

Abstract
PURPOSE
To examine the molecular genetic basis and phenotypic characteristics of an unusual late-onset autosomal dominant macular dystrophy with features of age-related macular degeneration (AMD) in a large family (SUNY901), by using linkage and mutation analyses.
METHODS
Blood samples were collected from 17 affected members, 17 clinically unaffected members, and 5 unrelated spouses. Clinical analyses included a review of medical history and standard ophthalmic examination with fundus photography, fluorescein angiography, and electroretinography. Linkage and haplotype analyses were performed with microsatellite markers. Mutation analysis was performed by amplification of exons followed by sequencing.
RESULTS
A wide spectrum of clinical phenotypes including exudative and nonexudative maculopathy was observed, with onset in the late fifth decade. Linkage analysis excluded most of the previously known maculopathy loci. Markers D6S1604 (Zmax of 3.18 at θ = 0), and D6S282 (Zmax of 3.18 at θ = 0) gave significant positive LOD scores and haplotype analysis localized the disease gene to a 9-centimorgan (cM) interval between markers D6S1616 and D6S459. Mutation analysis excluded the GUCA1A and GUCA1B genes and revealed a missense mutation in the RDS/peripherin gene leading to a Tyr141Cys substitution. A phenotype and haplotype comparison between this and a separate family with the Tyr141Cys mutation suggested the presence of a common ancestral haplotype.
CONCLUSIONS
The RDS mutation in codon 141 is associated with an unusual AMD-like late-onset maculopathy. An apparent selective bias was noted favoring the transmission of the mutant allele. These observations broaden the spectrum of phenotypes associated with RDS gene mutations.
Chorioretinal atrophy and choroidal neovascularization are sight-threatening complications that frequently result in permanent loss of central vision in patients with many macular disorders including age-related macular degeneration (AMD). The molecular pathogenesis of these severely disabling and poorly treatable conditions is not well understood. Recent evidence suggests that chorioretinal atrophy in patients with various retinal dystrophies can be traced to a wide array of mutations in photoreceptor-specific genes.1-4 Evidence tying choroidal neovascular disease to these same or other mutations in photoreceptor-specific genes has been weaker, although rare patients with familial age-related macular degeneration manifest both nonexudative atrophic and neovascular exudative phenotypes in association with sequence variants in the photoreceptor-specific gene ABCA4.5 Most other macular degenerative disorders studied to date with prominent neovascular exudative complications, including Sorsby dystrophy, Doyne honeycomb dystrophy, and Best vitelliform dystrophy, are caused by genes that are predominantly expressed in the retinal pigment epithelium and not in photoreceptors.6-9
Among dystrophies with neovascular features, Sorsby macular dystrophy10,11 stands out as the only late-onset autosomal dominant dystrophy in which choroidal neovascularization and disciform maculopathy are key defining clinical features, although atrophic features are also encountered. Mutations in the gene for tissue inhibitor of metalloproteinase (TIMP)-3, which is expressed primarily in the retinal pigment epithelium, were reported to cause this exudative disease.6 Most families and patients described with late-onset exudative phenotype either have been linked to a TIMP3 locus or carry a mutation in the TIMP3 gene, except for a family that we described earlier.12-14
We recently identified another large family (SUNY901) whose members have an unusual late-onset maculopathy with exudative and atrophic features. After excluding most of the previously described macular disease loci including the loci for exudative fundus dystrophies and AMD,5,7,15-17 we mapped the disease locus to a 9-centimorgan (cM) interval on chromosome 6. Mutation analysis of the RDS/peripherin, GUCA1A, and GUCA1B genes contained within the interval revealed a missense mutation resulting in a Tyr141Cys substitution in the RDS/peripherin gene product. Comparison of haplotypes between SUNY901 and a separate family (BCM-AD033) with the identical Tyr141Cys mutation suggests a possible common ancestral origin.18 Identification of Tyr141Cys establishes an association between an unusual exudative and nonexudative autosomal dominant late-onset macular degeneration with mutation in the photoreceptor-specific RDS/peripherin gene. Mutations in this essential outer segment structural gene have been associated with a wide array of central and peripheral human retinal dystrophies, but not typically the exudative phenotypes.19,20
METHODS
Clinical Studies
Informed consent was obtained from all participants before enrolling them in the molecular genetic studies that had been approved by the Institutional Review Boards for Human Subject Research at the State University of New York at Buffalo and Baylor College of Medicine (Houston). The research described in this study adhered to the tenets of the Declaration of Helsinki. A total of 39 individuals were recruited from SUNY901, including 17 with the disease, 17 without the disease, and 5 normal unrelated spouses. Five affected individuals were recruited from BCM-AD033. Blood samples and medical history were obtained from all participants. The ophthalmic examination included Snellen visual acuity measurements and ophthalmoscopy and/or fundus photography in most patients. Further ancillary testing including fluorescein angiography, visual field testing, electroretinography (ERG), and electro-oculography (EOG) were performed in selected participants.21,22
Genetic Analysis
Genotyping
Genomic DNA was isolated from leukocytes by standard techniques. Microsatellite marker analysis was conducted as described.23 Description of the polymorphic markers and genetic distances were obtained from Gènèthon (www.genethon.fr; provided in the public domain by the French Association against Myopathies, Evry, France).
Linkage Analysis
Two-point linkage analysis was performed between the disease locus and each microsatellite marker with the MLINK program of the LINKAGE package24 (http:www.hgmp.mrc.ac.uk/; provided in the public domain by the Human Genome Mapping Project Resources Center, Cambridge, UK). Linkage was assessed under the conditions of autosomal dominant inheritance of the disease trait with a frequency of 0.0001 for the disease-causing allele with the age-dependent penetrance and variable penetrance models and affected-only analysis.25 To calculate LOD scores using an age-dependent penetrance model, individuals in the pedigree were grouped into age-classes and the penetrance for each liability class was calculated as previously described.26
Mutation Analysis
The sequences containing exons and exon–intron boundaries of GUCA1A, GUCA1B, RDS/peripherin, and ROM1 genes were amplified with primers located in the flanking intronic regions as described.6,27-29 Sequence analysis was performed with amplified PCR products as templates and a 33P cycle sequencing reaction kit (Amersham, Arlington Heights, IL).
RESULTS
We recruited 39 participants from three living generations of the SUNY901 family. Thirty-four members were related through common ancestry. The other five were spouses who had married into the family but were unrelated to the members. Based on a well-established genealogy, the members of this family could be traced to a single ancestor who emigrated from Germany to North America.
To establish the inheritance pattern in SUNY901 pedigree, we traced the disease through five generations of descendants. In the pedigree shown in Figure 1, the disease appeared to have been passed sequentially from one generation to the next through affected individuals or unaffected carriers. In generation II, affected brothers II:1 and II:2 were reported to have passed the disease to several of their progeny, including III:2 and III:6 who are still living. In generation IV, two of the five siblings born to III:6 (IV:16 and IV:20) had the same atrophic macular disease found in their father, albeit at an earlier stage. All five children (IV:21-25), born to reportedly unaffected parents (III:7 and III:7S), had the disease either in an atrophic or exudative form, suggesting that their father (III:7), who died in his 60s, may have been a bearer of asymptomatic or minimally expressed disease. Altogether, 15 of the 25 family members examined who were born to affected or carrier parents appeared to be affected in generation IV, either with atrophic or neovascular disease (Fig. 1). The remaining 10 members in this generation are asymptomatic and could represent unaffected individuals, asymptomatic carriers, or even affected members who may not yet manifest the disorder. Several of our recruits from generation V were younger than 45 years and consequently may have been too young to have visual symptoms or show signs of the disease. Despite the ambiguities in the clinical status of diseased individuals in generation III, the observed pattern of inheritance is consistent with an autosomal dominant monogenic trait. Varying phenotypic features can be explained by variable expressivity, especially given the evidence for the presence of genetic carriers.
Figure 1.

Pedigree of SUNY901 showing the distribution of the phenotypes. Asterisks: participants for whom phenotype information was available. Cross-hatched and filled symbols: individuals affected with atrophic and exudative disease, respectively. Symbols with central dot: members who are deceased and were probable carriers of the disease without overt symptoms. Open symbols with asterisks: asymptomatic participants in the study. Unmarked open symbols: members who were not recruited into the study and whose phenotypes were not determined. Additional siblings in generation II were excluded from the pedigree because of space limitations. Pedigree was intentionally distorted to maintain confidentiality of participant's identity.
Further clinical assessment of the maculopathy in affected individuals in SUNY901 revealed some common and divergent features (Table 1, Figs. 2, 3). The patients in this family had undergone clinical evaluation by other ophthalmologists and were assigned numerous diagnoses including AMD, retinitis pigmentosa inversa, adult foveomacular dystrophy, and Stargardt disease. Their diseases usually manifested by the middle of the fifth decade and nearly all reported fading spots and relative scotoma and/or distortion near the center of vision, even early on. Most retained visual acuity of 20/50 or better and had limited mild atrophic changes, including subtle pigmentary disturbance and drusenlike deposits in the macula (Figs. 2A–E). Others with more severe involvement displayed choriocapillaris loss (Figs. 2F, 2G), geographic atrophy (Fig. 2H), or exudative changes (Figs. 2I–L). Among those with the most severe exudative disease, IV:4 and IV:27 had active choroidal neovascular disease and III:2, IV:21, and IV:22 had disciform scars representing involuted neovascular membranes (Table 1). The review of full-field ERG for IV:25 (Table 1, Fig. 3) showed only a mild generalized reduction in the amplitudes, despite the patient's report of night vision difficulty. The implicit times and the waveforms were unaffected in each eye of this subject. The ERG and EOG responses on IV:6 were entirely normal according to this subject's record, despite a similar self-report of nyctalopia. Visual field testing of IV:6 revealed pericentral scotomas but no peripheral constriction (data not shown). These visual functional studies suggest that the overall retinal function can be mildly affected in some cases despite the apparent confinement of the disease to macula.
TABLE 1.
Clinical Characteristics of the Maculopathy in Patients of Family SUNY901
| Member | Sex | A/On | Symptoms | VA | Fundus | ERG |
|---|---|---|---|---|---|---|
| III:2 | F | 81/6 | Poor vision OU | CF OU | Disciform scars OU; extensive macular atrophy | NA |
| III:6 | M | 80/6 | Central scotomas OU | 20/80 OD, 20/200 OS | Geographic atrophy: moderate OD, severe OS | NA |
| IV:3 | F | 55/5 | Central distortion OD, pericentral scotomas OS | 20/60 OD, 20/25 OS | Confluent drusen-like substance OU? Egg yolk lesion OU | NA |
| IV:4 | F | 59/6 | Poor vision OD; scotomas OS | CF OD, 20/25 OS | Occult subfoveal CNVM; Pericentral geographic atrophy OS | NA |
| IV:6 | F | 62/5 | Scotomas and distortion OU; night vision difficulty | 20/30 OD, 20/60 OS | Drusenlike deposits and mild pigment clumping OU | Normal |
| IV:9 | F | 54/5 | Scotomas OU | 20/30 OU | Diffuse geographic atrophy with extension beyond arcades | NA |
| IV:10 | M | 56/6 | Scotomas and distortion OU | 20/50+ OU | Dry AMD appearance with drusen and pigmentary changes | NA |
| IV:11 | M | 57/6 | Scotomas and distortion OU | 20/30+ OU | Drusenlike deposits OU | NA |
| IV:12 | M | 63/7 | Scotomas and distortion OD; poor vision OS | 20/50 OD, 20/200 OS | Drusenlike deposits OD; traumatic maculopathy OS | NA |
| IV:16 | M | 51/5 | Scotomas and distortion OU | 20/25 OU | Drusenlike deposits and mild pigment clumping OU | NA |
| IV:20 | F | 57/6 | Scotomas and distortion OU | 20/30+ OU | Drusenlike deposits and mild pigment clumping OU | NA |
| IV:21 | M | 58/5 | Poor vision OU | 20/50+ OD, CF OS | Drusenlike deposits and atrophy OD, disciform scar OS | NA |
| IV:22 | F | 60/5 | Poor vision OD; scotomas OS | CF OD, 20/30+ OS | Disciform scar OD; macular pigment atrophy OS | NA |
| IV:23 | F | 62/5 | Scotomas and distortion OU | 20/50+ (based on clinical records) | Dry AMD-like retinal degeneration (based on clinical records) | NA |
| IV:24 | F | 47/5 | Scotomas and distortion OU | 20/25 OU | Drusenlike deposits OU | NA |
| IV:25 | F | 51/5 | Scotomas and distortion OU; night vision difficulty | 20/30 OU | Choriocapillaris/RPE atrophy OU | Mildly reduced |
| IV:27 | F | 57/5 | Scotomas and distortion | 20/50 OU | Ill-defined CNVM OU; drusenlike deposits | NA |
A, age of assessment; On, decade of onset of disease; VA, visual acuities; CF, counting fingers; CNVM, choroidal neovascular membrane; RPE, retinal pigment epithelium; NA, not analyzed.
Figure 2.
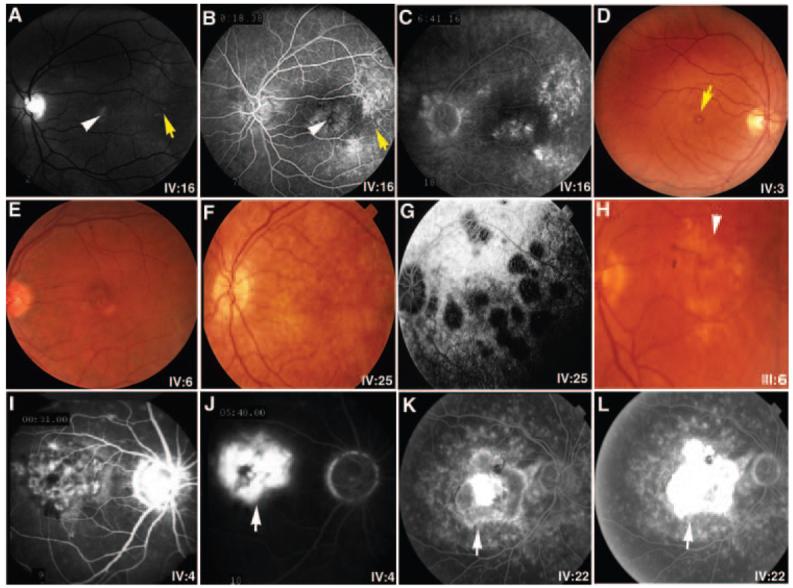
An assortment of representative fundus photographs and fluorescein angiograms from patients with maculopathy in SUNY901. Monochromatic fundus photographs (A) as well as the early (B) and late (C) fluorescein angiographic frames highlight the subtle features of early atrophic disease, including pigmentary changes (white arrowheads) and drusen (yellow arrows) in the macula of IV:16. (D, E) Fine yellowish drusenlike substance at the level of the retinal pigment epithelium with a small yolklike lesion (yellow arrow) in the foveas of IV:3 and IV:6, respectively. Photograph (F) and the late angiographic frame (G) show pigment epithelial atrophy and scalloped areas of choriocapillaris loss in later stages of atrophy in IV:25. (H) Extensive geographic atrophy of central macula encountered in III:6 is shown. Early (I, K) and late (J, L) angiographic frames highlight features of the exudative disease, showing subfoveal hyperfluorescence and leakage attributable to an ill-defined choroidal neovascular membrane in IV:4 (I, J) and a disciform scar in IV:22 (K, L). The choroid is especially dark in IV:4. White arrows: Choroidal neovascularization is indicated.
Figure 3.

Full-field ERG in patient IV:25. Dark-adapted (scotopic) and light-adapted (photopic) ERGs were recorded with bipolar Burian Allen contact lens electrodes in accordance with the 1999 International Society for Clinical Electrophysiology of Vision (ISCEV) standards. Representative responses under maximum (A) scotopic and (B) photopic conditions are shown. The included tables list absolute peak wave amplitudes for the maximum scotopic stimulus (threshold +4 log); scotopic a wave threshold (max. rod b) and maximum photopic responses. Percentages represent the fraction of the lower limit of normal (mean, −2.5 sec/d) for the listed stimulus condition.
Contemplating Sorsby fundus dystrophy as a possible first diagnosis in view of the propensity toward disciform disease, we set out to determine whether the TIMP3 locus might cause this disorder. We excluded this possibility by linkage analysis and by mutation screening of all five exons of the TIMP3 gene that contains nearly all Sorsby-associated mutations known to date. To determine whether the other retinopathy or maculopathy loci may be associated with the disease in SUNY901, we also tested Stargardt macular degeneration 1 (STGD1), STGD3, STGD4, age-related macular degeneration (ARMD1), Doyne honeycomb dystrophy (DHRD), North Carolina macular dystrophy (MCDR1), Best macular dystrophy (VMD2), cone-rod dystrophy 2 (CORD2), CORD5, CORD8, and the rhodopsin loci for possible linkage. Linkage analysis was performed on affected individuals only, to avoid inaccuracies introduced into the final results as a consequence of ambiguities in the assignment of phenotype in case of asymptomatic participants with normal examination results. LOD scores of −2.0 or less were obtained, with most markers linked to previously mapped macular disease loci excluding them from linkage.
In contrast, microsatellite marker D6S282 linked to RDS/peripherin and cone dystrophy 3 (COD3) gave a significant positive LOD score (Zmax = 3.18) at zero recombination fraction with an affected-only model. The pericentromeric region of chromosome 6 containing the marker D6S282 harbors at least five known retinal disease loci.30-34 Thus we analyzed additional markers linked to D6S282 to map precisely the disease locus in family SUNY901 in conjunction with the previously mapped disease loci in this region. Markers D6S1552, D6S1582, D6S271, and D6S1604 all had significantly positive LOD scores, thus localizing the disease gene to the short arm of chromosome 6, using only the affected individuals in linkage analysis (Table 2). LOD scores were also calculated by using age-dependent penetrance and variable penetrance models. The results obtained from different models did not vary significantly. Haplotype analysis localized the disease gene in SUNY901 family to a 9-cM interval between markers D6S1616 and D6S459 (Fig. 4A).
TABLE 2.
Two-Point LOD Scores of Markers on Chromosome 6 Versus Pedigree SUNY901
| LOD Scores at θ |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Markers | Distance* (cM) |
0 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | Zmax | θ |
| D6S1616 | 14 | −∞ | 0.27 | 0.37 | 0.33 | 0.18 | 0.06 | 0.37 | 0.1 |
| D6S426 | 1 | −∞ | 1.17 | 1.38 | 1.24 | 0.85 | 0.39 | 1.37 | 0.1 |
| D6S1552 | 3 | 1.91 | 1.84 | 1.66 | 1.21 | 0.73 | 0.29 | 1.91 | 0 |
| D6S1582 | 2 | 2.49 | 2.16 | 1.83 | 1.18 | 0.60 | 0.19 | 2.49 | 0 |
| D6S271 | 1 | 2.85 | 2.49 | 2.12 | 1.41 | 0.76 | 0.26 | 2.84 | 0 |
| D6S282 | 0 | 3.18 | 2.79 | 2.40 | 1.60 | 0.85 | 0.25 | 3.18 | 0 |
| D6S1604 | 0 | 3.18 | 2.78 | 2.38 | 1.59 | 0.86 | 0.30 | 3.18 | 0 |
| D6S459 | 2 | −∞ | 1.31 | 1.50 | 1.26 | 0.76 | 0.27 | 1.5 | 0.1 |
| D6S1651 | 3 | 1.21 | 1.13 | 1.02 | 0.72 | 0.41 | 0.14 | 1.21 | 0 |
| D6S1689 | 0 | −1.86 | 0.83 | 0.88 | 0.69 | 0.38 | 0.10 | 0.83 | 0.05 |
| D6S452 | 2 | −0.03 | 2.60 | 2.33 | 1.65 | 0.88 | 0.25 | 2.6 | 0.05 |
| D6S1714 | 1 | 0.94 | 0.84 | 0.72 | 0.47 | 0.24 | 0.06 | 0.94 | 0 |
| D6S1662 | 4 | −0.30 | 2.25 | 2.10 | 1.48 | 0.78 | 0.20 | 2.25 | 0.05 |
| D6S295 | 1 | −2.22 | 0.47 | 0.47 | 0.27 | 0.09 | 0.01 | 0.47 | 0.05 |
| D6S294 | 1 | 1.78 | 1.55 | 1.33 | 0.89 | 0.49 | 0.17 | 1.77 | 0 |
| D6S428 | 12 | 0.90 | 0.73 | 0.57 | 0.32 | 0.14 | 0.04 | 0.9 | 0 |
| D6S286 | 2 | −∞ | −0.53 | −0.29 | −0.09 | −0.03 | −0.003 | −0.003 | 0.4 |
| D6S445 | 1 | −0.86 | 1.29 | 1.29 | 0.98 | 0.56 | 0.20 | 1.29 | 0.05 |
| D6S1609 | 7 | −∞ | −0.84 | −0.38 | −0.06 | 0.01 | 0.01 | 0.01 | 0.3 |
| D6S1570 | 8 | −∞ | −1.08 | −0.65 | −0.32 | −0.12 | −0.02 | −0.02 | 0.4 |
| D6S1717 | −0.44 | 0.20 | 0.31 | 0.29 | 0.19 | 0.08 | 0.3 | 0.1 | |
Distance to the next distal marker.
Figure 4.

Edited version of pedigrees (A) SUNY901 and (B) BCMAD-033 with haplotypes constructed with eight markers on chromosome 6 flanking the RDS/peripherin locus. Individuals' numbers are same as noted in Figure 1 for SUNY901. Numbers shown at top right of each circle or square represent the age of the individual. Filled bars: disease haplotype. +/−, presence of RDS Tyr141Cys mutation in the heterozygous state; −/−, mutant alleles in the homozygous state. Questions marks in the haplotype denote PCR failure.
The critical interval for the disease gene in family SUNY901, defined by the linkage and haplotype analysis, included the retinal genes encoding peripherin/RDS, GUCA1A, and GUCA1B. Sequence analysis revealed no mutations in either the GUCA1A or GUCA1B genes. However, a heterozygous missense A-to-G mutation was found in codon 141 of the RDS/peripherin gene leading to the putative Tyr141Cys substitution in the encoded protein in all affected members of family SUNY901. This mutation was also present in the asymptomatic carriers as well, including IV:1 and 17-19 (Fig. 4A) but not in our control population of 50 normal individuals (100 chromosomes). This sequence variation was not detected in more than 1000 chromosomes surveyed in control populations in other reports in the literature.18,35
The protein product of the ROM1 gene localized to chromosome 11 has been reported to interact with RDS/peripherin.36 Mutations in this unlinked photoreceptor-specific gene have been reported to cause digenic retinitis pigmentosa.37,38 To evaluate the contribution of ROM1 to the variable phenotype observed in the SUNY901 family, we screened the gene for sequence alterations in affected individuals and carriers of the Tyr141Cys-RDS gene mutation. No sequence alterations were found in the coding region of ROM1 in either affected members or carriers.
A separate four-member pedigree BCM-AD033 with retinal dystrophy is the only other known family with the Tyr141Cys mutation.18 The four affected individuals examined from this family exhibited a late-onset dominant macular phenotype similar to the one found in SUNY901. One had disciform macular degeneration. A haplotype comparison between these two families showed that marker alleles were shared in common (Fig. 4B). Based on the population frequency of alleles, probability of occurrence of this particular haplotype by chance is estimated at 0.00001. Although the phase could not be determined conclusively for the construction of haplotype in family BCM-AD033, the presence of the SUNY901 disease-associated ancestral haplotype in the affected members of BCM-AD033 suggests that these two families probably share common ancestry.
DISCUSSION
In this report we describe an unusual dominantly inherited late-onset macular dystrophy and elucidate its molecular genetic basis in a large family, SUNY901. Through a series of linkage, haplotype, and mutation analyses, a rare Tyr141Cys mutation in the RDS gene was uncovered and found to cosegregate with the atrophic and exudative disease phenotype in this family. Haplotype analyses revealed comparable profiles among affected individuals from SUNY901 and an independent family BCM-AD033 with the Tyr141Cys mutation, further implying a possible common ancestral origin for this mutation. These findings support the pathogenic role of the RDS mutation in the AMD-like phenotype described.
The Tyr141Cys RDS/peripherin mutation is unique because it predisposes to exudative complications in addition to a range of maculopathies with primarily atrophic consequences. Approximately one in four patients (6/21 = 29% ± 20%, P = 0.05) in SUNY901 and BCM-AD033 carrying the mutation had choroidal neovascularization and exudative changes including hemorrhage and edema superimposed on a background of atrophic macular changes. The prevalence of this serious complication was quite high in this family, considering the rarity with which choroidal neovascularization has been reported previously in association with RDS or other photoreceptor-specific gene mutations. Only two isolated RDS-associated cases, one of a patient with a Pro210Arg mutation and adult foveomacular disease and another with a 4-bp insertion in codon 140 with a pattern dystrophy phenotype, have been reported to manifest choroidal neovascular maculopathy.39,40 Given the gravity of this complication for visual prognosis, as is evident in studies on age-related macular degeneration showing that 90% of all blindness due to this disease is caused by this complication, the above findings take on a special significance because the cause of choroidal neovascularization is currently unknown.
The RDS mutation described herein seems to be a rare mutation originating from a common ancestor in Europe. Although the RDS/peripherin gene has been screened extensively in populations worldwide,18,41 the only families known to carry this mutation are the two we have described. Both appear to have originated in Germany. The SUNY901 ancestors first immigrated to the east coast of North America in the 1700s with the early settlers, whereas the BCM-AD033 ancestors reportedly moved to the midwestern United States directly from Germany in the mid-1800s. No direct connections or overlaps were found between the detailed six-generation genealogy on SUNY901 and the limited genealogy available on BCM-AD033. However, haplotype analysis revealed the presence of shared markers among the affected individuals from each family, further supporting common lineage and possibly a founder haplotype.
A selective bias was noted in our families, suggesting apparent inheritance of mutant allele in favor of the wild-type copy from heterozygous parents. At least 18 of the 23 members in generation IV born to affected or obligate carrier parents also carried the mutant allele. In addition, at least four of the five children born to II:1 and three of the five children born to II:2 carried the mutation. In the major branches studied, the affected outnumbered the unaffected, even after we conservatively labeled the few with unknown phenotype or genotype as unaffected. The calculated odds of encountering the mutant versus the wild-type copy comes closer to 3:1 than to the 1:1 ratio expected based on simple unbiased meiotic sorting of the two alleles into two gametes (odds ratio: 1000:1, Z ∼3). The source of this bias is unclear and may not be solely attributable to an ascertainment bias or selective sampling, especially because the calculated proportions were based on the ratio of the affected or carrier phenotype relative to entire progenies of II:1, III:1, III:5, III:6, and III:7. Indeed, entire segments of the family were enrolled well before any linkage assignment or gene identification had been made, again reducing any intentional selection bias. Although persistent ascertainment bias remains a possibility, the markedly skewed ratio in favor of the mutant copies in SUNY901 may also suggest a selective advantage conferred by the mutant RDS allele or a linked gene. Such selective bias or meiotic drive has not been reported previously with any of the other RDS-associated alleles, although patients with X-linked retinitis pigmentosa and Usher syndrome have been known to manifest alterations in sperm morphology without known alterations in fertility.42,43
The variability of clinical phenotype among individuals carrying the Tyr141Cys mutation in the SUNY901 pedigree suggests potential involvement of other modifier genes or the possible contribution of environmental and other stochastic factors to the pathogenesis of the maculopathy. We investigated this possibility in our group by mutation analysis of the unlinked ROM1 gene.33 ROM1 is a structurally related protein partner of RDS which is thought to interact with the intradiscal D2 loop in the heterodimeric (i.e., compound tetrameric) complex of RDS-ROM1. An additive effect of mutations in RDS and ROM1 resulting in retinal dystrophies has been demonstrated previously.37,38 We did not find any sequence alterations in the ROM1 coding sequence among the affected individuals and carriers of the Tyr141Cys mutation. This negative finding suggests contributions from sequence alterations elsewhere, either in the ROM1 gene or other genes.
The Tyr141Cys mutation is likely to exert its pathogenic effect through disruption of the normal structure and metabolism of the RDS protein. The additional cysteine in the mutant protein could compete and interfere with the formation of the normal disulfide bond in the D2 loop. Disruption in the protein structure could then translate into a disorganization of the outer segments given the crucial role of RDS in maintaining the stacked configuration of outer segment disks.31 In addition, metabolic byproducts generated from processing the abnormal RDS protein by the retinal pigment epithelium could accumulate and change the composition of the extracellular matrix that normally forms part of the barrier against choroidal neovascularization.44 Alterations in physiology of extracellular matrix caused by mutations in TIMP3 and EFEMP1 have been associated with choroidal neovascular disease in Sorsby fundus dystrophy and Malattia Leventinese.6,7,45 The presence on fluorescin angiography of a dark choroid in several of the family members in the current study supports the possibility of accumulation of an abnormal metabolite in the pigment epithelium and underlying Bruch's membrane.
Insights gained from the molecular genetic study of this family and other families with inherited maculopathies could benefit our understanding the molecular basis of age-related macular degeneration in some fraction of the general population.46 The features encountered in the patients in this family mimic those in AMD, including late age of onset, variations in phenotypic expression, severity of the disease, differential age of expression, and the presence of drusen, geographic atrophy, and choroidal neovascularization. We would not be surprised if a fraction of patients with age-related macular degeneration harbored the RDS mutation described herein, especially given the clear evidence that this mutation and its associated haplotype can be selectively transmitted from one generation to next.
Acknowledgments
The authors thank all the members of the two families SUNY901 and BCM-AD033, whose willing and continued cooperation was essential to the success of these studies; all the physicians who kindly provided the clinical records and diagnostic studies for review; Paul Sieving for reviewing the clinical photographs and histories; Andrew Vine and Julia Richards for critical review of the manuscript; and Brydon Grant for assistance with statistical analysis.
Supported by Rockefeller Brothers Fund Culpepper Pilot initiative award 00-185 (SCK), the Foundation Fighting Blindness (RAL, RAy), the Michigan Life Sciences Corridor Fund (RAy), National Eye Institute Grant EY13198 (RAy), EY07142 (SPD) and Core Grant EY07003, Research to Prevent Blindness (RAL, RAy, SPD). RAL is a Senior Scientific Investigator for Research to Prevent Blindness.
Footnotes
Disclosure: S.C. Khani, None; A.J. Karoukis, None; J.E. Young, None; R. Ambasudhan, None; T. Burch, None; R. Stockton, None; R.A. Lewis, None; L.S. Sullivan, None; S.P. Daiger, None; E. Reichel, None; R. Ayyagari, None
References
- 1.Zack DJ, Dean M, Molday RS, et al. What can we learn about age-related macular degeneration from other retinal diseases? Mol Vis. 1999;5:30. [PubMed] [Google Scholar]
- 2.Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 3.Zhang K, Kniazeva M, Han M, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- 4.Ayyagari R, Demirici FY, Liu JF, et al. RPGR mutation in X-linked recessive atrophic macular degeneration. Genomics. 2002;80:166–171. doi: 10.1006/geno.2002.6815. [DOI] [PubMed] [Google Scholar]
- 5.Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- 6.Weber BH, Vogt G, Pruett RC, Stohr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby's fundus dystrophy. Nat Genet. 1994;8:352–356. doi: 10.1038/ng1294-352. [DOI] [PubMed] [Google Scholar]
- 7.Stone EM, Lotery AJ, Munier FL, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- 8.Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci USA. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blodi CF, Stone EM. Best's vitelliform dystrophy. Ophthalmic Paediatr Genet. 1990;11:49–59. [PubMed] [Google Scholar]
- 10.Sorsby A, Mason ME, Gardener J. A fundus dystrophy with unusual features. Br J Ophthalmol. 1949;33:67–97. doi: 10.1136/bjo.33.2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polkinghorne PJ, Capone MRC, Beringer T, Lyness AC, Shemi K, Bird AC. Sorsby fundus dystrophy: a clinical study. Ophthalmology. 1989;96:1763–1768. doi: 10.1016/s0161-6420(89)32654-6. [DOI] [PubMed] [Google Scholar]
- 12.Tabata Y, Isashiki Y, Kamimura K, Nakao K, Ohba N. A novel splice site mutation in the tissue inhibitor of the metalloproteinases-3 gene in Sorsby's fundus dystrophy with unusual clinical features. Hum Genet. 1998;103:179–182. doi: 10.1007/pl00008707. [DOI] [PubMed] [Google Scholar]
- 13.Assink JJ, de Backer E, ten Brink JB, et al. Sorsby fundus dystrophy without a mutation in the TIMP-3 gene. Br J Ophthalmol. 2000;84:682–686. doi: 10.1136/bjo.84.7.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ayyagari R, Griesinger IB, Bingham E, Lark KK, Moroi SE, Sieving PA. Autosomal dominant hemorrhagic macular dystrophy not associated with the TIMP3 gene. Arch Ophthalmol. 2000;118:85–92. doi: 10.1001/archopht.118.1.85. [DOI] [PubMed] [Google Scholar]
- 15.Klein ML, Schultz DW, Edwards A, et al. Age-related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Arch Ophthalmol. 1998;116:1082–1088. doi: 10.1001/archopht.116.8.1082. [DOI] [PubMed] [Google Scholar]
- 16.Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- 17.Small KW, Weber JL, Roses A, Lennon F, Vance JM, Pericak-Vance MA. North Carolina macular dystrophy is assigned to chromosome 6. Genomics. 1992;13:681–685. doi: 10.1016/0888-7543(92)90141-e. [DOI] [PubMed] [Google Scholar]
- 18.Sohocki MM, Daiger SP, Bowne SJ, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat. 2001;17:42–51. doi: 10.1002/1098-1004(2001)17:1<42::AID-HUMU5>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kajiwara K, Hahn LB, Mukai S, Travis GH, Berson EL, Dryja TP. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354:480–483. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- 20.Weleber RG, Carr RE, Murphey WH, Sheffield VC, Stone EM. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol. 1993;111:1531–1542. doi: 10.1001/archopht.1993.01090110097033. [DOI] [PubMed] [Google Scholar]
- 21.Sieving PA. Diagnostic issues with inherited retinal and macular dystrophies. Semin Ophthalmol. 1995;10:279–294. doi: 10.3109/08820539509063799. [DOI] [PubMed] [Google Scholar]
- 22.Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 update). International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol. 1998;97:143–156. doi: 10.1023/a:1002016531591. [DOI] [PubMed] [Google Scholar]
- 23.Griesinger IB, Sieving PA, Ayyagari R. Autosomal dominant macular atrophy at 6q14 excludes CORD7 and MCDR1/PBCRA loci. Invest Ophthalmol Vis Sci. 2000;41:248–255. [PubMed] [Google Scholar]
- 24.Lathrop GM, Lalouel JM. Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet. 1984;36:460–465. [PMC free article] [PubMed] [Google Scholar]
- 25.Terwilliger JD, Ott J. Handbook of Human Genetic Linkage. The Johns Hopkins University Press; Baltimore: 1994. [Google Scholar]
- 26.Ott J. Analysis of Human Genetic Linkage. The Johns Hopkins University Press; Baltimore: 1999. [Google Scholar]
- 27.Wells J, Wroblewski JJ, Keen J, et al. Mutations in human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3:213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- 28.Surguchov A, Bronson JD, Banerjee P, et al. The human GCAP1 and GCAP2 genes are arranged in a tail-to-tail array on the short arm of chromosome 6 (p21.1) Genomics. 1997;39:312–322. doi: 10.1006/geno.1996.4513. [DOI] [PubMed] [Google Scholar]
- 29.Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1997;18:1972–1982. [PubMed] [Google Scholar]
- 30.Payne AM, Downes SM, Bessant DA, et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet. 1998;7:273–277. doi: 10.1093/hmg/7.2.273. [DOI] [PubMed] [Google Scholar]
- 31.Travis GH, Sutcliffe JG, Bok D. The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron. 1991;6:61–70. doi: 10.1016/0896-6273(91)90122-g. [DOI] [PubMed] [Google Scholar]
- 32.Kelsell RE, Gregory-Evans K, Gregory-Evans CY, et al. Localization of a gene (CORD7) for a dominant cone-rod dystrophy to chromosome 6q. Am J Hum Genet. 1998;63:274–279. doi: 10.1086/301905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruiz A, Borrego S, Marcos I, Antinolo G. A major locus for autosomal recessive retinitis pigmentosa on 6q, determined by homozygosity mapping of chromosomal regions that contain gamma-aminobutyric acid-receptor clusters. Am J Hum Genet. 1998;62:1452–1459. doi: 10.1086/301866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dharmaraj S, Li Y, Robitaille JM, et al. A novel locus for Leber congenital amaurosis maps to chromosome 6q. Am J Hum Genet. 2000;66:319–326. doi: 10.1086/302719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Payne AM, Downes SM, Bessant DA, Bird AC, Bhattacharya SS. Founder effect, seen in the British population, of the 172 peripherin/RDS mutation-and further refinement of genetic positioning of the peripherin/RDS gene. Am J Hum Genet. 1998;62:192–195. doi: 10.1086/301679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bascom RA, Manara S, Collins L, Molday RS, Kalnins VI, McInnes RR. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron. 1992;8:1171–1184. doi: 10.1016/0896-6273(92)90137-3. [DOI] [PubMed] [Google Scholar]
- 37.Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 38.Kedzierski W, Nusinowitz S, Birch D, et al. Deficiency of rds/peripherin causes photoreceptor death in mouse models of digenic and dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 2001;98:7718–7723. doi: 10.1073/pnas.141124198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feist RM, White MF, Jr, Skalka H, Stone EM. Choroidal neovascularization in a patient with adult foveomacular dystrophy and a mutation in the retinal degeneration slow gene (Pro 210 Arg) Am J Ophthalmol. 1994;118:259–260. doi: 10.1016/s0002-9394(14)72913-7. [DOI] [PubMed] [Google Scholar]
- 40.Kim RY, Dollfus H, Keen TJ, et al. Autosomal dominant pattern dystrophy of the retina associated with a 4-base pair insertion at codon 140 in the peripherin/RDS gene. Arch Ophthalmol. 1995;113:451–455. doi: 10.1001/archopht.1995.01100040067029. [DOI] [PubMed] [Google Scholar]
- 41.Felbor U, Schilling H, Weber BH. Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Hum Mutat. 1997;10:301–309. doi: 10.1002/(SICI)1098-1004(1997)10:4<301::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 42.Hunter DG, Fishman GA, Kretzer FL. Abnormal axonemes in X-linked retinitis pigmentosa. Arch Ophthalmol. 1988;106:362–368. doi: 10.1001/archopht.1988.01060130388028. [DOI] [PubMed] [Google Scholar]
- 43.Hunter DG, Fishman GA, Mehta RS, Kretzer FL. Abnormal sperm and photoreceptor axonemes in Usher's syndrome. Arch Ophthalmol. 1986;104:385–389. doi: 10.1001/archopht.1986.01050150085033. [DOI] [PubMed] [Google Scholar]
- 44.Connell GJ, Molday RS. Molecular cloning, primary structure, and orientation of the vertebrate photoreceptor cell protein peripherin in the rod outer segment disk membrane. Biochemistry. 1990;29:4691–4698. doi: 10.1021/bi00471a025. [DOI] [PubMed] [Google Scholar]
- 45.Evans K, Gregory CY, Wijesuriya SD, et al. Assessment of the phenotypic range seen in Doyne honeycomb retinal dystrophy. Arch Ophthalmol. 1997;115:904–910. doi: 10.1001/archopht.1997.01100160074012. [DOI] [PubMed] [Google Scholar]
- 46.Gorin MB, Breitner JC, de Jong PT, et al. The genetics of age-related macular degeneration. Mol Vis. 1999;5:29. [PubMed] [Google Scholar]