

1. INTRODUCTION
Factors modifying clinical expression of inherited diseases are likely to be complex, involving genetic factors, environmental factors and stochastic effects. One way to reduce the complexity is to focus on individuals who share a dominant mutation identical by descent, thus eliminating variability in the underlying mutation and variation in cis to the mutation. A further simplification is to limit analysis to a single, extended family, which may reduce, though not eliminate, environmental effects.
Autosomal dominant retinitis pigmentosa (adRP) offers a number of opportunities for such studies. We are investigating factors which modify clinical features consequent to an Arg677ter stop mutation in the RP1 gene, a mutation which causes adRP with variable age-of-onset, progression and end stage consequences, that is, variable severity (Berson et al., 2002; Jacobson et al., 2000; Sullivan et al., 1999).
The RP1 locus was mapped by linkage testing to 8q13 in a large, extended adRP family, RP01, largely situated in southeastern Kentucky (Blanton et al., 1991; Field et al., 1982). Subsequently, the RP1 gene was identified by positional cloning and mutation screening in RP01 and additional adRP families (Guillonneau et al., 1999; Pierce et al., 1999; Sullivan et al., 1999). Mutations in the RP1 gene account for approximately 4% of adRP families in the United States. The RP1 Arg677ter mutation, found in RP01 and other families, accounts for approximately ½ of the total (Bowne et al., 1999).
The RP1 gene codes for a novel protein, 2,156 amino acids in length, with high sequence similarity over the first 10% of the amino terminus to doublecortin, a protein involved in cortical folding, but with very little sequence similarity elsewhere. The protein is photoreceptor-specific; localizes to the rod interconnecting cilium; stabilizes disc architecture in outer segments; and is most likely involved in ciliary matrix assembly and function (Gao et al., 2002; Liu et al., 2002; Liu et al., 2003; Liu et al., 2004; Pierce et al., 2004).
The Arg677ter mutation is in the final, 4th exon of the gene and probably escapes nonsense-mediated decay. If so, the product of the mutant message would be a severely truncated protein, retaining the doublecortin domain, but lacking most of the remaining sequence. Thus the pathophysiology probably involves a dominant negative or gain of function effect, rather than haploinsufficiency.
Factors modifying simple mendelian diseases, such as retinitis pigmentosa, may be just as complex as factors contributing to multifactorial diseases such as age-related macular degeneration (Haider et al., 2002; Nadeau, 2003). However, for dominant-acting diseases one factor is particularly likely: the “wild type” allele in trans to the mutation. For example, alleles in trans to alpha-spectrin mutations modify the severity of elliptocytisis (Gratzer, 1994; Randon et al., 1994). Also, of direct relevance to retinitis pigmentosa, variability in expression of the allele in trans to disease-causing mutations in PRPF31 modify penetrance of the RP11 form of adRP (McGee et al., 1997; Vithana et al., 2003). This is particularly relevant to RP1 because several polymorphic amino acid substitutions exist at the locus which result in at least 6 distinct amino acid haplotypes, i.e., distinct proteins, in Caucasian populations (Bowne et al., 1999). These protein haplotypes in trans are potential modifiers of clinical expression of the RP1 Arg677ter mutation.
2. CLINICAL AND MOLECULAR CHARACTERIZATION OF RP01
A published genealogy traces retinitis pigmentosa in the RP01 family to a single individual born in 1803 (Breeding, 1982). Over 100 living, affected family members have been identified by pedigree reconstruction (Blanton et al., 1991). The family has been the subject of clinical and genetic studies for more that 30 years, including field studies in Kentucky and ophthalmic evaluations at the Jules Stein Eye Institute, UCLA (Field et al., 1982; Heckenlively et al., 1982; Lehmer et al., 1992).
RP01 has classical type 2 adRP with late onset of night blindness, usually by the third decade of life, and slow progression. Characteristic findings include diffuse retinal pigmentation, progressive decrease in recordable ERGs, and concentric visual field loss. Fundoscopic findings include retinal atrophy, bone spicule-like pigment deposits, and vascular attenuation.
The family also shows substantial within-family variability. For example, three members, examined in their 50s, had only small areas of regional pigmentation while two of these had children with typical RP. Early changes in the children included focal depigmented spot atrophy with pigmentation of the edge and abnormal RPE granularity. By contrast, typical late changes in adults vary from diffuse atrophy with pavingstone-type changes to typical bone spicule pigmentation. Further, at least two instances of a “skipped generation” have been documented in RP01.
We have DNA samples, and in many cases, lymphoblastoid cell lines, from 96 members of the family. Of these, 66 have the Arg677ter mutation. Of the 66, we have at least minimal clinical data on 44 and detailed clinical records, including fundus photographs, on 35. For linkage and segregation analysis we use a pedigree structure with 150 individuals, including two inbreeding loops (Blanton et al., 1992).
3. DO GENETIC FACTORS PLAY A ROLE?
To answer this question we assigned an age-adjusted affectation status to the 44 individuals with at least minimal records and tested for segregation of severity using the non-parametric pedigree analysis program Loki (Daw et al., 1999; Daw et al., 1999a; Heath, 1997). Loki uses an iterated, Monte Carlo Markoff-chain approach to estimate the extent to which genetic differences play a role in variation of a trait (severity in this case) and the potential number of contributing genes. Based on this evaluation, 36% of the differences in severity in RP01 can be explained by variation at the RP1 locus (other than the mutation itself) and 18% can be explained by unlinked loci. For the unlinked loci, Loki estimated that there is an 80% probability that 6 or fewer loci are involved, and that one of these loci may account for at least 9% of the total variation (Figure 1; unpublished). These results are provisional, given the “soft” nature of the clinical phenotype, but they support the likelihood that genetic differences play a role in the variability of adRP caused by the Arg677ter mutation.
Figure 1.
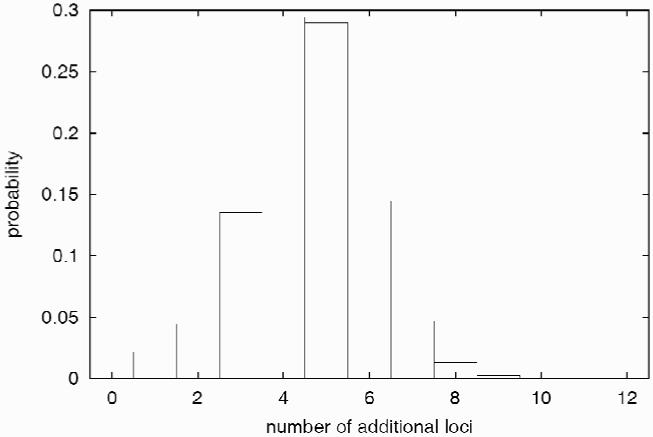
Number of genes estimated to contribute to RP1 Arg677ter severity based on Loki analysis in RP01.
4. POLYMORPHIC AMINO ACID HAPLOTYPES AT THE RP1 LOCUS
Numerous non-pathogenic amino acid substitutions have been reported at the RP1 locus (Berson et al., 2002; Bowne et al., 1999; Sohocki et al., 2001; Sullivan et al., 1999). Of these, at least 5 have high heterozygosity, that is, the lesser allele has a frequency of 10% or greater (Table 1). Although there is considerable linkage disequilibrium between these alleles, 6 distinct amino acid haplotypes were found in 100 unrelated CEPH parents, using offspring genotypes to reconstruct haplotypes, as shown in Table 1 (Sohocki et al., 2001; and unpublished). These polymorphic protein haplotypes are potential modifiers, in trans, of the Arg677ter mutation.
Table 1.
Polymorphic amino acid substitutions and haplotypes in RP1
| Substitution | Frequencies |
|---|---|
| Arg872His (CGT → CAT) | 75%, 25% |
| Asn985Tyr (AAT → TAT) | 54%, 46% |
| Ala1670Thr (GCA → ACA) | 78%, 22% |
| Ser1691Pro (TCT → CCT) | 77%, 23% |
| Cys2033Tyr (TGT → TAT) | 46%, 54% |
| Haplotype | Frequencies |
|---|---|
| 1: Arg - Tyr - Ala - Ser - Tyr | 41% |
| 2: Arg - Asn - Ala - Ser - Cys | 30% |
| 3: His - Asn - Thr - Pro - Cys | 26% |
| 4: Arg - Tyr - Ala - Ser - Cys | 1% |
| 5: His - Tyr - Ala - Ser - Tyr | 1% |
| 6: His - Asn - Ala - Pro - Cys | 1% |
5. DO THE HAPLOTYPES IN TRANS PLAY A ROLE?
To test this possibility, we determined the protein haplotype in trans in each of the 66 individuals carrying the Arg677ter mutation. Since a sequence of approximately 3 megabases on 8q13 is tracking with disease in the family, i.e., without recombination, there is effectively no variation in cis to the mutation (Blanton et al., 1992). (Not all Arg677ter mutations descend from a common ancestor [Bowne et al., 2002; Schwartz et al., 2003]). The haplotype in cis to the mutation in RP01 is haplotype 2 in Table 1; thus the allele in trans can be determined deductively from each individual’s genotype.
For this analysis we evaluated fundus photographs and clinical findings for the 33 individuals with detailed records. As dependent variables we considered age-of onset, other age-based landmarks, professionally graded fundus photographs and a composite, age-adjusted severity score. The independent variables in this case were the polymorphic amino acid alleles in trans, tested individually, and the protein haplotypes. Analytic methods included parametric significance testing and analysis of variance, and Kaplan-Meier survival modeling, implemented using the SAS statistical package (Allen, 1995).
In summary, a few combinations of dependent variables with independent variables show significant association, but are not significant if corrected for multiple comparisons. However, one of the significant associations, based on survival analysis, shows a protective effect of haplotype 3 in females (Figure 2). Because of the preliminary nature of the analysis, this should be taken as simply suggestive, but it is consistent with the Loki analysis and justifies further research.
Figure 2.
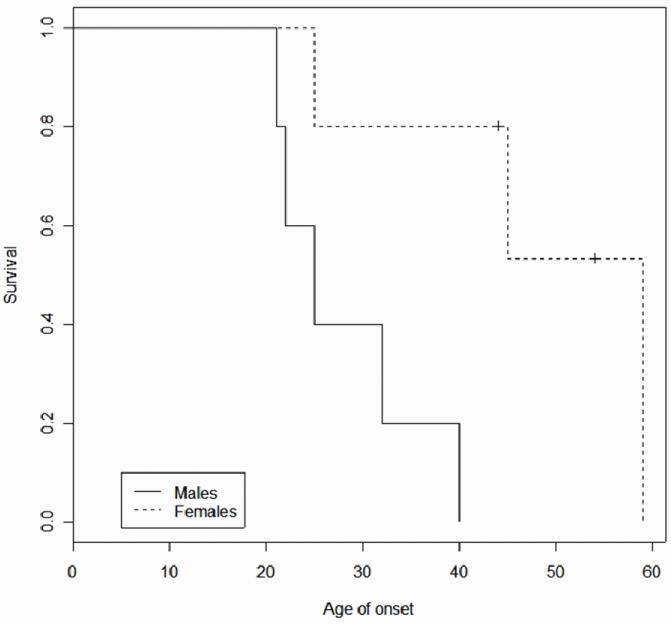
Kaplan-Meier survival analysis, RP1 protein haplotype 3 versus severity in males and females.
6. ACKNOWLEDGMENTS
Supported by grants from the Foundation Fighting Blindness and the Hermann Eye Fund, and by grants EY07142 and EY14170 from the National Eye Institute - National Institutes of Health.
7. REFERENCES
- Allen PD. Survival Analysis Using the SAS System: A Practical Guide. SAS Publishing; New York: 1995. [Google Scholar]
- Berson EL, Grimsby JL, Adams SM, McGee TL, Sweklo E, Pierce EA, Sandberg MA, Dryja TP. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1) Invest. Ophthalmol. Vis. Sci. 2002;42:2217–2224. [PubMed] [Google Scholar]
- Breeding C. Partial Ison Genealogy, 1650-1982. C. Breeding; Charleston, IN, USA: Private printing, copyright 1982. [Google Scholar]
- Blanton SH, Heckenlively JR, Cottingham AW, Friedman J, Sadler LA, Wagner M, Friedman LH, Daiger SP. Linkage mapping of autosomal dominant retinitis pigmentosa (RP1) to the pericentric region of human chromosome 8. Genomics. 1991;11:857–873. doi: 10.1016/0888-7543(91)90008-3. [DOI] [PubMed] [Google Scholar]
- Bowne SJ, Daiger SP, Hims MW, Sohocki MM, Malone KA, McKie AB, Heckenlively JR, Birch DR, Inglehearn CF, Bhattacharya SS, Bird A, Sullivan LS. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 1999;11:2121–2128. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw EW, Heath SC, Wijsman EM. Multipoint oligogenic analysis of age-at-onset data with applications to Alzheimer disease pedigrees. Am. J. Hum. Genet. 1999;64:839–851. doi: 10.1086/302276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw EW, Kumm J, Snow GL, Thompson EA, Wijsman EM. Monte Carlo Markov chain methods for genome screening. Genet. Epidemiol. 1999a;17:133–138. doi: 10.1002/gepi.1370170723. [DOI] [PubMed] [Google Scholar]
- Field LL, Heckenlively JR, Sparks RS, Garcia CA, Farson C, Zedalis D, Sparkes MC, Crist M, Tideman S, Spence MA. Linkage analysis of five pedigrees affected with typical autosomal dominant retinitis pigmentosa. J. Med. Genet. 1982;19:266–270. doi: 10.1136/jmg.19.4.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Cheon K, Nusinowitz S, Liu Q, Bei D, Atkins K, Azimi A, Daiger SP, Farber DB, Heckenlively JR, Pierce EA, Sullivan LS, Zuo J. Progressive photoreceptor degeneration, outer segment dysplasia and rhodopsin mis-localization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc. Natl Acad. Sci. USA. 2002;99:5698–5703. doi: 10.1073/pnas.042122399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratzer W. Human genetics. Silence speaks in spectrin. Nat. 1994;372:620–621. doi: 10.1038/372620a0. [DOI] [PubMed] [Google Scholar]
- Guillonneau X, Piriev NI, Danciger M, Kozak CA, Cideciyan AV, Jacobson SG, Farber DB. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum. Mol. Genet. 1999;8:1541–1546. doi: 10.1093/hmg/8.8.1541. [DOI] [PubMed] [Google Scholar]
- Haider NB, Ikeda A, Naggert JK, Nishina PM. Genetic modifiers of vision and hearing. Hum. Mol. Genet. 2002;10:1195–1206. doi: 10.1093/hmg/11.10.1195. [DOI] [PubMed] [Google Scholar]
- Heath SC. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 1997;61:748–760. doi: 10.1086/515506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckenlively JR, Pearlman JT, Sparkes RS, Spence MA, Zedalis D, Field L, Sparkes M, Crist M, Tideman S. Possible assignment of a dominant retinitis pigmentosa gene to chromosome 1. Ophthalmic Res. 1982;14:46–53. doi: 10.1159/000265173. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Iannaccone A, Weleber RG, Fishman GA, Maguire AM, Affatigato LM, Bennett J, Pierce EA, Danciger M, Farber DB, Stone EM. Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2000;41:1898–1908. [PubMed] [Google Scholar]
- Lehmer JM, Heckenlively JR, Stone EM, Kimura AE, Blanton SH, Daiger SP. Clinical characterization of chromosome 8 autosomal dominant retinitis pigmentosa (UCLA-RP01) Invest. Ophthal. Vis. Sci. 1992;33:1396. [Google Scholar]
- Liu Q, Lyubarsky A, Skalet JH, Pugh EN, Jr, Pierce EA. RP1 is required for the correct stacking of outer segment discs. Invest. Ophthalmol. Vis. Sci. 2003;44:4171–4183. doi: 10.1167/iovs.03-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhou J, Daiger SP, Farber DB, Heckenlively JR, Smith JE, Sullivan LS, Zuo J, Milam AH, Pierce EA. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest. Ophthal. Vis. Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zuo J, Pierce EA. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J. Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee TL, Devoto M, Ott J, Berson EL, Dryja TP. Evidence that the penetrance of mutations at the RP11 locus causing dominant retinitis pigmentosa is influenced by a gene linked to the homologous RP11 allele. Am. J. Hum. Genet. 1997;61:1059–1066. doi: 10.1086/301614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau J. Modifier genes and protective alleles in humans and mice. Curr. Opin. Genet. Dev. 2003;3:290–295. doi: 10.1016/s0959-437x(03)00061-3. [DOI] [PubMed] [Google Scholar]
- Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999;22:48–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- Randon J, Boulanger L, Marechal J, Garbarz M, Vallier A, Ribeiro L, Tamagnini G, Dhermy D, Delaunay J. A variant of spectrin low-expression allele alpha-LELY carrying a hereditary elliptocytosis mutation in codon 28. Brit. J. Haemat. 1994;88:534–540. doi: 10.1111/j.1365-2141.1994.tb05070.x. [DOI] [PubMed] [Google Scholar]
- Schwartz SB, Aleman TS, Cideciyan AV, Swaroop A, Jacobson SG, Stone EM. De novo mutation in the RP1 gene (Arg677ter) associated with retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2003;44:3593–3597. doi: 10.1167/iovs.03-0155. [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Daiger SP, Bowne SJ, Rodriquez JA, Northrup H, Heckenlively JR, Birch DG, Mintz-Hittner H, Ruiz RS, Lewis RA, Saperstein DA, Sullivan LS. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Heckenlively JR, Bowne SJ, Zuo J, Hide WA, Gal A, Denton M, Inglehearn CF, Blanton SH, Daiger SP. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat. Genet. 1999;22:248–251. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithana EN, Abu-Safieh L, Pelosini L, Winchester E, Hornan D, Bird AC, Hunt DM, Bustin SA, Bhattacharya SS. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: a molecular clue for incomplete penetrance? Invest. Ophthalmol. Vis. Sci. 2003;44:4204–4109. doi: 10.1167/iovs.03-0253. [DOI] [PubMed] [Google Scholar]