

Abstract
Purpose
The purpose of this study was to investigate retinal inosine monophosphate dehydrogenase 1 (IMPDH1) transcripts and proteins to gain an understanding of how mutations in IMPDH1 lead to retinal disease. Mutations in IMPDH1 cause the RP10 form of autosomal dominant retinitis pigmentosa (adRP) and are a rare cause of dominant Leber congenital amaurosis (LCA). IMPDH1 is a highly conserved, widely expressed housekeeping gene, the product of which catalyzes the rate-limiting step of de novo guanine synthesis. Despite its conservation and ubiquity, the clinical consequences of missense mutations in IMPDH1 are limited to the retina, and the disease mechanism is currently unknown.
Methods
A variety of methods were used to address the unique features of IMPDH1 in the retina, including Northern blot analysis, serial analysis of gene expression (SAGE), immunohistochemistry, transcript sequencing, and Western blot analysis.
Results
Results of the experiments showed that IMPDH1 levels are higher in the retina than in any other tissue tested. Specifically, IMPDH1 is found predominately in the inner segment and synaptic terminals of retinal photoreceptors. The predominant transcripts of IMPDH1 in human retina are the result of alternate splicing and alternate start sites of translation. They are significantly different from those in other tissues, and these variant transcripts encode distinct proteins. Further, the proportions of IMPDH1 transcripts and proteins in human retina are different from those in mouse retina.
Conclusions
Identification of unique retinal isoforms supports the existence of a novel IMPDH1 function in the retina, one that is probably altered by disease-causing mutations. This alone, or coupled with the high levels of IMPDH1 in the retina, may explain the retina-specific phenotype associated with IMPDH1 mutations. Elucidating the functional properties of these unique, human retinal isoforms is crucial to understanding the pathophysiology of IMPDH1 mutations.
Retinitis pigmentosa (RP) is a heterogeneous form of inherited retinal degeneration that affects 100,000 individuals in the United States and approximately 1.5 million individuals worldwide.1 Initial symptoms of RP include night blindness followed by loss of peripheral vision. Vision loss progresses as a result of photoreceptor death from the midperipheral retina toward the macula, culminating in tunnel vision, legal blindness, and all too often, total loss of sight.2
RP is caused by mutations in many distinct genes. To date, 16 autosomal dominant, 16 autosomal recessive, and 6 X-linked forms have been identified in addition to many other syndromic, systemic, and complex forms (RetNet; http//:www.sph.uth.tmc.edu/RetNet/ provided in the public domain by the University of Texas Houston Health Science Center, Houston, TX). Several forms of RP are caused by mutations in photoreceptor-specific or abundant proteins involved in phototransduction or the visual cycle. Understanding the pathophysiology of these is well advanced due to the large amount already known about the pathways involved.3 Other forms of RP are caused by mutations in more widely expressed genes whose mechanisms of action are largely unknown. One such gene is inosine monophosphate dehydrogenase 1 (IMPDH1). Mutations in IMPDH1 cause the RP10 form of autosomal dominant RP (adRP) and are also a rare cause of isolated Leber congenital amaurosis (LCA).4-6 IMPDH1 is located on chromosome 7, region q32.1, and codes for the enzyme IMPDH type 1.
Genes coding for IMPDH are found in all eukaryotes and most prokaryotes, and are highly conserved across species at both the gene and protein levels.7,8 Like most mammals, humans have an IMPDH1 and an IMPDH2 gene. These genes encode enzymes that are 84% identical at the amino acid level.9 All IMPDH proteins form active homotetramers that catalyze the rate-limiting step of de novo guanine synthesis by converting inosine monophosphate (IMP) to xanthosine monophosphate (XMP) with the reduction of NAD. Each IMPDH monomer is composed of an eight-stranded α/β barrel structure, which performs the enzymatic function, and a flanking subdomain, which is composed of two CBS regions similar to the cystathionine β-synthase gene. Human IMPDH1 and IMPDH2 enzymes have indistinguishable substrate affinities and catalytic activities, although they do show differences in inhibitor binding.10,11
Binding of single-stranded nucleic acids has recently been identified as a property of IMPDH proteins.12,13 In vitro and in vivo analysis demonstrates that several IMPDH species, including human IMPDH1 and IMPDH2, bind random pools of single-stranded nucleotides with nanomolar affinity.12 The biological role of the nucleic acid binding property of IMPDH is currently unknown.12
The expression of IMPDH in various tissues has been studied by several groups. Northern blot analyses show that IMPDH1 and IMPDH2 are expressed in most tissues, with the highest levels of IMPDH1 found in resting and activated peripheral blood lymphocytes. IMPDH2 levels are higher than IMPDH1 in all other tissues.7,14,15 IMPDH2 levels are even higher in cancerous cells, whereas IMPDH1 expression levels are not affected by transformation.16
Originally, three different IMPDH1 transcripts were identified and described in human cells and tissues.17 These transcripts differ in size (4.0, 2.7, and 2.5 kb) but contain identical coding sequences, derived from 14 exons, and identical 3′-untranslated regions (UTRs; for example, see Fig. 4A). The three transcripts differ only in splicing of three alternate untranslated 5′ exons, historically designated A, B, and C. Each of these transcripts encodes an identical protein that we refer to as “canonical IMPDH1,” a 55.6-kDa protein 514 amino acids in length. In contrast, the human IMPDH2 gene, though identical in exonic structure within the coding region, does not contain sequences homologous to the 5′ exons A, B, or C of IMPDH1, nor does it appear to have any additional exons preceding exon 1.
Figure 4.
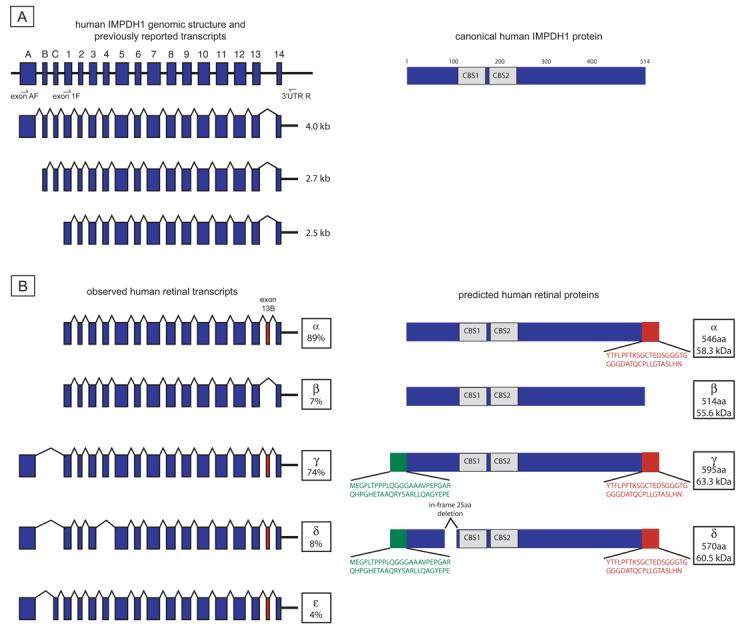
Human IMPDH1 transcripts and proteins. (A) Genomic structure and three IMPDH1 transcripts originally reported by Gu et al.17 Data suggested that all three transcripts encoded the canonical IMPDH1 protein shown on the right. (B) Observed retinal transcripts and predicted proteins. Transcripts α and β are from the exon 1F to 3′UTR R product subcloning. Transcripts γ, δ, and ε are from the exon A F to 3′UTR R product subcloning. Transcript percentages reflect results of separate subcloning experiments. Only transcripts seen in greater than 2% of clones from each subcloning experiment are reported. Green: novel protein regions from exon A. Red: residues resulting from exon 13b.
Known IMPDH1 mutations cause retinal degeneration only, despite a much wider expression pattern.4,5,18 The disease mechanism of these mutations and the reason that they manifest as a retina-specific phenotype are currently unknown. One possibility is that IMPDH1 mutations are null and cause a loss of enzyme activity that only affects photoreceptors or that is compensated for by IMPDH2 in other tissues. However, several studies have shown that IMPDH1 mutations do not affect enzyme activity and hence make this an unlikely mechanism for disease.6,19,20 Another possibility is that the IMPDH1 mutations confer a gain of function or alteration of a function unique to photoreceptors. In support of this possibility, IMPDH1 mutations alter the affinity and/or specificity of the nucleic acid binding property of IMPDH1.6,20 How, or even if, this effect on nucleic acid binding is related to retinal disease is currently unknown.
Although much is known about IMPDH1 expression and proteins in many human and mouse tissues, little is known about it in the retina. In this study, we used a variety of RNA and protein analyses to study retinal IMPDH1. Our expression analyses show that IMPDH1 is present at very high levels in the retina and that it is localized to the inner segment and synaptic terminals of photoreceptors. The predominant RNA and protein isoforms of IMPDH1 we identified in the retina are different from those that have been identified in other tissues. These unique isoforms are created via alternate splicing and/or the use of an in-frame, upstream initiation codon. Cross-species comparisons show that the unique protein regions of retinal IMPDH1 proteins are highly conserved, although our data from mice and humans indicate that the ratios of these unique isoforms vary between species. These experiments suggest that the pathophysiology of retinal degeneration is not caused by alteration of the canonical IMPDH1 protein, but rather acts through the unique retinal isoforms of IMPDH1.
Materials And Methods
Human and Animal Protocols
This study was performed in accordance with the Declaration of Helsinki, with informed consent obtained in all cases. Most subjects examined in this study were diagnosed at one of the following sites: (1) the Anderson Vision Research Center, Retina Foundation of the South-west (Dallas, TX); (2) the Jules Stein Eye Institute, UCLA School of Medicine (Los Angeles, CA); (3) the Kellogg Eye Center, University of Michigan (Ann Arbor, MI); (4) the Cullen Eye Institute, Baylor College of Medicine (Houston, TX); or the (5) Hermann Eye Center, University of Texas Health Science Center (Houston, TX). A few patients were also ascertained by ophthalmologists and genetic counselors in other institutions. The research at each academic institution was approved by the respective human subjects' review board. DNA was extracted from peripheral blood or bucal swabs by previously reported methods.6
This research adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the guidelines of the University of Pennsylvania for Animal Care and Use. Wild-type C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME). The Impdh1−/− mice were obtained from Beverly Mitchell at the University of North Carolina (Chapel Hill, NC).14
Northern Blot Analysis
Total RNA was prepared from human retina and C57BL/6J adult mouse tissues (TRIzol; Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. Total RNA from additional human tissues was purchased from BioChain (Hayward, CA). Total RNA (20 μg) from each tissue was electrophoresed on formaldehyde-agarose gels and transferred by capillary technique to nylon membrane (Biotrans; ICN, Aurora, OH). Hybridization was performed with 32P-labeled canonical human or mouse IMPDH1 cDNA probes, according to established methods.21 The cDNA for acidic ribosomal phosphoprotein PO (RPLPO) was used as a control probe. Membranes were washed then analyzed on a phosphorescence imager (PhosphorImager; GE Healthcare, Sunnyvale, CA).
Recombinant IMPDH1 and IMPDH2 Proteins
A pET-15b expression vector containing canonical human IMPDH1 sequence in the XhoI restriction site was kindly donated by Peter Humphries' laboratory (Trinity College, Dublin, Ireland). IMPDH2 cDNA was amplified from human peripheral blood leukocyte first-strand cDNA (BioChain) using the following primers: 5′-gacgacgacaagatggccgactacctgattagtgg-3′ and 5′-gaggagaagcccgtcagaaaagccgcttctcatacg-3′. PCR product was cloned into a pEK-LIC30 vector (Novagen, Madison, WI) using the manufacturer's protocol. Expression vectors were transfected into Escherichia coli (BL21) and induced using IPTG (isopropyl-β-d-thiogalactopyranoside; 1 mM final concentration). Recombinant proteins were isolated using an incorporated His tag. Bacterial pellets were sonicated in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 5 mM imidazole, and 10% glycerol [pH 8.0]), containing 1× protease inhibitor cocktail (Halt; Pierce, Rockford, IL). Total cell lysates were mixed with cobalt chelated discs (SwellGel; Pierce) for 2 hours and then decanted. Bound resin was washed four times (50 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole, 10% glycerol, pH 8.0) and then eluted (50 mM NaH2PO4, 300 mM NaCl, 300 mM imidazole, and 10% glycerol [pH 8.0]).
Antibodies
All antibodies were produced in conjunction with OpenBiosystems (Huntsville, AL), using a standard 90-day two-SPF rabbit protocol. Peptide sequences used to generate anti-N-IMPDH1, anti-N-IMPDH2, anti-C-IMPDH1, and anti-C-IMPDH1+2 were chosen to maximize the potential of distinguishing between type 1 and type 2 IMPDH (Table 1). Peptides used to generate anti-exon A-IMPDH1 and anti-exon 13b-IMPDH1 were chosen using the unique IMPDH1 protein sequences that result from transcript inclusion of exon A and exon 13b, respectively. Peptides were synthesized, conjugated to keyhole limpet hemocyanin and injected into rabbits. Antisera were affinity purified with the respective peptide antigens. At this writing, the affinity purification of anti-exon 13b-IMPDH1 was not complete, and so experiments performed with anti-exon 13b-IMPDH1, which recognizes the alternative carboxyl terminus encoded by the inclusion of exon 13b, were performed with antiserum.
Table 1.
Antibody Design and Reactivity
| Antibody | Designed to Recognize |
Peptide Sequence | Reactivity to Denatured IMPDH1 |
Reactivity to Denatured IMPDH2 |
Reactivity In Vivo |
|---|---|---|---|---|---|
| Anti-N-IMPDH1 | Canonical IMPDH1 residues 38–50 | GFIDFIADEVDLT | ++++ | — | — |
| Anti-N-IMPDH2 | IMPDH2 residues 38–50 | GYIDFTADQVDLT | — | +++ | — |
| Anti-C-IMPDH1 | Canonical IMPDH1 residues 426–438 | SQKRYFSEGDKVK | +++++ | + | +++ |
| Anti-C-IMPDH1+2 | IMPDH2 residues 426–438 | SQNRYFSEADKIK | ++++ | ++++> | +++ |
| Anti-exon A-IMPDH1 | IMPDH1 retinal isoform | EPGARQHPGHETAAQRYSAR | +++++ Exon A isoform |
— | ND |
| Anti-exon 13b-IMPDH1 | IMPDH1 retinal isoform | YTFLPFTRSGCTED | ++ Exon 13b isoform |
— | ND |
ND, not determined; + indicates binding affinity.
Immunohistochemistry
Preparation of mouse retinal sections and immunostaining was performed as described previously.22 The primary antibodies used were anti-N-IMPDH1, anti-N-IMPDH2, anti-C-IMPDH1, and anti-C-IMPDH1+2. Cy3- and Alexa 488–conjugated secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA) or Invitrogen (Eugene, OR). Hoechst dye was added with the secondary antibody to counterstain the cell nuclei. Stained sections were viewed with a confocal microscope (model LSM 510 Meta) and the images were processed with the accompanying software (Carl Zeiss MicroImaging, Thornwood, NY). The exposure time for wild-type and Impdh1−/− mouse retina sections were the same.
Analysis of Retinal Transcripts
Total RNA was isolated from mouse and human retinas (PARIS kit; Ambion, Austin, TX) using the manufacturer's protocol. Total RNA was reverse transcribed using a poly-T primer (18 bp; SuperScript III RNase H-Reverse Transcriptase and RNaseOUT Recombinant RNase Inhibitor; Invitrogen), according to the manufacturer's protocol. Alternatively, first-strand human retinal cDNA was purchased from BioChain.
Retinal transcripts were analyzed with a PCR subcloning approach. Human retina first-strand cDNA was amplified (with either AmpliTaq Gold polymerase; Applied Biosystems Inc. [ABI], Foster City, CA; or the Expand Long Template PCR system; Roche, Indianapolis, IN) and two of the following primers: exon AF-5′-caacacccgggacacgagac-3′; exon 1F-5′-catggcggactacctgatca-3′; or -3′UTR R-5′-ctggagaacccgtagtgc-3′. Mouse retina first-strand cDNA was amplified (Expand Long Template PCR system; Roche) with two of the following primers: mExon AF-5′-ggactggggctcttattg-3′; mExon 1F-5′-catggcggactacctgatca-3′; or m-3′UTR R-5′-ctggggaacccgtagtgc-3′.
PCR product was subcloned with a PCR cloning kit (TOPO XL; Invitrogen) and the manufacturer's protocol. Vectors were transfected into E. coli (strain BL21), then individual colonies were isolated and grown in Luria broth. Plasmids were isolated from overnight cultures (QIAprep Spin Miniprep kit; Qiagen, Valencia, CA) and sequenced (BigDye ver. 1.1; ABI). Sequencing reactions were purified with Sephadex columns (Princeton Separations, Adelphia, NJ), run on a genetic analyzer (3100 Avant Genetic Analyzer; ABI), and analyzed on computer (AutoAssembler software; ABI). A minimum of 50 clones were analyzed from each PCR reaction.
Semiquantitative PCR
To compare the relative levels of transcription initiation, human first-strand cDNA was amplified (Amplitaq Gold; ABI), with exon 2 reverse primer-5′-catcagctatgaagtctatgaatcc-3′, and either one or both of the following forward primers: exon AF2-5′-caacacccgggacacgagac-3′ and 5′-UTR F-5′-gtctcgcggcagcatggc-3′. Mouse first-strand cDNA was amplified in the same manner with mExon2 reverse primer 5′-tgggaggatcaggaagtcg -3′ and either one or both of the following forward primers: mExon A F-5′-caacacccgggacatgagac-3′ and m5′-UTR F-5′-gtctcccaggagcatggc-3′. Initially, PCR reactions were analyzed at many different amplification cycle points, to determine the best conditions for detectible linear amplification. In each species the forward primers were designed to distinguish between transcripts initiating in exon A and those initiating in the genomic sequence just 5′ of exon 1. PCR product was separated on a 2% agarose gel containing ethidium bromide and visualized (Gel Logic 200 system; Eastman Kodak, Rochester, NY). Each PCR experiment was performed again to ensure that the same ratio of bands was observed independently. The net intensity of each PCR band was measured with image-analysis software (1D Image; Eastman Kodak). An estimate of IMPDH1 transcript initiation site usage was calculated by comparing the net band intensities from each multiplex PCR.
Human IMPDH1 Retinal Isoform Recombinant Proteins
pEKLIC32 expression vectors corresponding to human retinal transcripts were constructed using ligation-independent cloning (LIC) technology and the following primers: 5′-gacgacgacaagatggcggactacctgatcagc-3′ (forward, transcripts α and β); 5′-gacgacgacaagatggaggggccactcactccac-3′ (forward, transcripts γ and δ); 5′-gaggagaagcccggtcagttatggagggaggctg-3′ (reverse, transcripts α, C, and δ); 5′-gaggagaagcccggttcagtacagccgcttttcg-3′ (reverse, transcript β). Corresponding plasmids sequenced during the transcript analyses were used as templates. Expression constructs were transfected into E. coli and induced with IPTG. The sequence of each expression construct was verified independently.
Bacterial pellets were lysed as described herein or with 8 M urea, 100 mM NaH2PO4, 10 mM Tris-HCl [pH 8.0], 8.0. rLysozyme (Novagen) and 1× protease inhibitor cocktail (Halt; Pierce) were added to the cell lysis buffer before sonification. Cleared total cell lysate was incubated with equilibrated Ni-NTA agarose (Qiagen) for 1 hour. Bound resin was isolated by centrifugation, washed four times (50 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole, and 10% glycerol [pH 8.0]), and the IMPDH1 proteins eluted (50 mM NaH2PO4, 300 mM NaCl, 300 mM imidazole, 10% glycerol [pH 8.0]). Isolated recombinant proteins were digested with enterokinase, to remove the purification tag (New England Biolabs, Ipswich, MA). The enterokinase also digested a degenerate amino acid sequence (DDK) in a percentage of each IMPDH1 recombinant protein preparation. This complicated the recombinant protein pattern, but still allowed for comparative analyses and antibody testing.
Western Blot Analysis
Normal human retinas were procured and flash frozen within 3 hours of death by the San Diego Eye Bank or were provided by the Foundation Fighting Blindness Eye Bank at the Scheie Eye Institute (Philadelphia, PA). Total protein lysates were made from tissues of human retina, from wild-type mouse, and from Impdh−/− mouse (PARIS protocol; Ambion). DNase (2.5 U/μL; Cal BioChem, La Jolla, CA), SDS (1% final concentration) and protease inhibitor cocktail (EDTA-free Halt; Pierce) were added to the cell disruption buffer before homogenization. Alternately, protein lysates were made by homogenization in NuPAGE sample buffer (Invitrogen). Proteins were quantified using a modified Lowry protocol (RC DC Protein Assay; Bio-Rad, Hercules, CA).
Protein lysates were separated on 8% SDS-PAGE gels (NuPAGE Novex, San Diego, CA) under reducing conditions and transferred electrophoretically to polyvinylidene difluoride (PVDF) membrane using a semidry transfer apparatus (Bio-Rad). Antibody hybridization was performed as previously described22 or as follows. The membranes were blocked for 1 hour in PBS-T solution (10 mM sodium phosphate, 150 mM sodium chloride, and 0.5% Tween) containing 0.5% nonfat dry milk (Bio-Rad). Primary antibodies were diluted in PBS-T and incubated for 1 hour with the membrane. Primary antibody binding was detected with either horseradish peroxidase (HRP)–conjugated goat anti-rabbit secondary antibodies (Bio-Rad) and an HRP chemiluminescence kit (Immun-Star; Bio-Rad) or with alkaline phosphatase–conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Bar Harbor, ME) and enhanced chemiluminescence (ECF) substrate (GE Healthcare, Piscataway, NJ). All incubations were performed at room temperature. Positive signals were visualized and quantified by fluorometry (Storm 860 PhosphorImager; GE Healthcare) or were visualized on x-ray film.
Analysis of Patients
The subjects examined in this study were described in detail previously.6 Genomic DNA was amplified (either AmpliTaq Gold polymerase; ABI; or HotStarTaq DNA polymerase; Qiagen) with the following primers: exon A forward: 5′-caggagacactggaaggtcc-3′, exon A reverse: 5′-ggggaagggtttgtgggg-3′, exon 13b forward: 5′-cgttggcagcagtagttgc-3′, exon 13b reverse: 5′-cctgctctactctacgcttgg-3′, long exon 14 forward: 5′-gggaaattttgggaag-3′, and long exon 14 reverse: 5′-gagctggagaacccgtagtg-3′. Q-solution (Qiagen) was added to facilitate the exon A amplification. PCR product was visualized with ethidium bromide on agarose gels before sequencing.
PCR products were treated with an exonnuclease 1 and shrimp alkaline phosphatase (ExoSapIt; USB, Cleveland, OH) and sequenced bidirectionally with dye termination chemistry (BigDye ver. 1.1; ABI) and the amplification primers. Sequence reactions were purified with Sephadex columns (Princeton Separations) and run on the genetic analyzer (3100 Avant Genetic Analyzer; ABI). Patient sequences were compared with the consensus IMPDH1 sequence, by computer (Seq-Scape software; ABI).
Results
IMPDH1 and IMPDH2 Retinal Expression
Expression data from the EyeSAGE database (http://neibank.nei.nih.gov/EyeSAGE/index.shtml/ provided in the public domain by the National Center for Biotechnology Information, Bethesda, MD) was examined to determine the levels of IMPDH1 and IMPDH2 in various retinal regions and to compare these levels with those from other regions of the body.23 Specifically, we compared the normalized serial analysis of gene expression (SAGE) data from (1) the pooled donor sections of peripheral retina and macular retina, (2) the single donor sections of peripheral and macular retina, and (3) representative normal tissues reported in EyeSAGE (Table 2). On average, IMPDH1 was expressed at a 10-fold higher level in the human retina than in other tissues. IMPDH1 in the retina is expressed at levels 10 times higher than IMPDH2. This level is in contrast to most other tissues where expression of IMPDH2 is more than two times higher than that of IMPDH1. Comparison of different retinal regions indicates that IMPDH1 is expressed at higher levels in the peripheral retina than in the macula.
Table 2.
SAGE Analysis of IMPDH1 and IMPDH2 in Multiple Human Tissues
| IMPDH1 | IMPDH2 | |
|---|---|---|
| Pooled macula retina* | 22 | 6 |
| Pooled peripheral retina† | 41 | 4 |
| Macula retina‡ | 21 | 2 |
| Peripheral retina§ | 30 | 0 |
| Peripheral retina∥ | 54 | 3 |
| Retina average | 34 | 3 |
| Brain fetal | 12 | 12 |
| Brain cerebellum | 4 | 12 |
| Spinal cord | 4 | 7 |
| Bone marrow | 0 | 11 |
| Breast | 0 | 11 |
| Breast epithelium | 29 | 8 |
| Colon | 8 | 4 |
| Heart | 2 | 5 |
| Kidney | 0 | 0 |
| Leukocytes | 4 | 8 |
| Liver | 0 | 6 |
| Lung | 2 | 9 |
| Muscle | 0 | 19 |
| Pancreas | 0 | 0 |
| Placenta | 2 | 7 |
| Prostate | 6 | 53 |
| Skin | 5 | 5 |
| Stomach | 0 | 23 |
| Stomach epithelium | 8 | 8 |
| Thyroid | 0 | 26 |
| Body average | 3.5 | 11.6 |
Tag counts per 200,000.
SAGE library prepared from five pooled 4-mm punches of human macula.23
SAGE library prepared from 5 pooled 4-mm punches of human peripheral retina from the same donor eyes as pooled macula retina.23
SAGE library prepared from a 6-mm punch of macula retina from one 44-year-old patient.24
SAGE library prepared from a 6-mm punch of peripheral retina from same 44-year-old patient as macula retina.24
SAGE library prepared from a 6-mm punch of macula retina from one 80-year-old patient.24
Northern blot analyses containing total RNA from a variety of human and mouse tissues, including total retina, were probed with canonical IMPDH1 probes (Fig. 1). As predicted by the SAGE data, IMPDH1 expression was highest in retinal tissue from both humans and mice. The major IMPDH1 mRNA band in human and mouse retina was approximately 2.5 kb, which is consistent with the size of the previously described transcripts.17 An additional minor band approximately 6 and 4 kb was detected in human and mouse retina, respectively. Moderate IMPDH1 expression was also seen in lung, muscle, and thymus. Faint bands were detected in all tissues analyzed, further confirming that IMPDH1 is a ubiquitously expressed housekeeping enzyme.
Figure 1.

Expression of IMPDH1 RNA in human and mouse tissues. Northern hybridizations were performed on 20 μg of total RNA for each tissue listed. Control corresponds to hybridization with acidic ribosomal phosphoprotein PO (RPLPO) cDNA. The highest levels of IMPDH1 RNA expression were present in retinal tissue of human and mouse (arrows).
Specificity of Anti-IMPDH Antibodies
Initially, four polyclonal antibodies, anti-N-IMPDH1, anti-N-IMPDH2, anti-C-IMPDH1, and anti-C-IMPDH1+2, were generated by OpenBiosystems, with synthetic peptides (Table 2). The specificity of these four antibodies was confirmed on Western analyses using recombinant proteins (Fig. 2, Table 1). Anti-N-IMPDH1 and anti-C-IMPDH1 were both specific for IMPDH1, although anti-C-IMPDH1 exhibits more cross-reactivity with other non-IMPDH proteins. Anti-N-IMPDH2 is specific for IMPDH2 and anti-C-IMPDH1+2 recognizes both IMPDH1 and IMPDH2, with relatively equal affinity.
Figure 2.

Sensitivity and specificity of IMPDH1 and IMPDH2 antibodies. Antibodies were tested against various concentrations of denatured recombinant canonical IMPDH1 and IMPDH2 proteins in standard Western analyses. Anti-N-IMPDH1 is IMPDH1 specific and anti-C-IMPDH2 is IMPDH2 specific. Anti-C-IMPDH1 had a much higher affinity (50×) for IMPDH1 than for IMPDH2. Anti-C-IMPDH1+2 can detect IMPDH1 and IMPDH2 with relative equal affinities. The distinguishable electrophoretic pattern of IMPDH1 and IMPDH2 recombinant proteins are due to variation in tag length.
Cellular Localization of IMPDH1 in Retina
Immunofluorescence staining was performed on frozen fixed retinas from wild-type and Impdh1-knockout mice using anti-N-IMPDH1, anti-N-IMPDH2, anti-C-IMPDH1, and anti-C-IMPDH1+2 (Fig. 3).14 No staining was seen in any of the retinas tested with anti-N-IMPDH1 or anti-N-IMPDH2, despite the high levels of IMPDH1 message found in the retina (Table 2). This result suggests that these two antibodies do not recognize native protein, but may only bind an epitope found on denatured IMPDH. Anti-C-IMPDH1 and anti-C-IMPDH1+2 staining showed that type 1 IMPDH was found predominantly in the inner segment, but some staining was also detected in the synaptic terminals of the photoreceptors (Figs. 3A, 3B, respectively). The inner segment signal was most clearly detected using the anti-C-IMPDH1+2 antibody, which showed little background staining in the Impdh−/− retinas (Fig. 3B). This pattern of protein localization is consistent with the in situ mRNA expression pattern reported previously by Aherne et al.19
Figure 3.

Localization of Impdh1 in mouse retina. Frozen fixed retinas were incubated with (A) anti-C-IMPDH1 or (B) anti-C-IMPDH1+2 followed by incubation with Cy3-conjugated goat anti-rabbit secondary antibody (red). Cell nuclei were stained with DAPI (blue). Impdh1 localized to the inner segments and synaptic terminals of photoreceptors (arrows). RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer.
IMPDH1 Retinal Transcripts
Early research on the regulation of the IMPDH1 gene by Gu et al.17 demonstrated the presence of three different IMPDH1 splice forms in the normal and tumor tissues they examined. These splice isoforms use three different transcription initiation sites and corresponding promoters, but encode the same 514 amino acid protein, canonical IMPDH1 (Fig. 4A). (Exonic nomenclature for both the noncoding and coding exons was established during this early research. For consistency, exon names used in this article will follow the historic precedent, despite divergence from current nomenclature recommendations.)
Given the large amount of IMPDH1 found in the retina compared with that in other parts of the body, and the existence of multiple IMPDH1 start sites of transcription and basal promoters, it was important to examine which of the start sites of transcription and corresponding isoforms were used predominantly in human retina.
PCR analysis of first-strand cDNA from human retina identified major transcripts originating from both exons A and 1. Exons B and C were only detected at very low levels, indicating that they are not major players in IMPDH1 retinal expression. Sequence analysis of the first strand cDNA PCR products identified novel splicing patterns. In this preliminary analysis transcripts were seen with exon A spliced directly to exon 1 with the exclusion of exons B and C and/or the inclusion of a novel exon near the 3′ end of the gene. We have named this novel exon 13b.
A subsequent search within the ACEview database (http://ncbi.nlm.nih.gov/IEB/Research/Acembly; provided in the public domain by NCBI) indicated that splicing of exon A to exon 1 was common in a range of tissues, including retina. Splicing from exons A to 1 creates an in-frame, alternate methionine in exon A. If translation start sites in exon A instead of in exon 1, an additional 49 amino acids are added to the amino terminus of the human protein. All other species analyzed contain exon A, although most of these exons are longer than the human exon and result in a larger protein addition (Fig. 5A).
Figure 5.

Conservation of novel IMPDH1 retinal protein sequences. Sequences were obtained for a range of organisms from genomic or EST databases and aligned using Clustal W (http://www.ebi.ac.uk/clustalw/ provided in the public domain by the European Bioinformatics Institute, European Molecular Biology Laboratory, Heidelberg, Germany). (A) Exon A; (B) exon 13b and exon 14.
However, published protein data suggest that not all transcripts with exon A spliced to exon 1 initiate translation in exon A. Western blot analyses of most human and mouse tissues failed to detect the larger protein that would result from translation initiation in exon A.7,17 Larger IMPDH1 proteins have previously been identified in mouse thymus and lung tissue and in human lung fibroblast cell lines.14,15
The presence of exon 13b, a 17-bp exon located in the genomic sequence between exons 13 and 14, was also seen in the ACEview database, but was only present in two of the 162 human cDNA clones, both of which were from retinal libraries. BLAST searches of expressed sequences show that exon 13b is found predominately in EST clones derived from retina and that it can be found in transcripts from mouse, frog, zebrafish, and human. Searches of genomic sequences showed that exon 13b is highly conserved in almost all eukaryotes. Inclusion of exon 13b in human IMPDH1 causes a reading frame shift that abolishes the stop codon in exon 14 and adds 32 amino acids to the carboxyl terminus of the protein (Fig. 5B).
Detection of these novel splicing patterns, which were likely to encode unique IMPDH1 proteins, prompted us to characterize IMPDH1 transcripts systematically in human and mouse retina. This characterization was achieved with a PCR and subcloning approach. PCR primers were designed to amplify from exon A to a 3′ UTR in exon 14, and from exon 1 to the same 3′ UTR. PCR product generated with primers exon AF and 3′ UTR and with primers exon 1F and 3′ UTR were subcloned, and 50 clones from each PCR reaction were isolated and sequenced in their entirety (Fig. 4A). Only transcript sequences occurring in greater than 2% of the clones are reported herein (Figs. 4, 6).
Figure 6.
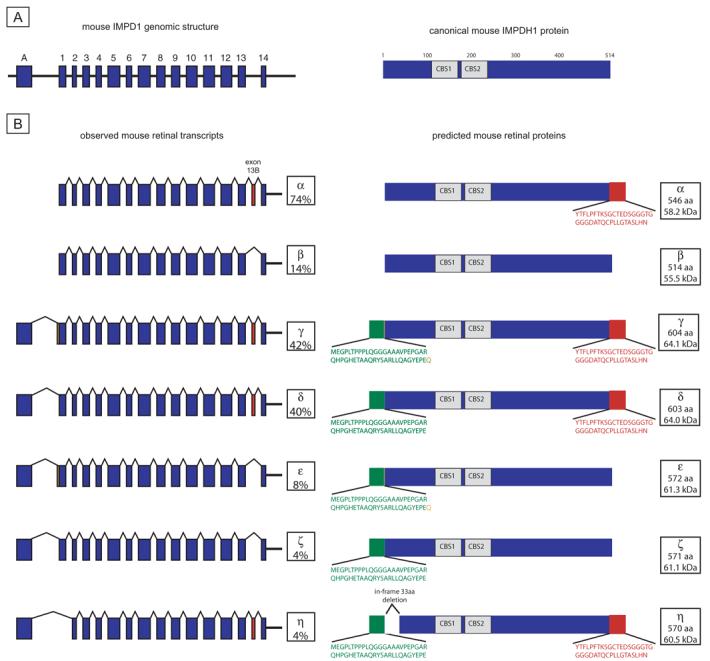
Mouse Impdh1 transcripts and proteins. (A) Genomic structure and canonical IMPDH1 protein. Unlike human, mouse does not have an Impdh1 exon B or exon C. (B) Observed retinal transcripts and predicted proteins. Transcripts α and β are from the exon 1 F to 3′UTR R product subcloning. Transcripts γ, δ, ε, ζ, and η are from the exon A F to 3′UTR R product subcloning. Transcript percentages reflect results of separate subcloning experiments. Only transcripts present in greater than 2% of clones from each subcloning experiment are reported. Green: novel protein regions from exon A. Orange: The additional amino acid, resulting from the use of 3 bp 5′ of exon 1. Red: residues resulting from exon 13b.
IMPDH1 Transcripts in Human Retina
Sequencing of clones containing transcripts amplified from exon 1 to the 3′ UTR identified one major and one minor human IMPDH1 retinal transcript. The major transcript contains exons 1 through 13 and includes exon 13b (Fig. 4B, transcript α and protein α). The minor human IMPDH1 retinal transcript amplified with these primers is homologous to the canonical IMPDH1 reported previously (Fig. 4B, transcript β and protein β).
Three different IMPDH1 transcripts emerged from sequence analysis of transcripts containing exon A (Fig. 4B). In the major transcript, transcript γ, exon A is directly spliced to exon 1 and includes exon 13b. Together, these two additional coding regions result in a predicted protein 85 amino acids longer than canonical IMPDH1 (Fig. 4B, protein γ). In addition, two minor IMPDH1 transcripts were detected by amplification with these primers. Transcript δ is identical with major transcript γ, except for the exclusion of exon 4. Splicing of exon 3 directly to exon 5 results in an in-frame deletion of 75 bp or 25 amino acids (Fig. 4B, transcript γ and protein γ). The other minor transcript included exon C. The inclusion of exon C results in a premature stop codon that is predicted to undergo nonsense-mediated decay if the initiation codon in exon A is used. Thus, at least five distinct IMPDH1 transcripts are found in the human retina.
Impdh1 Transcripts in Mouse Retina
Analysis of mouse retinal Impdh1 transcripts revealed a pattern similar to the expression detected in human retina (Fig. 6B). Analysis of all the exon 1–containing transcripts yielded the same two transcripts as in human retina. The major transcript, transcript α, included mouse exon 13b, which is 17 bp in length. Inclusion of exon 13b results in an alteration of the carboxyl terminus of the protein similar to the product of the equivalent human transcripts. The minor transcript seen in mouse retina, transcript β, also encodes canonical IMPDH1.
Mouse transcripts using exon A showed slightly more variability than that in the human transcripts. The two major transcripts, γ and δ, splice exon A to exon 1 and use exon 13b. The only difference in these two transcripts is inclusion of 3-bp 5′ of exon 1 in transcript δ, thereby creating a protein containing one additional amino acid. Inclusion of these three additional base pairs was also seen on one of the minor transcripts (Fig. 6B, transcript ε). Two of the minor Impdh1 mouse transcripts, ε and ζ, included the splicing of exon A to exon 1 and the exclusion of exon 13b. A third minor transcript, transcript η, excludes exon 1 such that exon A is spliced to exon 2. This results in an in-frame deletion of the 33 amino acids encoded by exon 1 (Fig. 6b, protein η).
Transcript Ratios
The exon 1F PCR primers used to analyze human and mouse retinal IMPDH1 transcripts cannot distinguish (1) transcripts containing exon 1 that initiate in exon A from (2) those that contain exon 1 but initiate 5′- of exon 1. That is, transcripts that initiate in exon A are indistinguishable from transcripts that initiate approximately 50 bp 5′ of exon 1 (initially described by Gu et al.,17 depicted in Fig. 4A). Semiquantitative PCR of human and mouse retinal first-strand cDNA was performed to examine the ratios of these transcripts. This analysis shows that most human transcripts initiate just upstream of exon 1, whereas most mouse transcripts initiate in exon A (Fig. 7). This difference suggests that the major human retinal IMPDH1 isoform is protein α (Fig. 4B), whereas the mouse major proteins are γ and δ (Fig. 6B).
Figure 7.

Semiquantitative PCR analyses of human and mouse retinal first-strand cDNA. Forward PCR primers that distinguish between transcripts initiating in exon A and those initiating in exon 1 were used individually and in combination. Human retina has approximately two times more transcripts that initiate in exon 1 than in exon A. Conversely, mouse retina has six times more transcripts initiating in exon A than in exon 1.
IMPDH1 Retinal Proteins
Human and mouse retinal cell lysates were probed for the presence of IMPDH1 isoforms using antibodies specific for either the amino or carboxyl termini of canonical IMPDH1. Anti-N-IMPDH1 detected three bands in human retina, approximately 63, 57, and 50 kDa in size. The same three bands were detected with the anti-C-IMPDH1 antibody, although the ratios of these bands varied between antibodies and retinal donors (Figs. 8A, 8B). Two Impdh1 bands were detected in mouse retina with each IMPDH1 antibody (Figs. 8A, 8B). These bands were approximately 65 and 59 kDa.
Figure 8.
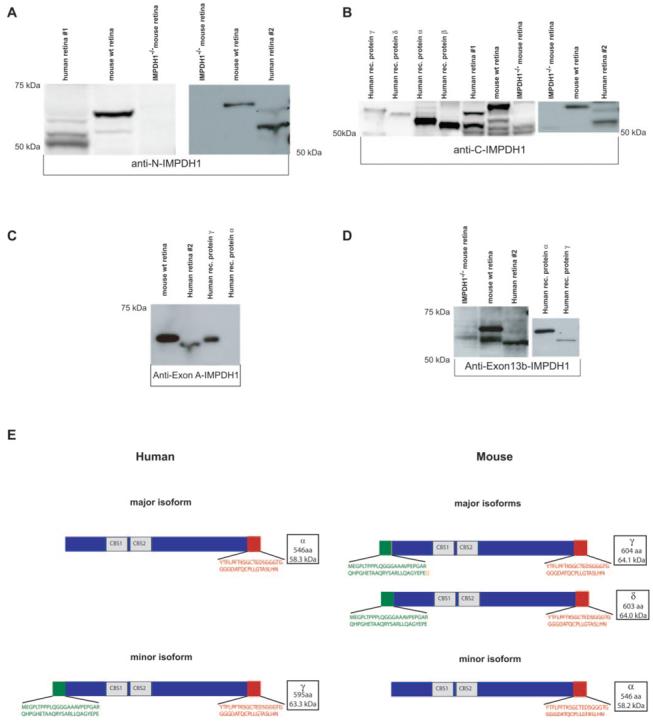
IMPDH1 isoforms in human and mouse retina contain residues encoded by exon A and exon 13b. (A) Anti-N-IMPDH1 detection of IMPDH1 in human and mouse retina. (B) Anti-C-IMPDH1 detection of IMPDH1 in human and mouse retina and comparison to recombinant human proteins. (C) Anti-A-IMPDH1 detection of proteins that contain the additional amino terminus sequence encoded by exon A. (D) Anti-exon 13b-IMPDH1 detection of proteins that contain additional carboxyl terminus encoded by exon 13b/14. (E) IMPDH1 protein isoforms detected in human and mouse retina.
The molecular weight of the mouse and human IMPDH1 proteins suggested that retinal IMPDH1 proteins contain the alternate carboxyl and/or amino termini predicted by the transcript analyses. Polyclonal antibodies specific to the predicted amino and carboxyl terminal sequences encoded by exon A (anti-exon A-IMPDH1) and exon 13b (anti-exon 13b-IMPDH1), respectively, were generated to further confirm the existence of these unique retinal proteins (Table 1). The specificity of these antibodies was established using recombinant proteins corresponding to human IMPDH1 transcripts α, β, γ, and ε. Anti-exon A-IMPDH1 is very sensitive and specific for IMPDH1 proteins containing the additional amino acid residues encoded by exon A (Fig. 8C). The serum generated for anti-exon13b-IMPDH1 recognizes proteins with the 13b-encoded residues, but also exhibits some general protein reactivity due to lack of affinity purification (Fig. 8D).
Western blot analysis with anti-exon A-IMPDH1 detected one distinguishable protein in both human and mouse retinas (Fig. 8C). Anti-exon 13b-IMPDH1 detected two protein bands in both human and mouse retina, although the larger-molecular weight human band was barely detectible due to high background levels (Fig. 8D). The larger molecular weight band detected in both human and mouse with anti-exon 13b-IMPDH1 corresponds to the same band seen with anti-exon A-IMPDH1, indicating that this protein contains both the unique amino terminus encoded by exon A and the unique carboxyl terminus from exon 13b. The smaller molecular weight IMPDH1 band recognized by anti-exon 13b-IMPDH1 is not detected with anti-exon A-IMPDH1, demonstrating that this protein does not contain the carboxyl residues encoded by exon A, but does contain the additional amino terminus residues encoded by exon 13b (Fig. 8D). These data are consistent with the transcript data and imply that the predominant, human IMPDH1 protein isoforms are proteins α and γ (Fig. 4B). The mouse Impdh1 isoforms are probably proteins γ and/or δ, which would not be distinguishable on Western blots and protein α (Fig. 8E).
Neither of the isoform-specific antibodies recognize the lowest IMPDH1 band (50 kDa) seen predominately in human retina 1 with anti-N-IMPDH1. The size of this band, its variable detection, and its low level in human retina 2, make it possible that this band corresponds to degraded protein which is present in higher quantities in human retina 1.
The high molecular weight human and mouse IMPDH1 retinal isoforms that, based on our analyses, correspond to predicted human protein γ (Fig. 4B) and to mouse proteins γ and δ (Fig. 6B), produce distinguishable electrophoretic patterns despite similar predicted sizes. The molecular mass of these different isoforms varies by less than 1 kDa which alone is not sufficient to result in distinct electrophoretic patterns. This disparity is also observed when comparing the small human and mouse IMPDH1 isoforms (Figs. 4B, 6B). The electrophoretic patterns of recombinant proteins α and γ are similar to the human retinal proteins, but not identical. Both recombinant proteins have slightly higher molecular weights than the native human retinal proteins (Figs. 8B, 8C). Together, these disparities suggest that the native IMPDH1 retinal isoforms are modified, possibly through posttranslational modification.
IMPDH1 Protein Isoforms
Semiquantitative PCR analyses suggested that the proportions of IMPDH1 retinal transcripts initiating in exon A, and the proportion of transcripts initiating upstream of exon 1, vary between human and mouse. Western blot analyses using antiexon A-IMPDH1, anti-C-IMPDH1, and anti-N-IMPDH1 (Fig. 8) further demonstrate these differences. The proportion of each unique IMPDH1 isoform is different in mouse and human retina. Mouse retina contains significantly more of the large IMPDH1 retinal isoforms that have the additional protein residues encoded by exons A and 13b. Conversely, human retina contains significantly more of the small isoforms which result from the addition of the exon 13b residues only.
Patient Screening
A cohort of patients with retinal degeneration (Table 3) was tested for possible disease-causing mutations in the newly identified coding regions of the unique IMPDH1 retinal isoforms. Each of these patients had been tested for mutations in the 14 previously described coding exons of canonical IMPDH1.6 Exons A and 13b and the additional coding region of exon 14 were analyzed for mutations by using PCR product sequencing. Only one amino acid substitution, Tyr45His (TAC3→CAC), was detected in the patients analyzed. Analysis of additional family members demonstrated that this variant did not segregate with disease and hence is a benign variant. No other protein altering mutations were seen in any of the 306 DNAs tested.
Table 3.
Patients with Retinal Degeneration Tested for Mutations in Exon A and Exon 13b/14
| Retinal Degeneration | Number of Probands Tested |
|---|---|
| Autosomal dominant retinitis pigmentosa (adRP) | 203 |
| Autosomal recessive retinitis pigmentosa (arRP) | 55 |
| Isolated retinitis pigmentosa | 7 |
| Macular degeneration (MD) | 17 |
| Multiplex Leber congenital amaurosis | 4 |
| Isolated Leber congenital amaurosis | 20 |
Discussion
IMPDH1 in Photoreceptors
Northern blot analysis and SAGE show that IMPDH1 is found at levels in the retina higher than in most other tissues. Immunofluorescence (IF) localizes most of the IMPDH1 to the inner segment and synaptic terminals of photoreceptors. Together, the IF and SAGE data suggest that IMPDH1 is present in both rod and cone photoreceptors, although mRNA levels are distinctly higher in rod-enriched regions, implying that photoreceptors have a unique requirement for IMPDH1, which is a plausible reason why mutations affect photoreceptors only.
Unique IMPDH1 Transcripts and Protein Isoforms in Photoreceptors
Human and mouse photoreceptors contain unique IMPDH1 isoforms. These unique isoforms arise from a combination of alternate transcription initiation sites, alternate splicing, and/or inclusion of a novel exon, exon 13b. Human retina contains two IMPDH1 isoforms which incorporate the additional carboxyl terminal residues that result from the inclusion of exon 13b. One of these isoforms also contains the additional 49 amino terminal residues that result from translation initiation in exon A (Fig. 8E).
Mouse retina contains the same isoforms as human retina, although the ratios of these protein isoforms are dramatically different from those in human retina (Fig. 8E). This large difference raises doubts as to the use of mice with a targeted disruption of the endogenous mouse gene as an appropriate animal model for the IMPDH1 form of human retinal degeneration. The Impdh1−/− mouse has very mild retinal degeneration, even at an advanced age.19 In contrast, the retinal degeneration seen in humans is characterized by an early age of onset and rapid progression.4,5,18,25,26 Alternatively, the expression differences in our studies make it possible that the reason the human disease is more severe is that the mutations affect only a subset of the retinal IMPDH1 isoforms whose concentrations differ between human and mouse.
Comparison of recombinant proteins with human retinal lysates and the electrophoretic differences between equivalent human and mouse isoforms suggests that the native human IMPDH1 retinal isoforms may undergo posttranslational modification or cleavage. Further study is needed to determine what this modification is and whether IMPDH1 retinal proteins from other species are also modified. Such studies may provide important information in choosing the appropriate animal model.
Possible Mechanisms for Photoreceptor Degeneration
Canonical IMPDH1 is not the major isoform of IMPDH1 expressed in human photoreceptors, making it unlikely that alterations in canonical protein function are the cause of retinal degeneration. It is more probable that retinal degeneration is the result of alteration(s) of the function of one or more of the unique retinal IMPDH1 isoforms. What this function may be is currently unknown. Mutations in canonical IMPDH1 do not alter enzymatic activity.6,19,20 It is possible, however, that mutations in the retinal isoforms alter enzyme activity in these unique proteins, if they still possess conventional enzyme activity.
Canonical IMPDH1 and IMPDH2 proteins both bind single-stranded nucleic acids.12 IMPDH can be found in both the cytoplasm and nucleus of cells and immunoprecipitation data suggest that it can bind both RNA and DNA.12 Each IMPDH tetramer binds sequences of approximately 100 nucleotides via an interaction with the CBS subdomains.12 The mutations that cause retinal degeneration alter this nucleic acid binding activity of canonical IMPDH1.4,20 It is possible that the retinal IMPDH1 isoforms bind a unique set of nucleic acids due to the additional amino and/or carboxyl terminal residues. Mutation studies have demonstrated that some core protein residues are also involved in nucleic acid binding, so it is conceivable that altering the amino or carboxyl terminus alters nucleotide binding.6,20 Determining whether and what nucleic acids the retinal isoforms bind and whether the disease-associated mutations alter this binding, will be important steps in understanding the retinal disease mechanism in patients with IMPDH1 mutations.
A final possibility is that these unique protein isoforms have a novel function in the retina not previously identified. Investigation of this possibility and of the other known functions described herein, are necessary before the disease mechanism of IMPDH1 mutations will be understood completely.
Acknowledgments
The authors thank Lizbeth Hedstrom for helpful discussion and critical reading of the manuscript; Beverly Mitchell (University of North Carolina) for providing the Impdh1−/− mice; and Peter Humphries, Avril Kennan, and Aileen Ahearne (Trinity College Dublin) for providing the canonical IMPDH1 vector.
Supported by grants from The Foundation Fighting Blindness, The William Stamps Farish Fund, The Hermann Eye Fund, Research to Prevent Blindness, the Rosanne Silberman Foundation, the F. M. Kirby Foundation, and National Eye Institute Grants EY014170 (SPD), EY12910 (EAP), and EY11286 (CBR).
Footnotes
Disclosure: S.J. Bowne, None; Q. Liu, None; L.S. Sullivan, None; J. Zhu, None; C.J. Spellicy, None; C. Bowes Rickman, None; E.A. Pierce, None; S.P. Daiger, None
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002:1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Heckenlively JR, Saiger SP. Hereditary retinal and choroidal degenerations. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin's Principals and Practices of Medical Genetics. 4th ed. Churchill Livingston; London: 2002. pp. 3555–3593. [Google Scholar]
- 3.Kennan A, Aherne A, Humphries P. Light in retinitis pigmentosa. Trends Genet. 2005;21:103–110. doi: 10.1016/j.tig.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Bowne SJ, Sullivan LS, Blanton SH, et al. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kennan A, Aherne A, Palfi A, et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho(−/−) mice. Hum Mol Genet. 2002;11:547–557. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- 6.Bowne SJ, Sullivan LS, Mortimer SE, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2006;47:34–42. doi: 10.1167/iovs.05-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Senda M, Natsumeda Y. Tissue-differential expression of two distinct genes for human IMP dehydrogenase (E.C. 1.1.1.205) Life Sci. 1994;54:1917–1926. doi: 10.1016/0024-3205(94)90150-3. [DOI] [PubMed] [Google Scholar]
- 8.Hedstrom L. IMP dehydrogenase: mechanism of action and inhibition. Curr Med Chem. 1999;6:545–560. [PubMed] [Google Scholar]
- 9.Natsumeda Y, Ohno S, Kawasaki H, Konno Y, Weber G, Suzuki K. Two distinct cDNAs for human IMP dehydrogenase. J Biol Chem. 1990;265:5292–5295. [PubMed] [Google Scholar]
- 10.Carr SF, Papp E, Wu JC, Natsumeda Y. Characterization of human type I and type II IMP dehydrogenases. J Biol Chem. 1993;268:27286–27290. [PubMed] [Google Scholar]
- 11.Hager PW, Collart FR, Huberman E, Mitchell BS. Recombinant human inosine monophosphate dehydrogenase type I and type II protein: purification and characterization of inhibitor binding. Biochem Pharmacol. 1995;49:1323–1329. doi: 10.1016/0006-2952(95)00026-v. [DOI] [PubMed] [Google Scholar]
- 12.McLean JE, Hamaguchi N, Belenky P, Mortimer SE, Stanton M, Hedstrom L. Inosine 5′-monophosphate dehydrogenase binds nucleic acids in vitro and in vivo. Biochem J. 2004;379:243–251. doi: 10.1042/BJ20031585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cornuel JF, Moraillon A, Gueron M. Participation of yeast inosine 5′-monophosphate dehydrogenase in an in vitro complex with a fragment of the C-rich telomeric strand. Biochimie (Paris) 2002;84:279–289. doi: 10.1016/s0300-9084(02)01400-1. [DOI] [PubMed] [Google Scholar]
- 14.Gu JJ, Tolin AK, Jain J, Huang H, Santiago L, Mitchell BS. Targeted disruption of the inosine 5′-monophosphate dehydrogenase type I gene in mice. Mol Cell Biol. 2003;23:6702–6712. doi: 10.1128/MCB.23.18.6702-6712.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain J, Almquist SJ, Ford PJ, et al. Regulation of inosine monophosphate dehydrogenase type I and type II isoforms in human lymphocytes. Biochem Pharmacol. 2004;67:767–776. doi: 10.1016/j.bcp.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 16.Nagai M, Natsumeda Y, Konno Y, Hoffman R, Irino S, Weber G. Selective up-regulation of type II inosine 5′-monophosphate dehydrogenase messenger RNA expression in human leukemias. Cancer Res. 1991;51:3886–3890. [PubMed] [Google Scholar]
- 17.Gu JJ, Spychala J, Mitchell BS. Regulation of the human inosine monophosphate dehydrogenase type I gene: utilization of alternative promoters. J Biol Chem. 1997;272:4458–4466. doi: 10.1074/jbc.272.7.4458. [DOI] [PubMed] [Google Scholar]
- 18.Kozma P, Hughbanks-Wheaton DK, Locke KG, et al. Phenotypic characterization of a large family with RP10 autosomal-dominant retinitis pigmentosa: an Asp226Asn mutation in the IMPDH1 gene. Am J Ophthalmol. 2005;140:858–867. doi: 10.1016/j.ajo.2005.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aherne A, Kennan A, Kenna PF, et al. On the molecular pathology of neurodegeneration in IMPDH1-based retinitis pigmentosa. Hum Mo Genet. 2004;13:641–650. doi: 10.1093/hmg/ddh061. [DOI] [PubMed] [Google Scholar]
- 20.Mortimer SE, Hedstrom L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem J. 2005;390:41–47. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arcellana-Panlilio MY, Schultz GA. Analysis of messenger RNA. Methods Enzymol. 1993;225:303–328. doi: 10.1016/0076-6879(93)25021-s. [DOI] [PubMed] [Google Scholar]
- 22.Liu Q, Zhou J, Daiger SP, et al. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- 23.Bowes Rickman C, Ebright JN, Zavodni ZJ, et al. Defining the human macula transcriptome and candidate retinal disease genes using EyeSAGE. Invest Ophthalmol Vis Sci. 2006;47:2305–2316. doi: 10.1167/iovs.05-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharon D, Blackshaw S, Cepko CL, Dryja TP. Profile of the genes expressed in the human peripheral retina, macula, and retinal pigment epithelium determined through serial analysis of gene expression (SAGE) Proc Natl Acad Sci USA. 2002;99:315–320. doi: 10.1073/pnas.012582799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schatz P, Ponjavic V, Andreasson S, McGee TL, Dryja TP, Abrahamson M. Clinical phenotype in a Swedish family with a mutation in the IMPDH1 gene. Ophthalmic Genet. 2005;26:119–124. doi: 10.1080/13816810500229090. [DOI] [PubMed] [Google Scholar]
- 26.Wada Y, Sandberg MA, McGee TL, Stillberger MA, Berson EL, Dryja TP. Screen of the IMPDH1 gene among patients with dominant retinitis pigmentosa and clinical features associated with the most common mutation, Asp226Asn. Invest Ophthalmol Vis Sci. 2005;46:1735–1741. doi: 10.1167/iovs.04-1197. [DOI] [PubMed] [Google Scholar]