Cdc42 to contribute to Salmonella entry into epithelial cells [27, 28]. SopE2 is a guanine nucleotide exchange factor (GEF) for the small Rho-family GTPase Cdc42 [27, 29]. Shigella does possess IpgB1 which acts upstream of Rac1 and Cdc42 to contribute to the bacterial invasion process [30], however, IpaC has been found to be sufficient for inducing filopodia formation [5, 12] [31]. There is no sequence homology between SopE2 or IpgB1 and IpaC, and there is no identified SopE2 homolog in Shigella. To thus determine the relationship between IpaC and Cdc42, fluorescence spectroscopy was used to assess the ability for these two proteins to directly interact.
Fluorescence polarization (FP) provides a measure of the rotational diffusion of a molecule in solution [32]. Because the rate of rotational diffusion is inversely related to its molecular volume, FP provides a sensitive measure of the changes in size that necessarily occur upon the binding of a fluorescently labeled protein to a nonfluorescent protein [32]. Therefore, FP was used to monitor the interaction between IpaC and Cdc42 that was labeled with FITC. Increasing amounts of IpaC was titrated with the FITC-labeled Cdc42 and the change in fluorescence polarization was measured (Fig. 1). SopE2 was used as a positive control in these experiments. SopE2 caused a significant increase in FITC-Cdc42 millipolarization (mP) with nanomolar concentrations of SopE2 being sufficient for seeing these polarization changes (Fig. 1). In contrast, regardless of the amount of protein added, IpaC did not cause a change in FITC-Cdc42 polarization. The protein concentrations used in these experiments were within the physiological range and fluorescence polarization provides a reliable measure of even weak protein-protein interactions, thus suggesting that IpaC may not act by direct association with Cdc42. The same result was obtained regardless of the nucleotide form bound to the Cdc42. Because IpaC was purified from bacterial inclusions in the presence of urea which requires that it undergo a refolding step, these experiments were repeated with IpaC that was prepared in a soluble form following co-expression with its chaperone IpgC as described by Birket et al. [23]. This this form of IpaC also failed to interact with Cdc42 by polarization analysis (data not shown). Furthermore, the fluorescence labeling of Cdc42 was performed as described by Nomanbhoy et al. [33], which leads to site-specific labeling on Lys150 and does not adversely affect Cdc42 activity. This did appear to be the case since the labeled Cdc42 could be incubated with GTP which caused a 32% increase in fluorescence intensity due to the proximity of the labeling site to the GTP-binding site [33].
Figure 1. Interaction of FITC-Cdc42 with IpaC or SopE2.
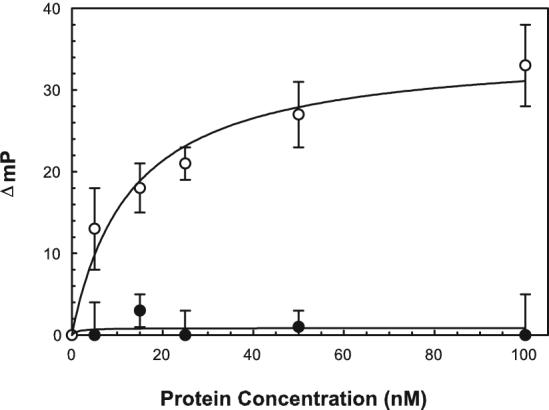
Protein-protein interactions between FITC-Cdc42 (10 nM) and IpaC (closed circles) or SopE2 (open circles) were measured by fluorescence polarization. The data are shown as the change in polarization (ΔmP). Because the ΔmP is influenced by the total change in molecular volume that occurs upon protein binding, it is worth noting that the molecular weights here are: Cdc42, ∼ 20 kDa; SopE2, ∼ 30 kDa; and IpaC, ∼ 40 kDa. The starting mP value for FITC-Cdc42 is approximately 125 mP units. The data shown are an average ± S.D. with n=3. The same assay could be carried out with IpaC that was prepared in the complete absence of urea (by initially purifying the protein in a soluble form in a complex with its chaperone IpgC), but the protein still failed to associate with the labeled Cdc42.
As a second test for the binding of IpaC to Cdc42, protein effects on Cdc42’s nucleotide binding pocket were monitored after labeling with the environmentally-sensitive fluorescent probe NBD on Lys150 [33]. As anticipated based on the data presented in Fig. 1, no change in NBD-Cdc42 intensity was seen following the addition of IpaC, suggesting that it does not affect the nucleotide binding pocket of Cdc42 with GDP bound (Fig. 2). In contrast, an increase in NBD-Cdc42 fluorescence intensity (with GDP bound) was seen upon adding SopE2 (Fig. 2). These results suggest that the impact of IpaC on host cells is not associated with its direct interaction with Cdc42.
Figure 2. Effect of IpaC binding on the emission of NBD-labeled Cdc-42.
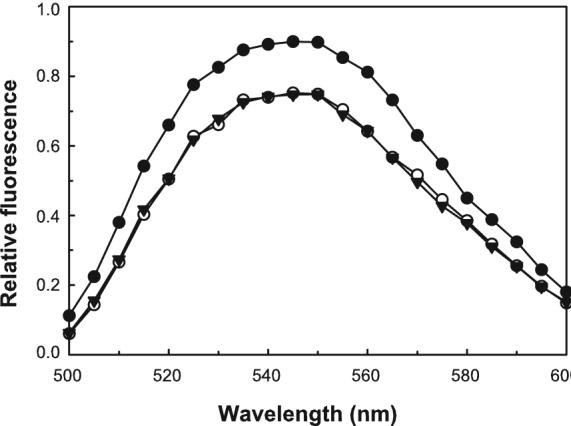
The effect of protein binding on the emission spectrum of Cdc42 labeled at Lys150 was determined using 30 nM NBD-Cdc42 with an excess (approximately 0.5 μM) nonfluorescent IpaC (open circles) or SopE2 (closed circles). The emission spectrum of NBD-Cdc42 alone is shown with the inverted closed triangles. The data shown are representative and were obtained with GDP prebound to NBD-Cdc42 as described in Methods. Nearly identical results were obtained with GTP or a nonhydrolyzable analog of GTP bound to the NBD-Cdc42.
To confirm the fluorescence-based findings, pull-down assays using SopE2 and IpaC with Cdc42 and Rac1 were performed. Because IpaC can be prepared with an N-terminal His6 tag, it could be incubated with Cdc42 or Rac1 and co-purification on nickel-chelation resin used as a measure of protein-protein interaction. SopE2 was purified as a GST-fusion protein which allowed co-purification using immobilized glutathione as a measure of interaction. Figure 3 shows the level of purity for the recombinant proteins that were purified for use here. When the pull-down assays were performed, an interaction between SopE2 and Cdc42 was observed, however, no such interactions were seen for IpaC with Cdc42 (Fig. 4). When Rac1 was specifically used, no interaction with IpaC or SopE2 was observed (Fig. 4). The absence of Rac1 binding by SopE2 appears to be consistent with results reported by Friebel et al. [29]. The protein concentrations used here were in the micromolar range, however, it is still possible that there could be an extremely weak interaction between IpaC and Cdc42 (or Rac1), however, the general consensus seems to be that IpaC does not directly interact with either of these proteins.
Figure 3. Purified IpaC, SopE2 and Cdc42.

IpaC was purified by nickel-chelation affinity chromatography as described in Methods. It could alternatively be co-purified with its chaperone IpgC and separated from the chaperone by lowering the pH as described in Methods to give a protein with similar purity (not shown). Both SopE2 and Cdc42 were purified as GST-fusions with the proteins shown here having the GST-tag removed by specific proteolysis.
Figure 4. Pull-down assays of SopE2 and IpaC (His6-tagged) with Cdc42 or Rac1.
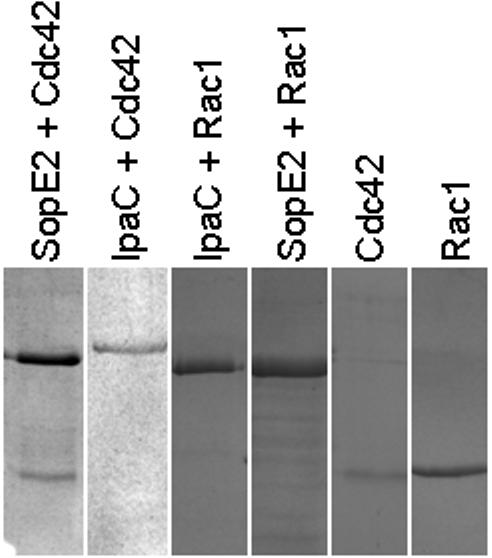
Based on this analysis, GST-tagged SopE2 was able to associate with both Cdc42 and Rac1 while His6-tagged IpaC did not appear to associate with either. In the first and fourth lanes, GST-SopE2 is the upper band and Cdc42 and Rac1, respectively, are the lower bands. In the second and third lanes, His6-IpaC is the only band that appears. The last two lanes shows only Cdc42 and Rac1, respectively. The assay could also be reversed to shown that a GST-tag on Rac1 or Cdc42 was not able to pull out IpaC on a glutathione-affinity column.
2.2. C-terminal deletions eliminate IpaC’s ability to nucleate actin
IpaC has been found to possess distinct functional regions that appear to have specific roles in its virulence function [20]. Within this structural and functional organization there is a putative “oligomerization” region (residues 300-344) that is predicted to participate in the formation of trimeric coiled-coils that may be required for translocon formation. Lastly, the C-terminal tail (residues 344-363) appears to contain a major effector function of IpaC since it is required for cellular invasion [5] [31, 34] but not for translocon pore formation [20, 34]. We previously showed that actin nucleation activity was associated with the C-terminal two thirds of IpaC [17], so we chose to more closely examine the importance of the IpaC C-terminus for its ability to cause actin nucleation in vitro. As shown in Fig. 5, deletion of the IpaC N-terminal amino acids 1-63 (IpaCΔI) do not eliminate its actin nucleation activity while deletion of residues 270-363 (IpaCΔIII) appear to block this activity. This is not necessarily surprising since this portion of IpaC is needed for important IpaC-IpaC interactions [20]. An even smaller deletion, IpaCΔ344-363 also eliminates actin nucleation activity, suggesting that it is the C-terminal tail of IpaC that is responsible for IpaC-induced actin nucleation.
Figure 5. Nucleation of actin by IpaC and IpaC mutants.
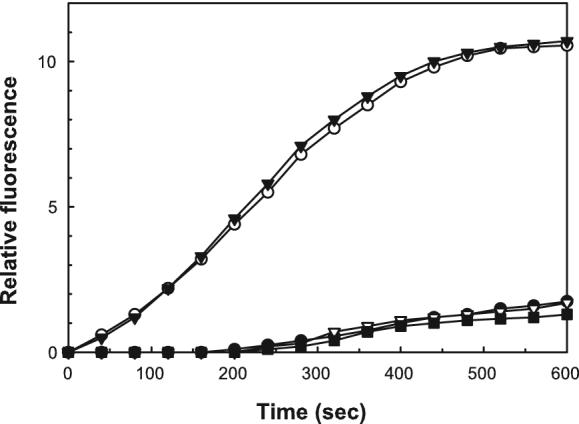
The nucleation of pyrene-labeled G actin was monitored as described in Methods. The plots shown are for actin alone (closed circles), IpaC (open circles), IpaCΔI(closed triangles), IpaCΔ344-363 (closed squares) and IpaCΔIII(open triangles). The IpaC proteins used here were at a final concentration of 0.4 μM and data the shown are for a representative set of experiments from an experiment that was repeated twice.
2.3. Correlation of changes at the IpaC C-terminus with in vivo function
The relationship between IpaC’s in vitro and in vivo functions was determined by examining in vitro actin nucleation relative to in vivo invasion activity and contact-mediated hemolysis which provides a putative measure of translocon pore formation (Table 1). IpaCΔI mutants exhibited actin nucleation activity similar to that of wild-type IpaC, but failed to show any ability to invade cultured cells or lyse red blood cells. IpaCΔI is not secreted because it lacks a functional TTSS secretion signal and thus cannot complement the extracellular functions necessary for invasion and hemolysis [20]. It nevertheless possesses all the in vitro functions of full-length IpaC [17, 20]. At the other extreme, while IpaCΔIII is secreted normally [20], deletion of the putative coiled-coil and the 19-residue C-terminal tail of the protein abolishes its in vivo invasion and hemolysis functions and its in vitro nucleation activity (Table 1). In contrast, although the deletion of the 19 amino acid C-terminal tail (IpaCΔ344-363) or the addition of a 15-residue tag (IpaCStag) has no effect on the contact-hemolysis activity of the resulting Shigella strains, these bacteria are completely noninvasive (Table 1). It is noteworthy that these two proteins also appear to be unable to nucleate actin in vitro (Fig. 5 and data not shown). This would suggest that while the deleted region in IpaCΔIII corresponding to the putative coiled-coil is required for translocon formation and is thus needed for the contact-hemolysis and invasion phenotypes of S. flexneri, along with actin nucleation in vitro, residues 344-363 are only required for the effector function-specific events (actin nucleation in vitro and invasion in vivo). These results also suggest that the effector function of IpaC is somehow related to its ability to contribute to actin nucleation in vitro, which may be a necessary step in Shigella invasion.
Table 1. Effect of C-terminal mutations on the in vitro actin nucleation activity and in vivo virulence functions of IpaC.
| Mutant | Actin nucleationa | Relative invasion (%)b | Contact hemolysis (%)c |
|---|---|---|---|
| IpaC | Yes | 100±9 | 100±1 |
| IpaCΔI | Yes | 0±0 | 8±3 |
| IpaCΔIII | No | 0±0 | 6±3 |
| IpaCΔ344-363 | No | 0±0 | 100±6 |
| IpaCStag | No | 0±0 | 100±5 |
| SipC | Yes | 0±0 | 10±5 |
This is an in vitro activity that involves purified protein and thus does not depend upon active type III secretion.
Invasion is relative to a positive control (SF621 complemented with ipaC), which gave 153±14 invading bacteria per well. (n=3)
Relative hemolysis is also related to SF621 complemented with ipaC. In this case, 100% is actually complete lysis of the RBCs. It is noteworthy that complete absence of ipaC still allows 5 to 10% hemolysis due to the residual activity of IpaB, which is inserted into the erythrocyte membranes even in the absence of IpaC.
2.4. Scanning mutagenesis of the IpaC C-terminal tail
Scanning mutagenesis was used to further investigate the importance of the 19-amino acid tail of IpaC. As would perhaps be expected based on the results of the 19-amino acid C-terminal deletion, all point mutations within this region resulted in levels of hemolysis similar to that seen for bacteria making wild-type IpaC (data not shown). This was not the case, however, with respect to restoring invasion to a Shigella ipaC null mutant (Table 2). Point mutations at residues S345, I346, N347, A354 and I357 resulted in a significant loss in invasion ability (Table 2). One important residue of note within the last 19 amino acids appears to be R362. This residue was identified as being potentially important when introduction of tryptophan at this site almost completely eliminated invasion functions [23] (Table 3). We thus changed this positively charged residue to lysine and found that invasion activity actually increased (though probably not to a statistically significant degree). In contrast, introduction of a similarly sized, nonpolar side chain (methionine) had a minor negative effect on IpaC invasion function (Table 3). Moreover, changing R362 to acidic glutamate had a much more significant negative effect on IpaC’s invasion function, but not to the same extent that conversion to Trp had caused (Table 3). None of these mutants had any negative effect on IpaC’s contribution to Shigella contact-mediated hemolysis (data not shown) and all the mutants with reduced invasiveness were able to make IpaC at or near levels seen in the wild-type bacteria (Fig. 6). These findings clearly indicate that the IpaC C-terminus is important for its invasion function.
Table 2. The effect of scanning mutagenesis on the IpaC C-terminal tail on invasion.
| 345 | 363 | Relative Invasiona |
|---|---|---|
| SINQSKNSTASQIAGNIRA (wt) | 100 ± 12 | |
| AINQSKNSTASQIAGNIRA | 23 ± 14 | |
| SSNQSKNSTASQIAGNIRA | 9 ± 7 | |
| SILQSKNSTASQIAGNIRA | 73 ± 8 | |
| SINLSKNSTASQIAGNIRA | 102 ± 5 | |
| SINQAKNSTASQIAGNIRA | 100 ± 9 | |
| SINQSINSTASQIAGNIRA | 95 ± 4 | |
| SINQSKASTASQIAGNIRA | 95 ± 6 | |
| SINQSKNATASQIAGNIRA | 93 ± 3 | |
| SINQSKNSAASQIAGNIRA | 93 ± 15 | |
| SINQSKNSTLSQIAGNIRA | 71 ± 15 | |
| SINQSKNSTASAIAGNIRA | 101 ± 2 | |
| SINQSKNSTASQSAGNIRA | 32 ± 15 | |
| SINQSKNSTASQISGNIRA | 82 ± 38 | |
| SINQSKNSTASQIASNIRA | 100 ± 10 | |
| SINQSKNSTASQIAGQIRA | 90 ± 36 | |
| SINQSKNSTASQIAGAIRA | 99 ± 5 | |
| SINQSKNSTASQIAGNSRA | 97 ± 8 | |
| SINQSKNSTASQIAGNIRI | 99 ± 10 | |
Invasion is relative to the positive control (SF621 + ipaC), which had 184±22 bacteria per well (n=3-5).
Table 3. Arginine 362 has a role in IpaC effector function.
| Mutant | Relative invasiona |
|---|---|
| Wild-type | 100 ± 12 |
| R362W | 3 ± 1 |
| R362K | 118 ± 17 |
| R362M | 66 ± 9 |
| R362E | 32 ± 5 |
Invasion was tested using a standard gentamycin protection assay with SF621 complemented with wild-type ipaC serving as the positive control. Invasiveness by the positive control was 132± 16 bacteria per well. (n=3)
Figure 6. Expression of IpaC point mutants by S. flexneri.
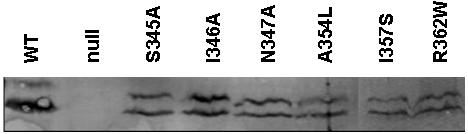
S. flexneri SF621 expressing specific ipaC point mutants that demonstrated reduced invasion function were collected and solubilized with SDS-PAGE buffer. Approximately equal numbers of bacteria were then subjected to SDS-PAGE and IpaC detected by immunoblot analysis using rabbit anti-IpaC antiserum. In this assay, the IpaC protein is often seen as a doublet as shown here.
3. Discussion
S. flexneri uses its type III secretion system (TTSS) to inject translocator and effector proteins into the membrane and cytoplasm of intestinal epithelial cells to promote bacterial invasion. Upon host cell contact IpaB and IpaC are first inserted into the host membrane where they form the translocon, a pore-like transmembrane channel through which other secreted effectors enter the host cytoplasm [9]. Among well-characterized TTSSs, IpaB and IpaC are rather unique translocators because they are clearly known to possess intrinsic effector functions. IpaC in particular has been implicated in triggering the host signaling events that promote the cytoskeletal rearrangements that allow Shigella uptake [11, 12]. While it is not known precisely how IpaC subverts host cell control of the actin cytoskeleton, one possibility is through the direct activation of Cdc42 [5]. Alternatively, IpaC may manifest its effector function through direct manipulation of the host cytoskeleton, such as through promoting localized actin nucleation [17].
We have demonstrated that IpaC possesses a distinct functional organization with the C-terminus possessing a putative coiled-coil domain and tail that are required for bacterial uptake [17, 20]. In this study, we further dissect the IpaC C-terminus to show that the coiled-coil region is essential for the in vivo translocator and effector functions of IpaC. In contrast, the 19-amino acid C-terminal tail does not appear to be required for translocon formation, but is essential for IpaC’s effector function. Other investigators have previously alluded to the importance of the IpaC C-terminus for its effector activities [5] [Barzu et al., Infect. Immun. 1997, 65:1599-1605]. Key deletions within this portion of the protein have no effect on Shigella contact-hemolysis activity, however, they completely eliminate invasion functions. In particular, amino acids at the coiled-coil/tail interface (residues 345 to 347) appear to be critical for effector IpaC function (along with residues I357 and R362). In the case of R362, a conserved mutation to similarly charged Lys does not interfere with effector function, however, conversion to an acidic Glu or bulky/aromatic Trp had major negative effects on this function. The fact that residue 362 is important for function fits well with the observation that either a five-residue deletion (data not shown) or 15-residue addition (IpaC-STag) completely eliminate effector function without having any effect on contact hemolysis. The mechanisms for why this tail is critical for IpaC invasion function still remain unclear.
When the tail region (with or without the coiled-coil segment) is deleted, not only are IpaC’s in vivo virulence functions lost, but the purified protein fails to nucleate actin in vitro. The point mutations introduced within the tail region, however, don’t appear to greatly affect actin nucleation (data not shown), so it is possible that the larger deletion mutants are inducing changes in overall protein structure that compromise the actin nucleation activity. Interestingly, Chang et al. previously proposed that it was not the immediate C-terminus of SipC, the IpaC homologue from Salmonella, but rather regions more to the N-terminal half of the protein that were responsible for actin-nucleating activity [19]. We do not yet have an explanation for the apparently contradictory result we see for IpaC. For SipC, it has been proposed that the C-terminus is needed for the protein’s translocation function [19], however, we see here that the even with mutations within the 19 amino acid tail of IpaC, the protein is still able to direct the formation of pores in red blood cells (as seen by contact hemolysis), suggesting that translocon structures are still able to form. It is possible that pore formation alone is not sufficient for effector translocation into the host cell cytoplasm, however, previous work showing that the C-terminus of IpaC is important for triggering host cytoskeletal changes in the absence of additional Shigella effector proteins suggests that this region is somehow key for IpaC-specific effector activity. It is likely, however, that deletions that extend beyond the 19 amino acid tail and into the putative coiled-coil region of IpaC (and presumably SipC) would have a major negative effect on many aspects of the protein’s function since these mutations are likely to disrupt yet to be identified protein-protein interactions.
Because of the possibility that IpaC may exert its effector function through direct interactions with Cdc42, fluorescence analyses were carried out to determine whether such interactions could be demonstrated in vitro. This would provide a starting point for examining the effects that point mutations have on Cdc42 activation. Unfortunately, no such interaction could be demonstrated in vitro, despite the fact that the assays employed effectively demonstrated a direct Cdc42-SopE2 interaction. The absence of an identifiable IpaC-Cdc42 interaction occurred regardless of the nucleotide that was bound to Cdc42 (GDP, GTP or nonhydrolyzable GTP).
It was anticipated that interactions between Cdc42 and IpaC would provide a foundation for explaining how function-altering point mutations introduced at the C-terminus of IpaC cause it to lose its effector function. Because it turns out that IpaC may not directly interact with Cdc42, alternative explanations for the mechanism behind IpaC’s effector function must be developed. It is attractive to speculate that these mutations might influence IpaC’s ability to nucleate actin, or perhaps IpaC is able to exert its effector function by acting upstream of Cdc42 and Rac1. Nevertheless, the experiments described here more precisely define the structural organization of IpaC and suggest that a key to its effector function may lie within a relatively small region at the C-terminus of the protein.
4. Materials and Methods
4.1. Materials
Shigella flexneri 2a strain 2457T was from A.T. Maurelli (Uniformed Services University of the Health Sciences, Bethesda, MD). S. flexneri strain SF621 containing a nonpolar null mutation in ipaC was from by P.J. Sansonetti (Institut Pasteur, Paris, France). A plasmid for the preparation of SopE2 from Salmonella was from by E.E. Galyov (Institute for Animal Health, Compton, Berkshire, UK). Cdc42 was from G. Bloom (University of Virginia, Charlottesville, VA). Rac1 was from E. Lundquist (University of Kansas) and GST-Rac1 was purchased from Cytoskeleton, Inc. (Denver, CO) Fluorescent probes were purchased from Molecular Probes (Eugene, OR). E. coli cloning strains were from Novagen (Madison, WI). The actin polymerization kit used here was from Cytoskeleton, Inc. and all other chemicals were of reagent grade.
4.2. Preparation of affinity-purified recombinant proteins
The plasmid used to prepare recombinant IpaC was previously described [35]. The ipaC mutant genes were subcloned from pWPsf into pET15b with the resulting plasmids transformed into E. coli BL21(DE3) for high level protein production. Recombinant proteins were purified via an N-terminal His6 tag by nickel-chelation chromatography in the presence of 6 M urea as described previously [20, 22]. Purified proteins were step-dialyzed against 10 mM NaPO4, pH 7.2, 150 mM NaCl (PBS) to remove the urea. In some cases, the urea concentration was reduced to 2M and experiments were carried out by rapid dilution into buffer without urea. This was possible because of the high IpaC concentration that could be maintained in the presence of 2 M urea. Alternatively, IpaC could be purified as a complex with its chaperone IpgC and subsequent separated from the chaperone by lowering the pH as recently described [23] to avoid any exposure to urea. Unfortunately this procedure results in IpaC being maintained at a much low stock protein concentration and so this form of IpaC was only used in confirmatory experiments.
Recombinant GST-Cdc42 and GST-SopE2 were purified after being synthesized in E. coli BL21(DE3) and purified by virtue of their GST fusion tags. Expression was induced by adding IPTG to log phase cultures. After an additional 3h incubation, the cells were harvested by centrifugation and resuspended in 10mM Tris, pH 7.4, 150mM NaCl (TBS). The cells were then frozen at -20°C overnight, thawed, and then sonicated. The insoluble material was removed by centrifugation with the GST fusion proteins remaining in the supernatant fraction. Chromatography using immobilized glutathione was used to affinity purify the GST fusion proteins with elution accomplished using 10 mM glutathione in TBS. Proteins were dialyzed against TBS to remove glutathione from the solution. The GST protein tag was removed by cleavage with thrombin followed by adsorption onto immobilized glutathione resin. All protein samples were stored at -80°C.
4.3. Introduction of mutations at the IpaC C-terminus
Mutations were introduced into ipaC by inverse PCR as described in detail previously [17, 20, 22]. The resulting plasmids were electroporated into S. flexneri SF621, an ipaC null strain [8]. Ampicillin selection ensured the presence of the plasmid while kanamycin resistance and Congo red binding were used as indicators of the presence of the Shigella virulence plasmid. Complementation of SF621 was assessed using a gentamycin protection assay and contact-mediated hemolysis as described [20, 36, 37]. In all cases, IpaC and its mutant derivatives were expressed and secreted by SF621 at comparable levels based on immunoblot analyses (data not shown).
4.4. Fluorescence labeling of Cdc42
Cdc42 was labeled with either succinimidyl 6-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino) hexanoate (NBD) or fluorescein isothiocyanate (FITC) as follows. NBD labeling was carried out by a published procedure that preferentially leads to modification of a single Lys residue (Lys150) of Cdc42 [33]. Modified protein was separated from unreacted dye by gel filtration using Sephadex G25 equilibrated with TBS. Confirmation that proper labeling had occurred was obtained by measuring the expected labeling efficiency of 1:1 by absorbance spectroscopy and determining that the binding of GDP and a nonhydrolyzable GTP analog differentially altered the fluorescence of the protein (data not shown). FITC labeling was carried out using the same conditions, however, its labeling occurred at slightly greater than a 1:1 molar ratio, indicating additional labeling sites. Because the FITC-labeled protein was only being used for fluorescence polarization experiments (see below), knowledge of the labeling sites in this case was not essential.
4.5. Monitoring protein binding to Cdc42 by fluorescence emission
The ability for IpaC to directly interact with NBD-labeled Cdc42 was initially tested by fluorescence emission changes [32] using a FluoroMax fluorescence spectrometer. The Salmonella guanine nucleotide exchange factor (SopE2) was used as a positive control in these experiments [28]. In the cuvette, NBD-labeled Cdc42 was added to buffer at a final concentration of 0.5 μM. EDTA (final concentration of 2 mM) was then added to chelate magnesium, thereby releasing the nucleotides remaining associated with Cdc42 from its purification. GTP, GDP, or GMPPNP was then added to the cuvette at a final concentration of 20 μM. The solution was thoroughly mixed, followed by the addition magnesium at 5 mM, which completely saturated any remaining EDTA and bound to the nucleotides to allow them to assume a proper binding conformation. The NBD-Cdc42 with nucleotide bound was then incubated with nonfluorescent IpaC or SopE2 and the fluorescence emission spectrum of the NBD was obtained.
4.6. Measurement of protein-protein interactions by fluorescence polarization
As another measure of possible IpaC interaction with Cdc42, the latter was labeled with FITC (see above) and its interaction with IpaC was monitored by fluorescence polarization (FP). SopE2 was again used as a positive control that is known to bind to Cdc42 [28]. A Beacon Fluorescence Polarimeter was then used to measure the binding events between IpaC and FITC-labeled Cdc42. In addition, FP was used to detect direct interactions between IpaC and Alexa Fluor 488-labeled actin or FITC-labeled heparin sulfate. In FP, the increase in molecular volume caused by the binding of a nonfluorescent species to a fluorescent species results in a slowing of the overall rotational diffusion rate [22, 32]. This results in an increase in FP for the fluorescent species.
Increasing concentrations of IpaC was added to either the FITC-Cdc42 or FITC-heparin sulfate and the FP value (in millipolarization or mP units) was obtained for each addition. For IpaC binding to Alexa Fluor 488-labeled actin, the interaction was detected using the Beacon Fluorescence Polarimeter using a single concentration of IpaC. The FP of the labeled actin (in the presence and absence of IpaC) was then monitored as a function of time so that IpaC binding to the actin could be distinguished from time-dependent autopolymerization of the actin.
4.7. Actin nucleation
For monitoring IpaC-mediated actin nucleation in vitro [17], pyrene G-actin was incubated at 4°C in G-actin buffer (5 mM Tris-HCl, pH 8.0, 0.1 mM ATP, 0.2 mM CaCl2). IpaC or an IpaC mutant was then added to the sample and pyrene fluorescence was monitored at 23°C. Pyrene fluorescence was measured using a time-based acquisition mode with an excitation wavelength of 330 nm and an emission wavelength of 385 nm. After 15 min, 50× actin polymerization buffer (100 mM MgCl2, 50 mM ATP, 2.5 M KCl) was added, and the change in pyrene fluorescence was monitored as a function of time. Negative controls either contained no added protein or contained IpaD, which has no actin-nucleating activity. SipC from Salmonella typhimurium, which nucleates actin in vitro, was used as a positive control.
4.8. Bacterial invasion of cultured cells
The level of S. flexneri invasion of cultured cells was monitored as described [22]. S. flexneri ipaC mutants harboring plasmids expressing ipaC or one of its mutants were grown in trypticase soy broth (TSB) with 100 μg/ml ampicillin and 50 μg/ml kanamycin to an A600 of 0.5. The bacteria were diluted into serum-free MEM containing 0.45% glucose (MEM-glc) and centrifuged onto pre-confluent Henle 407 cell monolayers (m.o.i. ∼ 10), and incubated with the cells for 30 min at 37° C. Free bacteria were removed by aspirating off the supernatant and the monolayers were washed with MEM containing 5% calf serum and 50 μg/ml gentamycin. The cells were incubated in the final gentamycin wash for 1 h to kill adherent, non-internalized bacteria and then rinsed with MEM-glc. The cells were then overlaid with 250 μl 0.5% agarose in water followed by 0.5% agar containing LB medium. After overnight incubation at 37°C, the internalized bacteria that formed subsurface colonies were counted.
4.9. Measurement of contact-mediated hemolysis
Hemolysis was measured according to the procedure of Sansonetti [22]. Bacteria were grown overnight on TSA-Congo red plates and a single red colony was used to inoculate TSB. Mid-log phase bacteria were collected by centrifugation and suspended in PBS at 2.5% of the original volume. Sheep red blood cells (RBC) were washed and resuspended in PBS at 1 × 1010 cells/ml. RBC and bacteria (50 μl each) were added to microtiter wells and the plates centrifuged at 2200 × g for 15 min at 20°C, and the plates were incubated at 37°C for 2 h. The cells were then suspended by adding 90 μl cold PBS and the plates were centrifuged again at 2200 × g at 15°C for 15 min. The supernatant fraction (100 μl) was transferred to a second microtiter plate and absorbance of the released hemoglobin measured at 545nm.
4.10. Immunoblot anaysis
To analyze the expression levels of IpaC in S. flexneri, bacteria from 1 ml of culture were suspended in SDS-sample buffer and boiled for 5 min. The proteins in the samples were separated on a 15% resolving SDS-PAGE gels and the gels were incubated in blotting buffer (25mM Tris, pH8.0, 192 mM glycine) for 20 min. The proteins were then blotted onto PVDF membranes nitrocellulose using a BioRad Transblot-SD Semidry Transfer Cell for 35 min at 15V. After blotting, the membrane was incubated in blotting buffer containing 3% nonfat dried milk for 45 min, following by incubation with rabbit anti-IpaC antiserum diluted 1:1000 at 4 °C overnight. The membranes were washed in PBS containing 0.1% Tween-20 three times for 5 min each. Membranes were incubated with infrared-labeled secondary antibody diluted 1:10,000 for 1 h at room temperature. Membranes were then washed an additional three times with PBS/Tween-20 and twice with PBS for 5min with shaking. Membranes were then scanned on a Li-Cor Odyssey™ Infrared scanner and imaged using Odyssey Software (LiCor Biosciences, Lincoln, NE).
4.11. Protein pull-down assays
Cdc42 or Rac1 (10-15 μg) was incubated with 0.8 μM GTPγS, and 50 μM EDTA at 37°C. MgCl2 was added to a concentration of 240 μM and the mixture was placed on ice. SopE2 or IpaC (10-15 μg) with or without a GST or His6 tag, respectively, was the added to the reaction which was maintained on ice for 30 min. Next, 60 μl of a 1:1 slurry of glutathione resin or charged nickel chelation resin, as appropriate, with TBS was added to proteins and reaction was incubated on ice for 30 min. The reaction mixture was centrifuged at 8,000 rpm at 4 °C for one min to collect the resin. The supernatant fraction was discarded and the resin was washed twice in 500 μl TBS with 30 μM MgCl2. After the second wash, 60 μl of TBS containing 10 μM glutathione (or 1 mM EDTA when the nickel-chelation resin was used) was added to the resin and reaction was incubated on ice for 10 min. The resin was then pelleted by centrifugation at 8,000 rpm at 4 °C for 5 min and the supernatant was removed without disturbing the pellet. The supernatant fraction was then analyzed using SDS-PAGE with Coomassie blue staining. Different permutations of the pull-down were accomplished by using GST-tagged GTPases to also examine the possibility that IpaC may be able to associate with them.
Acknowlegements
This work was supported by PHS grant AI 034428 and KU Research Development Fund to W.D.P. as well as the NIH COBRE Program, PHS grant RR017708, to the University of Kansas.
Abbreviations
- Ipa
invasion plasmid antigen
- Sip
Salmonella invasion protein
- TTSS
type III secretion system
- TTSA
type III secretion apparatus
- GST
glutathione S-transferase
- NBD
6-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)hexanoate
- FITC
fluorescein isothiocyanate
- TBS
Tris-buffered saline
- PBS
phosphate-buffered saline
- FP
fluorescence polarization
- mP
millipolarization units
- RBC
red blood cells
- GEF
guanine nucleotide exchange factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hale TL. Bacillary dysentery. In: Hansler M, editor. Topley and Wilson’s Microbiology and Microbial Infections. Vol. 3. London: 1998. pp. 47–493.pp. 479–493. [Google Scholar]
- [2].Mohle-Boetani JC, Stapleton M, Finger R, Bean NH, Poundstone J, Blake PA, Griffin PM. Communitywide shigellosis: control of an outbreak and risk factors in child day-care centers. Am J Public Health. 1995;85:812–816. doi: 10.2105/ajph.85.6.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Parsot C, Sansonetti PJ. Invasion and the pathogenesis of Shigella infections. Curr Top Microbiol Immunol. 1996;209:25–42. doi: 10.1007/978-3-642-85216-9_2. [DOI] [PubMed] [Google Scholar]
- [4].Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–169. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- [5].Tran Van Nhieu G, Caron E, Hall A, Sansonetti PJ. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. Embo J. 1999;18:3249–3262. doi: 10.1093/emboj/18.12.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sasakawa C, Adler B, Tobe T, Okada N, Nagai S, Komatsu K, Yoshikawa M. Functional organization and nucleotide sequence of virulence Region-2 on the large virulence plasmid in Shigella flexneri 2a. Mol Microbiol. 1989;3:1191–1201. doi: 10.1111/j.1365-2958.1989.tb00269.x. [DOI] [PubMed] [Google Scholar]
- [7].Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Menard R, Sansonetti PJ, Parsot C. Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J Bacteriol. 1993;175:5899–5906. doi: 10.1128/jb.175.18.5899-5906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Blocker A, Gounon P, Larquet E, Niebuhr K, Cabiaux V, Parsot C, Sansonetti P. The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J Cell Biol. 1999;147:683–693. doi: 10.1083/jcb.147.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Buttner D, Bonas U. Port of entry--the type III secretion translocon. Trends Microbiol. 2002;10:186–192. doi: 10.1016/s0966-842x(02)02331-4. [DOI] [PubMed] [Google Scholar]
- [11].Marquart ME, Picking WL, Picking WD. Soluble invasion plasmid antigen C (IpaC) from Shigella flexneri elicits epithelial cell responses related to pathogen invasion. Infect Immun. 1996;64:4182–4187. doi: 10.1128/iai.64.10.4182-4187.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tran Van Nhieu G, Bourdet-Sicard R, Dumenil G, Blocker A, Sansonetti PJ. Bacterial signals and cell responses during Shigella entry into epithelial cells. Cell Microbiol. 2000;2:187–193. doi: 10.1046/j.1462-5822.2000.00046.x. [DOI] [PubMed] [Google Scholar]
- [13].De Geyter C, Vogt B, Benjelloun-Touimi Z, Sansonetti PJ, Ruysschaert JM, Parsot C, Cabiaux V. Purification of IpaC, a protein involved in entry of Shigella flexneri into epithelial cells and characterization of its interaction with lipid membranes. FEBS Lett. 1997;400:149–154. doi: 10.1016/s0014-5793(96)01379-8. [DOI] [PubMed] [Google Scholar]
- [14].Osiecki JC, Barker J, Picking WL, Serfis AB, Berring E, Shah S, Harrington A, Picking WD. IpaC from Shigella and SipC from Salmonella possess similar biochemical properties but are functionally distinct. Mol Microbiol. 2001;42:469–481. doi: 10.1046/j.1365-2958.2001.02654.x. [DOI] [PubMed] [Google Scholar]
- [15].Tran N, Serfis AB, Osiecki JC, Picking WL, Coye L, Davis R, Picking WD. Interaction of Shigella flexneri IpaC with model membranes correlates with effects on cultured cells. Infect Immun. 2000;68:3710–3715. doi: 10.1128/iai.68.6.3710-3715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tran Van Nhieu G, Sansonetti PJ. Mechanism of Shigella entry into epithelial cells. Curr Opin Microbiol. 1999;2:51–55. doi: 10.1016/s1369-5274(99)80009-5. [DOI] [PubMed] [Google Scholar]
- [17].Kueltzo LA, Osiecki J, Barker J, Picking WL, Ersoy B, Picking WD, Middaugh CR. Structure-function analysis of invasion plasmid antigen C (IpaC) from Shigella flexneri. J Biol Chem. 2003;278:2792–2798. doi: 10.1074/jbc.M208383200. [DOI] [PubMed] [Google Scholar]
- [18].Hayward RD, Koronakis V. Direct nucleation and bundling of actin by the SipC protein of invasive Salmonella. Embo J. 1999;18:4926–4934. doi: 10.1093/emboj/18.18.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chang J, Chen J, Zhou D. Delineation and characterization of the actin nucleation and effector translocation activities of Salmonella SipC. Mol Microbiol. 2005;55:1379–1389. doi: 10.1111/j.1365-2958.2004.04480.x. [DOI] [PubMed] [Google Scholar]
- [20].Picking WL, Coye L, Osiecki JC, Barnoski Serfis A, Schaper E, Picking WD. Identification of functional regions within invasion plasmid antigen C (IpaC) of Shigella flexneri. Mol Microbiol. 2001;39:100–111. doi: 10.1046/j.1365-2958.2001.02210.x. [DOI] [PubMed] [Google Scholar]
- [21].Harrington A, Darboe N, Kenjale R, Picking WL, Middaugh CR, Birket S, Picking WD. Characterization of the Interaction of Single Tryptophan Containing Mutants of IpaC from Shigella flexneri with Phospholipid Membranes. Biochemistry. 2006;45:626–636. doi: 10.1021/bi0512593. [DOI] [PubMed] [Google Scholar]
- [22].Harrington AT, Hearn PD, Picking WL, Barker JR, Wessel A, Picking WD. Structural characterization of the N terminus of IpaC from Shigella flexneri. Infect Immun. 2003;71:1255–1264. doi: 10.1128/IAI.71.3.1255-1264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Birket SE, Harrington AT, Espina M, Smith ND, Terry CM, Darboe N, Markham AP, Middaugh CR, Picking WL, Picking WD. Preparation and characterization of translocator/chaperone complexes and their component proteins from Shigella flexneri. Biochemistry. 2007;46:8128–8137. doi: 10.1021/bi700099c. [DOI] [PubMed] [Google Scholar]
- [24].Pallen MJ, Dougan G, Frankel G. Coiled-coil domains in proteins secreted by type III secretion systems. Mol Microbiol. 1997;25:423–425. doi: 10.1046/j.1365-2958.1997.4901850.x. [DOI] [PubMed] [Google Scholar]
- [25].Adam T, Arpin M, Prevost MC, Gounon P, Sansonetti PJ. Cytoskeletal rearrangements and the functional role of T-plastin during entry of Shigella flexneri into HeLa cells. J Cell Biol. 1995;129:367–381. doi: 10.1083/jcb.129.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Adam T, Giry M, Boquet P, Sansonetti P. Rho-dependent membrane folding causes Shigella entry into epithelial cells. Embo J. 1996;15:3315–3321. [PMC free article] [PubMed] [Google Scholar]
- [27].Stender S, Friebel A, Linder S, Rohde M, Mirold S, Hardt WD. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol Microbiol. 2000;36:1206–1221. doi: 10.1046/j.1365-2958.2000.01933.x. [DOI] [PubMed] [Google Scholar]
- [28].Bakshi CS, Singh VP, Wood MW, Jones PW, Wallis TS, Galyov EE. Identification of SopE2, a Salmonella secreted protein which is highly homologous to SopE and involved in bacterial invasion of epithelial cells. J Bacteriol. 2000;182:2341–2344. doi: 10.1128/jb.182.8.2341-2344.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Friebel A, Ilchmann H, Aepfelbacher M, Ehrbar K, Machleidt W, Hardt WD. SopE and SopE2 from Salmonella typhimurium activate different sets of RhoGTPases of the host cell. J Biol Chem. 2001;276:34035–34040. doi: 10.1074/jbc.M100609200. [DOI] [PubMed] [Google Scholar]
- [30].Ohya K, Handa Y, Ogawa M, Suzuki M, Sasakawa C. IpgB1 is a novel Shigella effector protein involved in bacterial invasion of host cells. Its activity to promote membrane ruffling via Rac1 and Cdc42 activation. J Biol Chem. 2005;280:24022–24034. doi: 10.1074/jbc.M502509200. [DOI] [PubMed] [Google Scholar]
- [31].Menard R, Prevost MC, Gounon P, Sansonetti P, Dehio C. The secreted Ipa complex of Shigella flexneri promotes entry into mammalian cells. Proc Natl Acad Sci U S A. 1996;93:1254–1258. doi: 10.1073/pnas.93.3.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum Press; New York: 1983. [Google Scholar]
- [33].Nomanbhoy TK, Leonard DA, Manor D, Cerione RA. Investigation of the GTP-binding/GTPase cycle of Cdc42Hs using extrinsic reporter group fluorescence. Biochemistry. 1996;35:4602–4608. doi: 10.1021/bi951743d. [DOI] [PubMed] [Google Scholar]
- [34].Barzu S, Benjelloun-Touimi Z, Phalipon A, Sansonetti P, Parsot C. Functional analysis of the Shigella flexneri IpaC invasin by insertional mutagenesis. Infect Immun. 1997;65:1599–1605. doi: 10.1128/iai.65.5.1599-1605.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Picking WL, Mertz JA, Marquart ME, Picking WD. Cloning, expression, and affinity purification of recombinant Shigella flexneri invasion plasmid antigens IpaB and IpaC. Protein Expr Purif. 1996;8:401–408. doi: 10.1006/prep.1996.0117. [DOI] [PubMed] [Google Scholar]
- [36].Picking WL, Nishioka H, Hearn PD, Baxter MA, Harrington AT, Blocker A, Picking WD. IpaD of Shigella flexneri is independently required for regulation of Ipa protein secretion and efficient insertion of IpaB and IpaC into host membranes. Infect Immun. 2005;73:1432–1440. doi: 10.1128/IAI.73.3.1432-1440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Espina M, Olive AJ, Kenjale R, Moore DS, Ausar SF, Kaminski RW, Oaks EV, Middaugh CR, Picking WD, Picking WL. IpaD Localizes to the Tip of the Type III Secretion System Needle of Shigella flexneri. Infect Immun. 2006;74:4391–4400. doi: 10.1128/IAI.00440-06. [DOI] [PMC free article] [PubMed] [Google Scholar]