

Abstract
Huntington’s disease (HD) is associated with transcriptional dysregulation, and multiple studies with histone deacetylase (HDAC) inhibitors suggest that global approaches for restoring transcriptional balance and appropriate protein acetylation are therapeutically promising. To determine whether more targeted approaches might be effective, we have tested the impact of all the HDACs in Drosophila on Huntingtin (Htt)-induced pathology. Among the zinc-dependent or ‘classic’ HDACs, we find that neurodegeneration is most sensitive to levels of Rpd3. We also find that among the NAD+-dependent class III deacetylases, genetic or pharmacological reduction of either Sir2 or Sirt2 provides neuroprotection to Htt-challenged animals and that even greater neuroprotection is achieved when Rpd3 and Sir2 are simultaneously reduced. Our experiments suggest that longevity promoting strategies may be distinct from those that protect against neurodegeneration in Drosophila challenged with mutant human Htt. These results highlight a novel therapeutic approach for HD in the form of Sir2 inhibition and possible combinatorial inhibition of Sir2 and Rpd3.
INTRODUCTION
Huntington’s disease (HD) is a devastating neurodegenerative disorder caused by an expanded polyglutamine (polyQ) repeat in the Huntingtin (Htt) protein, and is one of many diseases fitting into the broader category of protein misfolding diseases (1–4). Transcriptional dysregulation is one of the early phenotypes seen in HD (for review see 5). Initial studies in Drosophila (6) followed by studies in mammalian (7,8) and other (9) model systems have demonstrated that global reduction of histone deacetylase (HDAC) activities slows the rate of neurodegeneration in in vivo models of HD and related polyQ diseases.
A number of cellular processes, including modulation of DNA accessibility for transcription, replication and repair, are regulated by posttranslational protein modifications including acetylation and deacetylation of proteins, most notably histones. Acetylated histones are generally correlated with gene activity, whereas deacetylated histones are associated with chromatin structure that is less accessible to transcriptional activation (10). Deacetylases are divided into two mechanistic groups: (i) the zinc-dependent or classic HDACs, which include the Rpd3-like proteins (class I; in humans HDACs 1, 2, 3 and 8), the Hda-1-like proteins (class II; HDACs 4, 5, 6, 7 and 9) and the class IV HDAC11; and (ii) the NAD+-dependent Sir2-like group of sirtuins (class III deacetylases) (Table 1). The deacetylases are highly conserved across species, suggesting non-redundant roles in biological processes (11–13). Despite the label of HDACs and histone acetyltransferases, these enzymes also regulate the activity of non-histone protein targets such as p53 (deacetylated by HDAC1) (14,15) or tubulin (by HDAC6 and Sirt2) (16). The sirtuins, typified by yeast and Drosophila Sir2 and the human ortholog SIRT1, are reported to affect a number of genes that influence neuronal survival (17–19) and are also reported to promote lifespan extension in several organisms (20).
Table 1.
HDAC orthologs in yeast, worm, fly and human are shown for reference
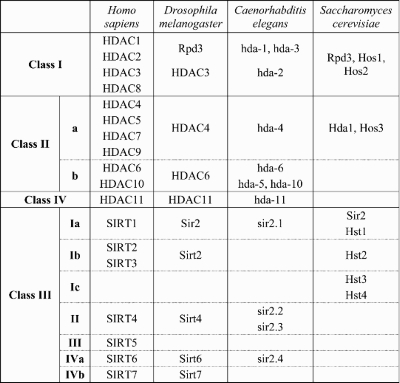 |
To explore the possibility of modulating specific HDACs as a therapeutic strategy for treating HD-mediated neurodegeneration, we used both genetic and pharmacological strategies to examine the contribution of members of each deacetylase class to growth, survival and neurodegeneration in a Drosophila model of HD that expresses mutant human Htt exon 1 protein (Httex1p Q93) in all neurons. We find that Httex1p-induced neurodegeneration in Drosophila is most readily impacted by inhibition of selected HDACs (i.e. Rpd3; Sir2) either individually or in combination. These studies indicate highly restricted roles for the different HDACs in their contribution to mutant Htt-mediated pathology in flies.
RESULTS
Neuronal survival of HD flies is most sensitive to levels of Rpd3 among the classic HDACs
Previous studies revealed that broad-based inhibition of classic HDACs (either genetically or pharmacologically) is protective for neurodegeneration in Httex1p-challenged Drosophila (6) and other animals (7,8,21). To investigate the potential specificity of these HDACs in neurodegeneration, we tested all members of the Drosophila class I, II and IV HDACs for effects on Htt-mediated degeneration using two alleles for each locus, including classic loss of function alleles and short hairpin RNA (shRNA) silencing constructs.
Drosophila Rpd3 is a class I HDAC, that is equally homologous to human HDACs 1/2, and HDAC3 (Table 1). Flies expressing the mutant human Httex1p Q93 in all neurons, exhibit reduced eclosion rates, progressive neuronal degeneration and early lethality (6,22). To determine the consequences of altered Rpd3 activity to this pathology, we compared Httex1p Q93-expressing animals with normal levels of Rpd3 with those with partially reduced levels of Rpd3. Using two independent mutations, heterozygous reduction in the levels of Drosophila Rpd3 markedly increases survival of Htt-challenged flies (Fig. 1A), and neuronal survival is also increased, as measured by the number of remaining photoreceptor neurons in the eye (Fig. 1B).
Figure 1.

Rpd3 is unique among the classic deacetylases in impacting degeneration: (A) heterozygous reduction of Rpd3 improves survival and (B) reduces neurodegeneration of flies challenged with Httex1p Q93; (C) the effect of heterozygous loss of each of the HDACs in Drosophila on survival to eclosion of Httex1p Q93-challenged animals was determined using multiple heterozygous mutations and/or shRNA constructs (one allele from each is shown here, for the full set of allele data see Supplementary Material, Fig. S1A and B). Only reduction of Rpd3 led to an increase in survival. Relative survival was calculated as (HDACHtt/HDACwt)/(ctrlHtt/ctrlwt), where ‘HDACHtt’ and ‘ctrlHtt’ is the number of eclosed elav > Httex1p Q93 flies with or without the HDAC mutation, and ‘HDACwt’ and ‘ctrlwt’ the eclosion number of the Htt non-expressing siblings with or without the HDAC mutation; (D) the effect of reduced HDAC levels on survival of neurons in Httex1p Q93-challenged animals. The results are shown as the difference in photoreceptor number compared with internal controls with a normal genetic background. Only Rpd3 leads to improved neuronal survival. The null allele of Rpd3 shown here, Rpd3[m5-5], reduces the extent of neuronal loss even more than a weaker hypomorphic allele (see panel B and Supplementary Material, Fig. S1B).
The contribution of other HDACs to pathology was also investigated using loss of function mutations or shRNA constructs that reduce the endogenous messenger ribonucleic acid (mRNA) levels in the nervous system (Fig. 1C and D and Supplementary Material, Fig. S1A and B). We do not observe any suppression of lethality or of neuronal degeneration in animals with reduced levels of HDAC3, in animals with reduced levels of either of the class II HDACs (HDAC4 or 6), nor in Drosophila with reduced levels of HDAC11 (CG31119). We conclude that among the class I, II and IV HDACs, neuronal survival is uniquely sensitive to Rpd3 levels in Drosophila challenged with mutant Httex1p.
Reduction of Sir2 activity is neuroprotective in mutant Httex1p-expressing Drosophila
Sirtuins have been implicated in diverse processes, such as aging, neurodegeneration and overall cellular metabolism (23), and they are capable of deacetylating histones. Therefore, we investigated the potential impact of the Sir2-ortholog genes of Drosophila on HD pathology. Httex1p Q93-challenged flies that are also heterozygous for Sir2 null mutations exhibit improved survival compared with animals with normal Sir2 doses (Fig. 2A). The number of photoreceptor neurons remaining, a measure of neuronal loss (24), is also improved when the Sir2 dose is reduced by 50% (Fig. 2B).
Figure 2.

Reduced sirtuin levels are neuroprotective: A 50% reduction of Sir2 (i.e. heterozygous null) leads to (A) greater survival to adulthood and (B) an increased number of photoreceptor neurons of Httex1p Q93-expressing flies; (C) heterozygous loss of Sirt2 also reduces photoreceptor neuron survival; (D) sirtinol, (E) nicotinamide and (F) niacin administration result in reduced neuronal death; (G) pseudopupil images of a normal eye and 7-day-old Httex1p Q93 flies is shown; niacin reduces the rhabdomere loss in Htt flies.
Recent studies find that SIRT2, a second member of the NAD+-dependent HDAC family which is most widely known for its ability to interact with tubulin, is also found in the nucleus (25) and in neurons of mice (G. Bates et al., personal communication), thus identifying SIRT2 as a putative therapeutic target for affecting HD-mediated transcriptional dysregulation. Accordingly, we tested Sirt2 in Drosophila. We find that reduction in the level of Sirt2 in Drosophila leads to greater survival of photoreceptor neurons (Fig. 2C), although it does not suppress lethality (Supplementary Material, Fig. S1A).
The Httex1p Q93 Drosophila model has been used extensively to study the effects of various pharmacological agents on HD pathogenesis (6,24). At least two HDAC inhibitors, namely butyrate and suberoylanilide hydroxamic acid (SAHA), have been shown to suppress neurodegeneration of these flies. We next explored whether pharmacological modulation of sirtuin activity could mimic the effects observed with genetic modulation. Sirtinol is an inhibitor of SIR2 (and possibly other sirtuins) that exhibits no activity against classic HDACs such as Rpd3 (26). Feeding Httex1p Q93-challenged flies on sirtinol-containing food increases survival of photoreceptor neurons with maximal rescue at 100 µm (Fig. 2D). Nicotinamide is the first product of the deacetylation reaction catalyzed by sirtuins and has been implicated as a physiologically relevant inhibitor of SIR2 activity (27,28). We fed flies nicotinamide to inhibit the deacetylation reaction and observed a reduced loss of photoreceptor neurons compared with control siblings (Fig. 2E). Niacin, a conveniently available vitamin supplement that can readily exchange with nicotinamide, also exhibits a similar rescue (Fig. 2F and G). Similar results with nicotinamide feeding have been reported for a Drosophila model of SCA3 (29).
Combinatorial reduction of HDACs is additive
To determine whether Rpd3 and Sir2 affect distinct mechanisms, we tested whether combinatorial inhibition of these two activities is additive. Animals doubly heterozygous for loss of function mutations of Sir2 and Rpd3, show a greater reduction in Htt-induced lethality than animals heterozygous for single mutants (Fig. 3A) and they exhibit less neuronal degeneration than single mutants (Fig. 3B). To determine whether this genetic synergy can be replicated pharmacologically, animals were fed the Rpd3 inhibitor, butyrate, or a Sir2 inhibitor, nicotinamide, or both. To prevent toxicity in this experiment, we used low concentrations that produce no effect on pathogenesis when fed alone and found that feeding these compounds in combination produces a significant rescue of photoreceptor neurons (Fig. 3C).
Figure 3.

Combinatorial reduction of Rpd3 and Sir2 shows increased neuroprotection in Httex1p Q93 flies: (A) genetic reduction of either Rpd3 or Sir2 in heterozygotes is protective. The survival to adulthood of control flies (Httex1p Q93 alone) is increased when double heterozygous for Rpd3 and Sir2; (B) the level of neuroprotection of Httex1p Q93-challenged flies is also significantly increased by concurrent reduction of both Rpd3 and Sir2 compared with either alone; (C) pharmacological treatment using butyrate (B) and nicotinamide (N) is additive. Although each of these compounds can be neuroprotective at 10 times higher concentrations, here we fed animals low levels of nicotinamide (2 mm) and butyrate (10 mm) and combinatorial synergy is observed leading to neuroprotection; (D) reduced levels of Rpd3 do not lead to increased levels of Sir2 expression. Animals of the genotype Rpd3[04556]/TM3 were crossed to either wild-type (Ore-R) or w flies (to control for background effects if present), RNA was extracted from whole males and real time quantitative RT–PCR used to evaluate the levels of Sir2 expression.
The observation that heterozygous loss of either Rpd3 or Sir2 alone is neuroprotective and that heterozygous loss of both together is additive is most consistent with a parallel effect of these two genes on transcription. However, because these HDACs have opposing effects on lifespan, one study suggested that Rpd3 may suppress Sir2 expression (30), while another found that chemical inhibitors of class I, II HDACs (e.g. Rpd3) did not increase Sir2 levels (31). To resolve this conflict, we re-examined the response of Sir2 levels to reduced Rpd3. Using the same allele as previously reported (30), we find that Sir2 mRNA levels are normal and unchanged in Rpd3 heterozygotes in two different genetic backgrounds, indicating that Rpd3 does not regulate Sir2 expression levels (Fig. 3D). This lack of change in Sir2 levels in response to lowered Rpd3 was also observed in Htt-challenged animals (data not shown). Taken together, we conclude that Rpd3 and Sir2 act independently and in parallel and that combinatorial reduction of both deacetylase activities produces an improved rescue over any single treatment alone. Importantly, the improvement in neurodegenerative phenotypes of flies heterozygous for the Rpd3 or Sir2 mutation is not due to altered transcription levels of the Httex1p Q93 transgene itself, as measured by reverse transcription polymerase chain reaction (RT–PCR) (Supplementary Material, Fig. S2A and B).
Effects of deacetylase levels on lifespan
HDACs have been implicated in regulating longevity. Reduced levels of Rpd3 have been reported to extend the lifespan of Drosophila (30,32) while caloric restriction (CR) has been shown to increase Sir2 levels and lead to increased longevity in Drosophila and other metazoans (20,33,34). Since flies expressing Httex1p Q93 have a significantly shorter lifespan than normal flies and levels of both of these deacetylases are important in HD pathogenesis, we designed experiments to monitor the lifespan of Htt-expressing Drosophila mutant for Rpd3 or Sir2. We found that the lifespan of Htt non-expressing control flies was extended from a median of 49 to 56 days by reducing Rpd3, but the early death phenotype of Htt-challenged flies was unaffected (Fig. 4A).
Figure 4.

Relationship between lifespan altering treatments and neurodegeneration: (A) reduced Rpd3 does not extend the lifespan of Htt-challenged flies (solid lines) but does extend the lifespan of control flies (dashed lines). Black lines are flies with reduced Rpd3, gray lines are controls with normal Rpd3 levels; (B) the reduction in Sir2 levels does not alter the longevity of either the Htt-challenged animals (solid lines – black: reduced Sir2; gray: control) or Htt non-expressing siblings (dashed lines – black: reduced Sir2; gray: control); (C and D) caloric restriction (CR) does not increase longevity of Httex1p Q93-expressing animals (solid lines) (C) nor is it neuroprotective (D). control animals not expressing Htt reared on the calorie-restricted diet (dashed black line) showed an increase in lifespan compared with fully fed (FF) siblings (dashed gray line) as has been reported; (E–G) overexpression of Sir2 (oe) does not increase the percent of animals surviving to adulthood (E) nor does it alter the survival of photoreceptor neurons (F). As previously described, overexpression of Sir2 (G) does increase the longevity of normal flies (dashed lines – black: overexpression; gray: controls) and the longevity of diseased flies is slightly increased by elevated Sir2 (solid lines – black: overexpression; gray: normal Sir2 controls).
Increased Sir2 activity is reported to lead to increased longevity (35,36), and CR is one of several manipulations that up-regulates Sir2. To determine whether CR can affect ongoing HD pathogenesis, adult virgin flies expressing Httex1p Q93 in their nervous systems were reared on standard media or media containing half the amount of glucose and yeast (37). Although the lifespan of Htt non-expressing flies was increased when reared on diluted food (median lifespan 72 versus 59 days), the early death observed in Htt-challenged animals was unaffected by CR (Fig. 4C). There was also no change in photoreceptor neuron survival under these conditions (Fig. 4D). Thus, animals challenged with Htt and undergoing active neurodegeneration do not derive benefit from dietary restriction either with respect to neuronal survival or with respect to longevity.
We also monitored the effect of genetically increasing Sir2 on neuronal survival, lethality and lifespan, using an enhancer/promoter insertion that increases expression of the downstream Sir2 gene (34). We find that overexpression of Sir2 does not reduce the lethality caused by Htt (Fig. 4E), nor do increased levels of Sir2 reduce the level of neuronal degeneration observed in Httex1p Q93 flies (Fig. 4F). However, overexpression of Sir2 significantly extends the lifespan of Htt non-expressing flies while slightly increasing the lifespan of Htt-expressing siblings (Fig. 4G). Conversely, when animals are heterozygous for a null mutation in Sir2, lifespan is not significantly altered in either wild-type siblings (Fig. 4B and 38) or in Htt-expressing flies (Fig. 4B). Thus, Sir2 has distinct effects on lifespan and on neuronal survival in mutant Httex1p-expressing flies. Taken together, these studies indicate that manipulations that positively impact aging do not necessarily positively impact the effects of neurodegeneration.
Since it has been suggested that resveratrol extends lifespan in flies (39), can be neuroprotective (40), and may act by modulating SIRT1 activity (41), we tested this drug in our Htt model by feeding resveratrol to Htt-challenged Drosophila. We find that resveratrol can rescue neuronal degeneration in Htt-challenged flies in a dose-dependent manner (Fig. 5A), although it does not alter the early death phenotype of flies expressing Httex1p Q93 (Fig. 5B). We also find that Htt-challenged flies homozygous for a Sir2 null mutation are rescued to a similar extent (Fig. 5C), indicating that the ability of resveratrol to suppress neurodegeneration does not depend on Sir2.
Figure 5.

Resveratrol feeding: (A) resveratrol exhibits a dose-dependent increase in neuronal survival of Httex1p Q93 animals; (B) 10 µm resveratrol does not rescue the early death phenotype of Htt-challenged flies; (C) 10 µM resveratrol improves the survival of neurons in Sir2 homozygous null Httex1p Q93 flies.
DISCUSSION
This study indicates that selective targeting of specific HDACs can provide much of the neuroprotection seen with general HDAC inhibition in Htt-challenged Drosophila. Early studies using broadly acting pharmacological agents (SAHA, butyrate) (6–8,21), or genetic cofactors that act with multiple HDAC complexes such as Sin3A (6,42) demonstrated that reducing HDACs can be neuroprotective for flies challenged by expanded Htt. Here we show that reducing Rpd3 can provide levels of protection comparable with those achieved with more broadly acting class I/II agents (Fig. 1; 6). These observations suggest that specific pharmacological agents that target Rpd3 would be therapeutically preferable. Our observations do not mean that other HDACs may not play a role in neuronal survival but rather that Rpd3 is the most rate-limiting among them and that the bulk of therapeutic potential may be achieved by targeted inhibition of this particular classical HDAC. For example, overexpression of HDAC6 is seen to be beneficial for a spinobulbar muscular atrophy model in Drosophila (43) and complete knockdown of HDAC6 by RNAi increases GFP-HttQ150 inclusion bodies in neuro2a cells (44) although neuronal degeneration was not evaluated. The lack of a demonstrable change in response to a 50% reduction of HDAC6 in our HD model may indicate that this enzyme is not rate-limiting or that there are disease-specific differences in the response to protein.
In addition to Rpd3, specific targeting of the NAD+-dependent class III HDACs, Sir2 and Sirt2, is also beneficial. Comparing the effect of reduced Sir2 on neurodegeneration with the effect of increased Sir2 in the modulation of lifespan in other settings suggests that the role of Sir2 in cellular protection may be complex. Yeast studies indicate that increasing Sir2 positively regulates replicative aging, while negatively impacting chronological aging (45–47). Studies in flies and worms suggest that lifespan can be extended by increasing Sir2 (Fig. 4G, 34,48), but the corollary, that Sir2 reduction might shorten lifespan, is not necessarily true since we and others find that heterozygous loss of Sir2 has no effect on lifespan (Fig. 4B; 38,49). We find that reducing Sir2 improves neuronal survival in mutant Httex1p-expressing flies without affecting lifespan (Fig. 2A and B, Fig. 4B). These results are consistent with the recent observation that reduction of Sirt1 in cultured neurons can be neuroprotective in the face of oxidative challenge (50). Also, consistent with our observations that both niacin and nicotinamide suppress degeneration in Htt-challenged animals, pharmacological inhibition of Sir2 with nicotinamide suppresses neuronal degeneration of Kenyon cells in a Drosophila model expressing mutant Ataxin-3 (29), while increased Sir2 enhances degeneration in Drosophila models of SCA1 (Ataxin-1) (29,42). Although a NAD synthase (nicotinamide mononucleotide adenylytransferase) has been found to be protective in a SCA1 model, this effect was independent of its enzymatic activity but rather reflected a novel chaperone function for this protein (51,52). Studies in both Caenorhabditis elegans and Drosophila melanogaster suggest that complete loss of Sir2 is deleterious in the worm (53) and is deleterious compared with heterozygous loss in Htt-challenged flies (Supplementary Material, Fig. S1C). In worms overexpressing Sir2, Htt-induced degeneration of the tail mechanosensory neuron (53) and of the ASH neurons (21) is suppressed, whereas overexpression in flies confers no significant neuroprotection. In contrast, heterozygous loss of Sir2 is protective in flies (Fig. 2B) and although heterozygous loss of Sir2 in worms was not examined, partial loss of sirtuins using SIRT2 inhibitors leads to rescue of Htt-induced degeneration in both flies and worms (our unpublished results and C. Neri et al., personal communication). These SIRT2 inhibitors, while potentially affecting other sirtuins with different affinities, are also protective in Drosophila and cell models of Parkinson’s disease (54) further supporting a role for sirtuin inhibitors as potential therapeutic agents for several neurodegenerative diseases including HD.
There are multiple mechanisms whereby altered Sir2/SIRT1 levels may affect neuronal survival. HD is associated with progressive transcriptional dysregulation, particularly involving decreased expression of important neuronal proteins such as the D2 dopamine receptor (55), NMDAR subunits (NR1; NR2B) (56), PGC1α-regulated genes (57) and brain-derived neurotrophic factor (58). Lowered Sir2 may positively impact mutant Htt-induced transcriptional changes both by affecting histones directly and by potentially affecting specific other proteins in several ways. For example, the acetyltransferase activities of CREB-binding protein (CBP) and p300, which acetylate histones and other cellular proteins, are inhibited both by mutant Htt in vitro (6) and by the normal action of SIRT1, which inhibits p300/CBP by directly deacetylating CBP (18). Increased CBP appears protective in several models of polyQ repeat disease (59,60) suggesting that decreased Sir2 deacetylation is relevant to pathology. Expression of the neuroprotective mitochondrial uncoupling protein UCP2 (61–64), which is reduced in HD and may protect against oxidative stress, can be directly suppressed by SIRT1 (17). Also, SIRT1 is seen to deacetylate PGC1α in several cell types (19,65), although depending upon the cell type this may either increase or decrease PGC1α’s effect on transcription. Interestingly, PGC1α-regulated transcription is suppressed in HD patient and mouse striatum (66); and SIRT1 can inhibit PGC1α in neuron-like PC12 cells (19). In addition, recent reports indicating that overexpression of Sir2 activates the c-jun terminal protein kinase pathway and leads to apoptosis in the Drosophila eye while loss of Sir2 results in a partial suppression of UV irradiation-induced apoptosis, provide another avenue for possible neuroprotection (67).
Resveratrol is a well-studied compound reported to be protective to mammalian neurons (68) as well as to suppress aging in yeast (41), Htt-mediated neurodegeneration in worms (53) and 3NP-induced toxicity in rats (69). Our studies confirm the value of resveratrol (Fig. 5A). However, resveratrol has previously been suggested to act by elevating SIRT1 activity (41,70) although the basis for this conclusion has been questioned (71,72). We find that the rescuing ability of resveratrol is similar in Sir2 homozygous null flies to that observed in flies with normal levels of Sir2 (Fig. 5C). Thus, while effective in suppressing Htt-mediated pathology in Drosophila, resveratrol, which is known to affect many cellular targets (73), is likely to be capable of operating through mechanisms that are independent of Sir2.
Notably, we find that combinatorial targeting of Rpd3 and Sir2 provides additive benefit in this model. This observation indicates that in mature adult neurons of the diseased fly, Rpd3 and Sir2 exert parallel influences on neuronal survival. Our gene expression study (Fig. 5D) also demonstrates that Rpd3 does not repress Sir2 expression as previously suggested (30). The discrepancy between our observations and earlier ones may be due to improved sensitivity of RNA detection methods in recent years; or due to the means of identifying the mutant versus control genotypes. Specifically, we identified non-mutant segregants by the presence of a dominant mutation while previous studies used a variable variegation phenotype of Rpd3 as a selection marker, leaving open the possibility for bias.
We find that in Drosophila, the parameters that allow neurons to survive in the face of polyQ challenge and the parameters that affect organism longevity can respond differently to Rpd3 and Sir2 manipulations; that is, reduction of both is synergistic with respect to neuronal degeneration whereas Rpd3 and Sir2 are antagonistic with respect to lifespan extension in wild-type flies. Our study suggests that therapeutic strategies designed to target both Rpd3 and Sir2 activities specifically are likely to result in improved neuronal benefit with no reduction in longevity.
MATERIALS AND METHODS
Drosophila crosses
To compare phenotypes of Htt-expressing animals in a normal versus an HDAC-altered background, an X chromosomal elav-Gal4[C155] driver stock heterozygous for an HDAC mutation and with a marked second or third chromosome was crossed to UAS-Httex1p Q93 (line P463; 6) at 25°C on a standard medium. Eclosion data from ≥1000 segregants was calculated as percent of elav > Htt/mutation versus elav > Htt/marker, normalized to the ratio of Htt non-expressing male siblings. The shRNA lines were generated by transforming w[1118] embryos with modified pWIZ vector constructs. Pseudopupil analysis was performed as described (6). For longevity experiments, the crosses were performed at 18°C and the freshly eclosed virgin flies aged at 25°C in groups of 25–30 animals. Longevity was determined by counting the number of surviving animals daily and the flies were passed every 2–3 days; at least 150 flies were used. Drugs were tested by placing freshly eclosed virgin females into vials containing the drug at different concentrations and aged for pseudopupil analysis for 7 days; except for the combinatorial feeding experiment, which was performed on 4-day-old animals. Fresh food was provided daily. Reagent sources were as follows: Butyrate, Aldrich; Nicotinic acid, Niacinamide, Resveratrol, Sigma; Sirtinol, Calbiochem. CR was accomplished by diluting the food ∼50% as described (37).
Cloning
UAS-shRNA lines were generated by cloning a fragment of the gene in question in both the reverse and forward orientation into a modified pWIZ vector on either side of an intron from the wg gene (74) and transgenic lines established. Transcript levels in different shRNA lines driven by elav > Gal4 were determined by real time quantitative RT–PCR (qRT–PCR) and lines with at least 50% suppression were selected for further study (Supplementary Material, Fig. S2C–E).
Quantitative RT–PCR
Total RNA was extracted from heads or whole animals using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s recommendations. Reverse transcriptase reactions were performed using the SuperScript III kit (Invitrogen) with random hexamer primers and the PCR was performed using the SYBRGreen reagent (Applied Biosystems, Foster City, CA, USA) in a DNA engine opticon real time PCR machine (MJ research/Bio-Rad, Hercules, CA, USA). All samples were run in at least triplicates (three independent RNA extractions). Primers were designed to overlap an intron to prevent amplification from genomic DNA.
Statistical analysis
Statistical analyses were performed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health. awards [NS045283 to J.L.M., NS52789 to L.M.T.], the Hereditary Disease Foundation [HDF-24085], and the Huntington’s Disease Society of America [35326].
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Douglas Bournemann (UCI) for the HDAC3 alleles, Stefan Åström (Stockholm University) for the Sir2[17] allele and Bruce Blumberg (UCI) for sharing the qPCR equipment. We thank the Bloomington Drosophila Stock Center and Szeged Drosophila Stock Centre for providing several fly stocks.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Bates G., Harper P., Jones L. Huntington’s Disease. Oxford: Oxford University Press; 2002. [Google Scholar]
- 2.Shao J., Diamond M.I. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum. Mol. Genet. 2007;16(Spec no. 2):R115–R123. doi: 10.1093/hmg/ddm213. [DOI] [PubMed] [Google Scholar]
- 3.Weydt P., La Spada A.R. Targeting protein aggregation in neurodegeneration – lessons from polyglutamine disorders. Expert Opin. Ther. Targets. 2006;10:505–513. doi: 10.1517/14728222.10.4.505. [DOI] [PubMed] [Google Scholar]
- 4.Marsh J., Thompson L. Can flies help humans treat neurodegenerative diseases? Bioessays. 2004;26:485–496. doi: 10.1002/bies.20029. [DOI] [PubMed] [Google Scholar]
- 5.Cha J. Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steffan J., Bodai L., Pallos J., Poelman M., McCampbell A., Apostol B., Kazantsev A., Schmidt E., Zhu Y., Greenwald M., et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 7.Ferrante R.J., Kubilus J.K., Lee J., Ryu H., Beesen A., Zucker B., Smith K., Kowall N.W., Ratan R.R., Luthi-Carter R., et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hockly E., Richon V.M., Woodman B., Smith D.L., Zhou X., Rosa E., Sathasivam K., Ghazi-Noori S., Mahal A., Lowden P.A., et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl Acad. Sci. USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadri-Vakili G., Cha J.H. Histone deacetylase inhibitors: a novel therapeutic approach to Huntington’s disease (complex mechanism of neuronal death) Curr. Alzheimer Res. 2006;3:403–408. doi: 10.2174/156720506778249407. [DOI] [PubMed] [Google Scholar]
- 10.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Gregoretti I.V., Lee Y.M., Goodson H.V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Frye R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 13.Yang X., Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito A., Kawaguchi Y., Lai C., Kovacs J., Higashimoto Y., Appella E., Yao T. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juan L., Shia W., Chen M., Yang W., Seto E., Lin Y., Wu C. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 2000;275:20436–20443. doi: 10.1074/jbc.M000202200. [DOI] [PubMed] [Google Scholar]
- 16.Glozak M.A., Sengupta N., Zhang X., Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Bordone L., Motta M.C., Picard F., Robinson A., Jhala U.S., Apfeld J., McDonagh T., Lemieux M., McBurney M., Szilvasi A., et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4:e31. doi: 10.1371/journal.pbio.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouras T., Fu M., Sauve A.A., Wang F., Quong A.A., Perkins N.D., Hay R.T., Gu W., Pestell R.G. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 2005;280:10264–10276. doi: 10.1074/jbc.M408748200. [DOI] [PubMed] [Google Scholar]
- 19.Nemoto S., Fergusson M.M., Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J. Biol. Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 20.Guarente L. Calorie restriction and SIR2 genes–towards a mechanism. Mech. Ageing Dev. 2005;126:923–928. doi: 10.1016/j.mad.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Bates E.A., Victor M., Jones A.K., Shi Y., Hart A.C. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J. Neurosci. 2006;26:2830–2838. doi: 10.1523/JNEUROSCI.3344-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsh J., Pallos J., Thompson L. Fly models of Huntington’s disease. Hum. Mol. Genet. 2003;12(Spec no. 2):R187–R193. doi: 10.1093/hmg/ddg271. [DOI] [PubMed] [Google Scholar]
- 23.Haigis M.C., Guarente L.P. Mammalian sirtuins – emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2920. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 24.Agrawal N., Pallos J., Slepko N., Apostol B., Bodai L., Chang L., Chiang A., Thompson L., Marsh J. Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc. Natl Acad. Sci. USA. 2005;102:3777–3781. doi: 10.1073/pnas.0500055102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.North B.J., Marshall B.L., Borra M.T., Denu J.M., Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 26.Grozinger C.M., Chao E.D., Blackwell H.E., Moazed D., Schreiber S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 27.Bitterman K.J., Anderson R.M., Cohen H.Y., Latorre-Esteves M., Sinclair D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 28.Landry J., Slama J.T., Sternglanz R. Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 2000;278:685–690. doi: 10.1006/bbrc.2000.3854. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh S., Feany M.B. Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum. Mol. Genet. 2004;13:2011–2018. doi: 10.1093/hmg/ddh214. [DOI] [PubMed] [Google Scholar]
- 30.Rogina B., Helfand S.L., Frankel S. Longevity regulation by Drosophila Rpd3 deacetylase and caloric restriction. Science. 2002;298:1745. doi: 10.1126/science.1078986. [DOI] [PubMed] [Google Scholar]
- 31.Kyrylenko S., Kyrylenko O., Suuronen T., Salminen A. Differential regulation of the Sir2 histone deacetylase gene family by inhibitors of class I and II histone deacetylases. Cell. Mol. Life Sci. 2003;60:1990–1997. doi: 10.1007/s00018-003-3090-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang H., Benzer S., Min K. Life extension in Drosophila by feeding a drug. Proc. Natl Acad. Sci. USA. 2002;99:838–843. doi: 10.1073/pnas.022631999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Partridge L., Piper M.D., Mair W. Dietary restriction in Drosophila. Mech. Ageing Dev. 2005;126:938–950. doi: 10.1016/j.mad.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 34.Rogina B., Helfand S. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl Acad. Sci. USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolf G. Calorie restriction increases lifespan: a molecular mechanism. Nutr. Rev. 2006;64:89–92. doi: 10.1301/nr.2006.feb.89-92. [DOI] [PubMed] [Google Scholar]
- 36.Guarente L., Picard F. Calorie restriction – the SIR2 connection. Cell. 2005;120:473–482. doi: 10.1016/j.cell.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 37.Mair W., Goymer P., Pletcher S.D., Partridge L. Demography of dietary restriction and death in Drosophila. Science. 2003;301:1731–1733. doi: 10.1126/science.1086016. [DOI] [PubMed] [Google Scholar]
- 38.Astrom S.U., Cline T.W., Rine J. The Drosophila melanogaster sir2+ gene is nonessential and has only minor effects on position-effect variegation. Genetics. 2003;163:931–937. doi: 10.1093/genetics/163.3.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wood J.G., Rogina B., Lavu S., Howitz K., Helfand S.L., Tatar M., Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 40.Tang B., Chua C. SIRT1 and neuronal diseases. Mol. Aspects Med. 2008;29:187–200. doi: 10.1016/j.mam.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Howitz K.T., Bitterman K.J., Cohen H.Y., Lamming D.W., Lavu S., Wood J.G., Zipkin R.E., Chung P., Kisielewski A., Zhang L.L., et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 42.Fernandez-Funez P., Nino-Rosales M.L., Gouyon B., She W.C., Luchak J.M., Martinez P., Turleganos E., Benito J., Capovilla M., Skinner P.J., et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 43.Pandey U.B., Nie Z., Batlevi Y., McCray B.A., Ritson G.P., Nedelsky N.B., Schwartz S.L., DiProspero N.A., Knight M.A., Schuldimer O., et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 44.Iwata A., Riley B.E., Johnston J.A., Kopito R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 45.Fabrizio P., Pozza F., Pletcher S.D., Gendron C.M., Longo V.D. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- 46.Fabrizio P., Longo V.D. The chronological life span of Saccharomyces cerevisiae. Aging Cell. 2003;2:73–81. doi: 10.1046/j.1474-9728.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- 47.Fabrizio P., Gattazzo C., Battistella L., Wei M., Cheng C., McGrew K., Longo V.D. Sir2 blocks extreme life-span extension. Cell. 2005;123:655–667. doi: 10.1016/j.cell.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 48.Tissenbaum H.A., Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 49.Newman B.L., Lundblad J.R., Chen Y., Smolik S.M. A Drosophila homologue of Sir2 modifies position-effect variegation but does not affect life span. Genetics. 2002;162:1675–1685. doi: 10.1093/genetics/162.4.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y., Xu W., McBurney M., Longo V. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab. 2008;8:38–48. doi: 10.1016/j.cmet.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhai R., Cao Y., Hiesinger P., Zhou Y., Mehta S., Schulze K., Verstreken P., Bellen H. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhai R., Zhang F., Hiesinger P., Cao Y., Haueter C., Bellen H. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parker J.A., Arango M., Abderrahmane S., Lambert E., Tourette C., Catoire H., Neri C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet. 2005;37:349–350. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- 54.Outeiro T.F., Kontopoulos E., Altmann S.M., Kufareva I., Strathearn K.E., Amore A.M., Volk C.B., Maxwell M.M., Rochet J.C., McLean P.J., et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 55.Augood S.J., Faull R.L., Emson P.C. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington’s disease. Ann. Neurol. 1997;42:215–221. doi: 10.1002/ana.410420213. [DOI] [PubMed] [Google Scholar]
- 56.Arzberger T., Krampfl K., Leimgruber S., Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease – an in situ hybridization study. J. Neuropathol. Exp. Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- 57.Cui L., Jeong H., Borovecki F., Parkhurst C.N., Tanese N., Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 58.Zuccato C., Ciammola A., Rigamonti D., Leavitt B.R., Goffredo D., Conti L., MacDonald M.E., Friedlander R.M., Silani V., Hayden M.R., et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 59.Taylor J.P., Taye A.A., Campbell C., Kazemi-Esfarjani P., Fischbeck K.H., Min K.T. Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev. 2003;17:1463–1468. doi: 10.1101/gad.1087503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang H., Poirier M.A., Liang Y., Pei Z., Weiskittel C.E., Smith W.W., DeFranco D.B., Ross C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 2006;23:543–551. doi: 10.1016/j.nbd.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 61.Andrews Z.B., Diano S., Horvath T.L. Mitochondrial uncoupling proteins in the CNS: in support of function and survival. Nat. Rev. Neurosci. 2005;6:829–840. doi: 10.1038/nrn1767. [DOI] [PubMed] [Google Scholar]
- 62.Fridell Y.W., Sanchez-Blanco A., Silvia B.A., Helfand S.L. Targeted expression of the human uncoupling protein 2 (hUCP2) to adult neurons extends life span in the fly. Cell Metab. 2005;1:145–152. doi: 10.1016/j.cmet.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 63.Sanchez-Blanco A., Fridell Y.W., Helfand S.L. Involvement of Drosophila uncoupling protein 5 in metabolism and aging. Genetics. 2006;172:1699–1710. doi: 10.1534/genetics.105.053389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bechmann I., Diano S., Warden C.H., Bartfai T., Nitsch R., Horvath T.L. Brain mitochondrial uncoupling protein 2 (UCP2): a protective stress signal in neuronal injury. Biochem. Pharmacol. 2002;64:363–367. doi: 10.1016/s0006-2952(02)01166-8. [DOI] [PubMed] [Google Scholar]
- 65.Rodgers J.T., Lerin C., Haas W., Gygi S.P., Spiegelman B.M., Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 66.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 67.Griswold A., Chang K., Runko A., Knight M., Min K. Sir2 mediates apoptosis through JNK-dependent pathways in Drosophila. Proc. Natl Acad. Sci. USA. 2008;105:8673–8678. doi: 10.1073/pnas.0803837105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sinclair D. Toward a unified theory of caloric restriction and longevity regulation. Mech. Ageing Dev. 2005;126:987–1002. doi: 10.1016/j.mad.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 69.Kumar P., Padi S., Naidu P., Kumar A. Effect of resveratrol on 3-nitropropionic acid-induced biochemical and behavioural changes: possible neuroprotective mechanisms. Behav. Pharmacol. 2006;17:485–492. doi: 10.1097/00008877-200609000-00014. [DOI] [PubMed] [Google Scholar]
- 70.Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F., Messadeq N., Milne J., Lambert P., Elliott P., et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 71.Kaeberlein M., McDonagh T., Heltweg B., Hixon J., Westman E.A., Caldwell S.D., Napper A., Curtis R., DiStefano P.S., Fields S., et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]
- 72.Kaeberlein M. The ongoing saga of sirtuins and aging. Cell Metab. 2008;8:4–5. doi: 10.1016/j.cmet.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 73.Granados-Soto V. Pleiotropic effects of resveratrol. Drug News Perspect. 2003;16:299–307. doi: 10.1358/dnp.2003.16.5.829318. [DOI] [PubMed] [Google Scholar]
- 74.Lee Y., Carthew R. Making a better RNAi vector for Drosophila: use of intron spacers. Methods. 2003;30:322–329. doi: 10.1016/s1046-2023(03)00051-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.