

Abstract
Post utero development of the mammary gland is a complex developmental process characterized by states of rapid cell proliferation (branching morphogenesis) followed by functional differentiation (lactation) and the consequent apoptosis (involution) of the secretory mammary epithelial cell. This process is cyclical, such that involution returns the mammary gland to a near-virgin-like state capable of responding to morphogenic cues with each consecutive pregnancy. Importantly, many of the regulatory processes which oversee mammary gland development are corrupted or otherwise compromised during the development of breast cancer. For example, Interferon Regulatory Factor 6 (IRF6) is a novel protein with growth inhibitory properties that was initially identified in mammary epithelial cells through its interaction with maspin, a known tumor suppressor in normal breast tissue. Recent findings from our laboratory suggest that IRF6 functions synergistically with maspin to regulate mammary epithelial cell differentiation by acting on the cell cycle. This perspective focuses on the possible involvement of IRF6 in promoting differentiation by regulating exit from the cell cycle and entry into the G(0) phase of cellular quiescence, and how these new findings shed light on normal mammary gland development and the initiation and progression of breast cancer.
Keywords: IRF6, Maspin, Cell Differentiation, Mammary Gland, Breast Cancer
Introduction
In 2002, Kondo and colleagues reported a link between Van der Woude Syndrome (VWS, OMIM #119300) and mutations in the gene cluster at 1p34 encoding the gene for Interferon Regulatory Factor 6. Since this seminal publication, multiple reports have been published demonstrating novel mutations in the Irf6 gene locus that cause VWS or its more severe counterpart Popliteal Pterygium Syndrome (PPS, OMIM #119500), but little has been reported regarding the possible functions of IRF6 in the development of PPS and VWS. Shortly thereafter, our laboratory reported a protein-protein interaction between IRF6 and maspin.1 Maspin (Mammary Serine Protease Inhibitor) is a class II tumor suppressor in normal breast tissue with anti-invasive properties in vitro and in vivo.2 Maspin is also known to play an important role during mammary gland development.3 Hence, we sought to define the function of IRF6 in cooperation with maspin in mammary epithelial cells and thereby shed light on the involvement of IRF6 in disease states such as VWS, PPS, and also breast cancer. We have since demonstrated that IRF6 expression follows an expression pattern in mammary epithelial cells similar to that of maspin. Both IRF6 and maspin are differentially expressed throughout differentiation of the mammary gland, with maximal expression of both proteins occurring during lactation, when the secretory mammary epithelial cells are quiescent and functionally differentiated.4 Furthermore, we have demonstrated that, similar to maspin, IRF6 is reduced or absent in a majority of breast carcinomas.1 These findings led us to hypothesize that maspin and IRF6 function in a coordinated manner to promote mammary epithelial cell differentiation, and that the loss of the maspin-IRF6 interaction may promote neoplastic transformation. Because coordination between differentiation and proliferation is critical for proper mammary gland development and function, and awry in breast cancer, we examined the regulation of IRF6 by the cell cycle. Our findings demonstrate the post-translational regulation of IRF6 expression and phosphorylation by the cell cycle and suggest that IRF6, in collaboration with maspin, may regulate mammary epithelial cell differentiation by promoting exit from the cell cycle and entry into the G(0) state of differentiated quiescence.
IRF6 and the Cell Cycle: Cause or Effect?
The cell cycle is an intricate, temporally organized system that allows for the supervised and tightly regulated process of cell division. The exquisite control of expression and regulation of factors involved in progression through the cell cycle is mediated in part by the proteasome, which regulates the timely expression of many cell cycle regulators, both protagonistic and antagonistic. The current literature contains many reports describing the various phases and checkpoints of the cell cycle throughout which these cell cycle regulators are differentially expressed. Upon completion of a successful cellular division, mammalian cells either reenter the cell cycle or exit the cycle to enter a physiological arrest known as G(0), or quiescence, which is a necessary step in cellular differentiation and cellular senescence. In many cases, such as terminal keratinocyte differentiation, commitment to differentiation is irreversible and results in eventual cell death. Likewise, during mammary gland differentiation, many secretory mammary epithelial cells succumb to cell death with the onset of post-lactational involution as the gland is remodeled to a virgin-like state. However, certain conditions, such as morphogenesis or wound repair, require that quiescent cells exit their state of dormancy and reenter the cell cycle. Although much is understood about the cell cycle with its various phases and checkpoints, less is known about the signals responsible for entry into or exit from the cell cycle in response to signals driving proliferation or differentiation.
Recent findings from our laboratory show the unique regulation of IRF6 phosphorylation and expression by the cell cycle. Cell cycle arrest, induced either by serum starvation, cell contact inhibition, or pharmacological cell cycle arrest, results in a significant increase in total IRF6 protein levels in cultured mammary epithelial cells (MCF-10A cells), with non-phosphorylated IRF6 serving as the prominent isoform. However, instigation of the cell cycle is associated with rapid (within 15 minutes) phosphorylation and subsequent ubiquitination and degradation of phosphorylated IRF6, such that by 1 hour following serum stimulation, IRF6 protein levels are significantly reduced.4 In comparison, cyclin D3 expression is detectable at two hours, with maximal expression occurring four hours following serum stimulation. This rapid IRF6 phosphorylation only occurs following the addition of complete growth medium (5% serum, insulin, hydrocortisone, EGF), whereas neither insulin, hydrocortisone and EGF, nor the serum replacement supplement MitoPLUS were able to induce rapid cellular proliferation and the concomitant phosphorylation of IRF6. These data suggest that IRF6 phosphorylation is induced as an important step toward entry into the cell cycle rather than activation of a mitogenic signal cascade. Following phosphorylation IRF6 is ubiquitinated and degraded in a proteasome-dependent manner and the regulation of IRF6 protein stability, via phosphorylation, appears to be the primary mechanism for regulating IRF6 expression. This was confirmed by pulse-chase experiments which demonstrated decreased protein stability in proliferating cells versus quiescent cells. Changes in IRF6 mRNA levels do not account for the increased expression of IRF6 protein in quiescent cells, demonstrating that increased transcription does not likely account for the increase in total IRF6 protein levels observed in these cells.4 Involvement of the cell cycle would suggest that cyclin dependent kinase (cdk) activity may be involved in IRF6 phosphorylation. Indeed, recent studies have demonstrated that cdk3 in association with cyclin C is involved in regulating exit from G(0).5 However, treatment of serum starved MCF-10A cells with the cdk inhibitor flavopiridol prior to the addition of serum failed to inhibit IRF6 phosphorylation following serum addition, suggesting that cdks are not involved (Fig. 1). Importantly, cdk3 function is not well understood, and although flavopiridol is considered a pan cdk inhibitor, a direct inhibitory effect of flavopiridol on cdk3 has not been reported and must be investigated further.
Figure 1. IRF6 phosphorylation is unaffected by the cdk inhibitor flavopiridol.
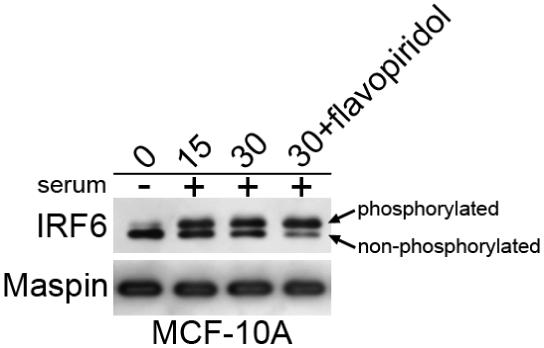
MCF-10A mammary epithelial cells were serum starved for 48 hours. Serum-starved cells were pre-treated with flavopiridol for one hour at a 400 nanomolar concentration prior to the addition of serum. Cells were harvested at 15 and 30 minutes following serum addition according to standard techniques.4 Phosphorylated and non-phosphorylated forms of IRF6 are demarcated by the arrows. Maspin levels were unchanged and used as a loading control.
Together, our findings support two distinct models which implicate IRF6 in either entry into the G(0) phase or exit from G(0) and entry into G(1). Support for a role for IRF6 in promoting entry into G(1) from G(0) stems from the paradigm established by other IRF family members. Generally speaking, IRFs such as IRF3 are expressed latently within the cytosol in a non-phosphorylated state. Activation signals (from Toll-like receptor signaling cascades) result in their phosphorylation and subsequent nuclear translocation. For several IRFs, their phosphorylation not only renders them active by allowing dimerization and translocation to the nucleus, but also promotes their polyubiquitination and targeted proteasomal degradation.6-8 If IRF6 followed this established paradigm, then its early phosphorylation (and putative activation) as the cell prepares to enter the cell cycle would suggest that IRF6 functions to promote exit from the quiescent state in preparation for cell division. However, the fact that phosphorylated IRF6 is not readily observed in the nucleus at this time would argue that IRF6 does not follow stereotypical IRF activation pathways. Also, it is unclear whether IRF6 phosphorylation results in its activation or whether it is merely a signal for ubiqitination and subsequent degradation. Furthermore, the re-expression of IRF6 in breast cancer cells promotes cell cycle arrest, which supports the hypothesis that a buildup of IRF6 induces entry into the G(0) phase. Further support for this model stems from two recent reports demonstrating the inability of keratinocytes to terminally differentiate in the absence of functional IRF6.9, 10 Mice which fail to express functional IRF6 die shortly after birth due to complications from a severe skin defect. Histological analysis of the epidermis demonstrates the lack of granular and cornified epidermal layers while p63 expression and BrdU staining are visible throughout the suprabasal layer, indicating that these keratinocytes are unable to exit the cell cycle, effectively blocking terminal differentiation.
Similarities between IRF6, 14-3-3σ, and IKKα Knockout Mice
The hallmark phenotype of the IRF6 knockout mouse is the severe skin abnormality along with abnormal limb and craniofacial development, likely resulting from the skin phenotype. Very similar phenotypes are shared by at least two other knockout mouse models: the repeated epilation (Er/Er) mouse recently identified as an insertion mutation in the Stratifin gene (also called 14-3-3σ), and mice deficient in nuclear IκB kinase α (IKKα).11-14 Embryos from each of these groups exhibit shiny, taut skin stemming from the dysfunctional terminal differentiation of the epidermal keratinocytes, and similar limb and skeletal defects. Histological examination of the epidermis of Irf6 null embryos revealed an expanded spinous layer which aberrantly expressed Keratin 14 and p63, both markers of the basal layer which are normally silenced during cell cycle arrest and terminal differentiation of the desquamating keratinocytes. Furthermore, BrdU incorporation into cells throughout the spinous layer demonstrated severe hyperplasia and aberrant cellular proliferation, which are also evident in (Er/Er) and IKKα(-/-) knockout mice.
IKKα is one of two catalytic subunits of the IκB kinase involved in the regulation of NF-κB activation. IKKα is a critical regulator of keratinocyte differentiation and it has been demonstrated that this function is independent of IKKα kinase function, suggesting an NF-κB-independent pathway.15 Although the exact mechanism through which IKKα regulates keratinocyte differentiation is unclear, its involvement in regulating multiple cell cycle-related proteins has been sufficiently demonstrated.15-18 Importantly, a link has recently been identified between IKKα and 14-3-3σ, in which IKKα prevents the epigenetic silencing of 14-3-3σ by blocking 14-3-3σ promoter methylation.19 14-3-3σ is a member of the 14-3-3 family of phosphoserine/phosphothreonine binding proteins which bind and sequester a wide range of phosphorylated proteins, thereby regulating their localization and function.20 14-3-3σ expression is specific for keratinocytes and other epithelial cells and is often down-regulated in epithelial-derived cancers.21-23 14-3-3σ can be induced by p53 and acts specifically as a G(2)/M cell cycle checkpoint gene by sequestering CDC2-cyclin B complexes in the cytoplasm.24, 25
The similarities between mice mutant for IRF6, 14-3-3σ, and IKKα prompted researchers to examine the possible existence of linear pathways involving IRF6 and either 14-3-3σ or IKKα. Microarray analysis of Irf6-null embryos demonstrated normal expression of IKKα and slightly increased 14-3-3σ expression, suggesting that IRF6 is not necessary for their proper expression.10 Furthermore, no detectable protein interactions were identified between IRF6, 14-3-3σ and IKKα.10 However, a cross between mice heterozygous for mutated Irf6 and 14-3-3σ (Irf6+/R84C Sfn+/Er) induced a phenotype not observed in either of the heterozygous strains but which was akin to, albeit less severe than, the phenotype observed in homozygous null embryos. These embryos exhibit smooth, taut skin, syndactyly of the fore- and hindpaws, and similar cranio- and orofacial abnormalites, from which the authors hypothesize a genetic interaction between IRF6 and 14-3-3σ in regulating keratinocyte differentiation.9 Because 14-3-3σ plays a primary role in the regulation of cdc2 and cyclin B1 activity, these results sustain our findings implicating IRF6 in cell cycle regulation and support our hypothesis that IRF6 advances cellular differentiation by promoting entry into the G(0) stage of the cell cycle.
IRF6 as a Transcription Factor
Perhaps the greatest conundrum regarding IRF6 is our inability to consistently detect IRF6 in the nucleus. IRF6 contains a stereotypical IRF DNA binding domain which consists of a helix-turn-helix or “winged-helix” motif centered around a highly conserved spatial repeat of five tryptophan residues.1 This winged-helix domain recognizes the consensus DNA sequence 5′-A/GNGAAANNGAAACT-3′, termed the Interferon Stimulated Response Element (ISRE).26 Crystal structure analysis of the IRF-DNA interaction has shown that the repeating sequence GAAA of the ISRE is the primary site of interaction, and that the IRFs typically must form dimers in order to properly bind DNA.27 With no known target genes, the current lack of evidence for nuclear IRF6 suggests that IRF6 may not function as a transcription factor. However, support for an IRF6-DNA interaction originates from the fact that mutations in the Irf6 gene affecting amino acids that are predicted to interact with DNA tend to result in a more severe phenotype associated with PPS, compared to VWS.28
Because we do not yet understand the full scope of IRF6 function, it is possible that our inability to identify nuclear IRF6 reflects our limited understanding of IRF6 activation. The focus of IRF6 research to date has emphasized its role in the development of specific epidermal derived tissues. However, very little is known about how IRF6 might respond to the canonical signaling of Toll-like receptors (TLR) following antigen presentation, which is the primary means of IRF activation.29 Thought initially to solely regulate immune cell differentiation and function, TLR signaling pathways, commonly through TLR3 and TLR4, have been identified as crucial for proper maintenance and function of a host of epithelial cell types including gut, airway, kidney epithelium and epidermal keratinocytes.30-33 Although there is currently little information available with respect to TLR expression or function in human mammary epithelial cells, recent findings have demonstrated a role for TLR function in regulating cell cycle progression and it is possible that TLRs may function similarly in mammary epithelial cells. It is well established that type I interferons (IFNα and IFNβ) can exert a direct antiproliferative effect through control of the expression of cell cycle protagonists, such as c-myc, and antagonists, such as the CDK inhibitor p27.34, 35 Work by Hasan and colleagues has suggested that signaling through TLR3 and TLR4 following stimulation with their respective ligands polyI:C (a synthetic double stranded RNA) and lipopolysaccharide (LPS) can induce entry into the cell cycle, but only in the absence of type I IFN signaling, which is also regulated through TLR signaling pathways.36, 37 Because IRFs are known regulators of type I IFN, it is possible that IRF6 functions through canonical TLR-IRF signaling pathways to induce type I IFN as a means of promoting quiescence and differentiation. To examine this, we tested whether TLR3 mRNA is expressed in MCF-10A cells and if TLR3 activation by polyI:C induces changes to IRF6 expression, phosphorylation, or localization. We show that TLR3 mRNA is expressed in MCF-10A cells and that TLR3 mRNA levels increase in quiescent cells versus proliferating cells (Fig. 2a). Treatment with polyI:C resulted in an increase in TLR3 mRNA, demonstrating a positive feedback mechanism regulating TLR3 expression in these cells. Importantly, treatment with polyI:C also induced the translocation of a small fraction of the total IRF6 to the nucleus (Fig. 2b). Notably, the predominant IRF6 isoform observed in the nucleus is the phosphorylated form, although the non-phosphorylated form is also present. Changes in cytoplasmic IRF6 phosphorylation can also be observed. Hence, signaling through TLR3 can induce IRF6 translocation to the nucleus, where IRF6 may act to promote quiescence and differentiation, perhaps through the regulation of type I IFN or IFN-associated genes. Once in the nucleus, phosphorylated IRF6 would be targeted for ubiquitination and subsequent proteasomal degradation. Such a scenario portrays the regulation of IRF6 through canonical IRF signaling pathways as a means of regulating cellular growth and differentiation.
Figure 2. IRF6 responds to treatment with the TLR3 agonist polyI:C.
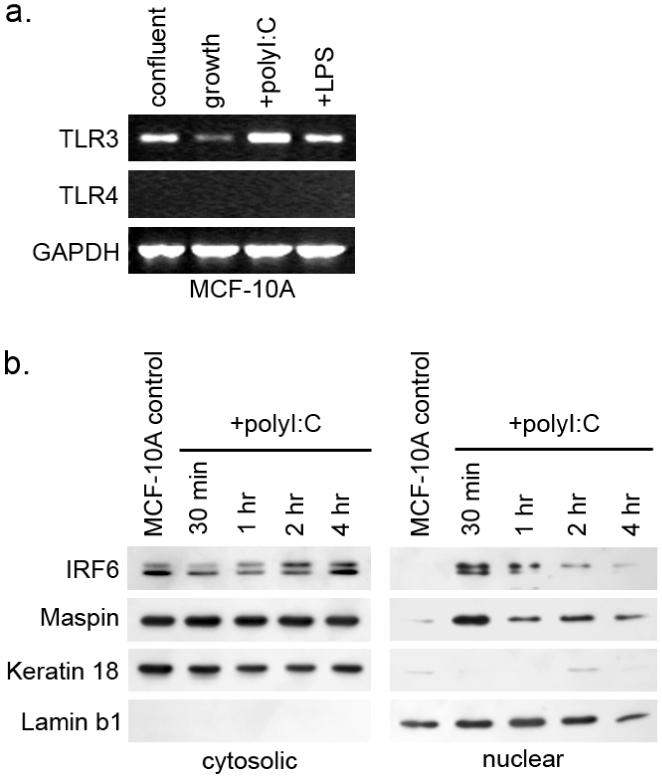
A) TLR3 mRNA is detectable by RT-PCR in MCF-10A cells whereas TLR4 mRNA was undetectable. Expression of TLR3 mRNA was reduced in log-phase proliferating cells (growth) and increased in confluent cells. Treatment of MCF-10A cells with the TLR3 agonist polyI:C (a synthetic double-stranded RNA construct) induced an increase in TLR3 mRNA whereas lipopolysaccharide (LPS), an agonist for TLR4, did not affect TLR3 mRNA expression. B) Western blot analysis of MCF-10A cytosolic and nuclear fractions following treatment with polyI:C. IRF6 is maximally expressed in the nuclear fraction 30 minutes after polyI:C treatment. Keratin 18 was used as a cytosolic marker and Lamin b1 was used as a nuclear marker. Fractionation was performed as previously described.1
Another possible link exists between IRF6, TLR signaling and cell cycle regulation. As mentioned previously, IKKα is a critical regulator of keratinocyte differentiation. As a catalytic subunit of the IKK complex, IKKα activation is an important step in TLR-mediated NF-κB activation. It has been reported that IKKα can reduce overall p27 levels independent of IFN signaling and the TLR-NF-κB signaling pathway may represent an additional TLR-mediated pathway involved in regulating cell cycle entry and exit.37 Therefore, because IKKα is a known regulator of 14-3-3σ, it is possible that IRF6, 14-3-3σ, and IKKα function through similar pathways, perhaps in coordination with TLR3 signaling, to regulate specific cell cycle events that are necessary for exit from the cell cycle and entry into the G(0) differentiation phase.
IRF6 and Maspin
Our identification of the protein interaction between IRF6 and maspin may be critical to our understanding and the eventual elucidation of IRF6 function. Maspin was first reported in 1994 as a serpin with tumor suppressive properties. Maspin was initially isolated through subtractive hybridization and differential display analysis as a 42kDa protein that is expressed in normal mammary epithelial cells but reduced or absent in breast carcinomas.2 Further research led to maspin’s characterization as a class II tumor suppressor based on its ability to inhibit cell invasion, promote apoptosis and inhibit angiogenesis.38-40 Recent insight into maspin function has been gained through microarray analysis of breast cancer cells following the re-expression of maspin, which revealed several distinct protein networks which are altered following the ectopic re-expression of maspin in metastatic breast cancer cells.41 Additionally, phenotypic changes characterized by a reversion from a fibroblast-like phenotype (characteristic of an epithelial-to-mesenchymal transformation) back to a more epithelial-like phenotype, concomitant with changes in cytoskeletal organization, were also observed.42, 43 In addition, many of the highly induced genes identified by microarray can be linked to pathways involved in response to cellular stress or injury. Cumulatively, these data suggest an important role for maspin in mediating the cellular stress response by regulating aspects of cell growth and differentiation. In support of this, maspin re-expression results in the up-regulation of multiple genes directly associated with cellular proliferation and the cell cycle, including the cyclin-dependent kinase inhibitors p18 and CDKN3, the E2F transcription factor 1 (E2F1) and BRCA1b, which has been shown to associate with E2F transcription factors in vitro.41, 44
Interestingly, one of the most highly induced genes following maspin re-expression in MDA-MB-231 breast cancer cells is Brm (Smarca2), a catalytic subunit of the SWI/SNF chromatin remodeling complex.41 Although SWI/SNF function is most commonly associated with transcriptional regulation via chromatin remodeling, recent data indicate a clear role for Brm in regulating certain aspects of the cell cycle, namely entry into the G(0) quiescent state.45, 46 Embryonic fibroblasts from mice lacking a functional Brm gene fail to show proper growth arrest at confluence and are unable to enter the canonical G(0) state as evidenced by the aberrant expression of c-myc and several cyclins following serum starvation.46 Intriguingly, Brm expression in wild-type murine fibroblasts during cellular proliferation mirrors our reported expression of IRF6 in proliferating MCF-10A mammary epithelial cells in that Brm is highly expressed in growth arrested cells, but significantly reduced in proliferating cells.47 Furthermore, Brm is only induced upon differentiation of murine embryonic ES and F9 cells, and Brm expression is generally lost during transformation.47, 48 Because both Brm and IRF6 follow a similar expression pattern during progression through the cell cycle, and both proteins are thought to be involved in cellular differentiation and are lost during cellular transformation, there may be an unidentified link between the functions of these two proteins, and it is possible that maspin may act as the functional link promoting differentiation and inducing quiescence.
IRF6 and Cancer
Because the pathways controlling the decision between cell cycle progression and cell cycle exit are defective in most human tumors, our ultimate goal is to evaluate the potential role of IRF6 in the development and progression of breast cancer.49 Several IRF family members have been reported to possess tumor suppressive functions, in part through the regulation of growth-suppressive cellular processes such as cell cycle regulation and induction of apoptosis.50-52 Although aberrant expression and regulation of some IRFs have been studied in the context of cancer initiation and progression, the clinical relevance of these findings is unclear. Our studies have demonstrated that IRF6 is differentially expressed in normal versus neoplastic mammary tissue.1 These studies indicate that IRF6 is significantly reduced at both the mRNA and protein levels in poorly aggressive human breast cancer cell lines (MCF-7, T47-D) and completely absent in aggressive and metastatic breast cancer cell lines (MDA-MB-231 and HS578T). This expression pattern is corroborated in situ as assessed by immunohistochemistry of patient samples, which demonstrate a reduction in IRF6 immunoreactivity in ductal carcinoma in situ (DCIS) lesions and a complete loss of IRF6 in invasive ductal carcinoma. This stepwise decline in IRF6 message and protein levels throughout the initiation of a neoplastic lesion and progression toward an aggressive, metastatic phenotype has led us to speculate that the loss of IRF6 is an important step in cancer development, perhaps by allowing uncontrolled cellular proliferation and dedifferentiation of the neoplastic cell. Thus, it is possible that the pathway(s) which regulate IRF6 expression may represent pathways which could be therapeutically targeted to induce cell cycle arrest and promote differentiation, thus reverting the cell to a less aggressive, quiescent phenotype. Indeed, the ectopic re-expression of IRF6 (via adenoviral infection) significantly reduced cellular proliferation in both poorly invasive and highly invasive breast cancer cell lines and this effect was synergistically augmented in the presence of maspin.4 These data highlight a potentially important means of regulating tumor cell growth and support our hypothesis that IRF6 is a key regulator of mammary epithelial cell differentiation which functions by promoting quiescence through entry into the G(0) phase of the cell cycle.
Perspective
IRF6 is a unique member of the IRF transcription factor family. To date, its expression has only been reported in epithelial-derived tissues such as keratinocytes, the medial edge epithelium of the fusing palate, and mammary and prostate epithelial cells. IRF6 appears to be involved in the regulation of the cell cycle, specifically by promoting entry into the G(0) state (illustrated in Fig. 3). Because of its putative role in quiescence and differentiation, pathways which regulate IRF6 expression and function may represent valid therapeutic and/or diagnostic targets for breast cancer.
Figure 3. A model illustrating the involvement of IRF6 in promoting quiescence and differentiation by instigating entry into the G(0) phase of cellular quiescence.

Increasing IRF6 levels promote exit from the cell cycle. As cells proliferate, IRF6 is ubiquitinated and targeted for proteasomal degradation.
Acknowledgements
The authors wish to thank Dr. Zhila Khalkhali-Ellis, Dr. Daniel Abbott and Dr. Brian Schutte for helpful scientific discussions. This research was supported by NIH CA75681.
List of Abbreviations
- IRF
Interferon Regulatory Factor
- EGF
epidermal growth factor
- cdk
cyclin dependent kinase
- BrdU
5-Bromo-2-deoxyuridine
- VWS
van der woude syndrome
- PPS
popliteal pterygium syndrome
References
- 1.Bailey CM, Khalkhali-Ellis Z, Kondo S, Margaryan NV, Seftor RE, Wheaton WW, Amir S, Pins MR, Schutte BC, Hendrix MJ. Mammary serine protease inhibitor (Maspin) binds directly to interferon regulatory factor 6: identification of a novel serpin partnership. J Biol Chem. 2005;280:34210–7. doi: 10.1074/jbc.M503523200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou Z, Anisowicz A, Hendrix MJ, Thor A, Neveu M, Sheng S, Rafidi K, Seftor E, Sager R. Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science. 1994;263:526–9. doi: 10.1126/science.8290962. [DOI] [PubMed] [Google Scholar]
- 3.Zhang M, Magit D, Botteri F, Shi HY, He K, Li M, Furth P, Sager R. Maspin plays an important role in mammary gland development. Dev Biol. 1999;215:278–87. doi: 10.1006/dbio.1999.9442. [DOI] [PubMed] [Google Scholar]
- 4.Bailey CM, Abbott DE, Margaryan NV, Khalkhali-Ellis Z, Hendrix MJ. Interferon regulatory factor 6 promotes cell cycle arrest and is regulated by the proteasome in a cell cycle-dependent manner. Mol Cell Biol. 2008;28:2235–43. doi: 10.1128/MCB.01866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren S, Rollins BJ. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117:239–51. doi: 10.1016/s0092-8674(04)00300-9. [DOI] [PubMed] [Google Scholar]
- 6.Nakagawa K, Yokosawa H. Degradation of transcription factor IRF-1 by the ubiquitin-proteasome pathway. The C-terminal region governs the protein stability. Eur J Biochem. 2000;267:1680–6. doi: 10.1046/j.1432-1327.2000.01163.x. [DOI] [PubMed] [Google Scholar]
- 7.Xiong H, Li H, Kong HJ, Chen Y, Zhao J, Xiong S, Huang B, Gu H, Mayer L, Ozato K, Unkeless JC. Ubiquitin-dependent degradation of interferon regulatory factor-8 mediated by Cbl down-regulates interleukin-12 expression. J Biol Chem. 2005;280:23531–9. doi: 10.1074/jbc.M414296200. [DOI] [PubMed] [Google Scholar]
- 8.Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol. 2006;7:598–605. doi: 10.1038/ni1347. [DOI] [PubMed] [Google Scholar]
- 9.Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot-Handford RP, Shore P, Whitmarsh A, Dixon MJ. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat Genet. 2006;38:1329–34. doi: 10.1038/ng1894. [DOI] [PubMed] [Google Scholar]
- 10.Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, Murray JC, Schutte BC. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat Genet. 2006;38:1335–40. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herron BJ, Liddell RA, Parker A, Grant S, Kinne J, Fisher JK, Siracusa LD. A mutation in stratifin is responsible for the repeated epilation (Er) phenotype in mice. Nat Genet. 2005;37:1210–2. doi: 10.1038/ng1652. [DOI] [PubMed] [Google Scholar]
- 12.Holbrook KA, Dale BA, Brown KS. Abnormal epidermal keratinization in the repeated epilation mutant mouse. J Cell Biol. 1982;92:387–97. doi: 10.1083/jcb.92.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guenet JL, Salzgeber B, Tassin MT. Repeated epilation: a genetic epidermal syndrome in mice. J Hered. 1979;70:90–4. doi: 10.1093/oxfordjournals.jhered.a109223. [DOI] [PubMed] [Google Scholar]
- 14.Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M. Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999;284:316–20. doi: 10.1126/science.284.5412.316. [DOI] [PubMed] [Google Scholar]
- 15.Descargues P, Sil AK, Sano Y, Korchynskyi O, Han G, Owens P, Wang XJ, Karin M. IKKalpha is a critical coregulator of a Smad4-independent TGFbeta-Smad2/3 signaling pathway that controls keratinocyte differentiation. Proc Natl Acad Sci U S A. 2008;105:2487–92. doi: 10.1073/pnas.0712044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blazkova H, von Schubert C, Mikule K, Schwab R, Angliker N, Schmuckli-Maurer J, Fernandez PC, Doxsey S, Dobbelaere DA. The IKK inhibitor BMS-345541 affects multiple mitotic cell cycle transitions. Cell Cycle. 2007;6:2531–40. doi: 10.4161/cc.6.20.4807. [DOI] [PubMed] [Google Scholar]
- 17.Prajapati S, Tu Z, Yamamoto Y, Gaynor RB. IKKalpha regulates the mitotic phase of the cell cycle by modulating Aurora A phosphorylation. Cell Cycle. 2006;5:2371–80. doi: 10.4161/cc.5.20.3359. [DOI] [PubMed] [Google Scholar]
- 18.Schneider G, Saur D, Siveke JT, Fritsch R, Greten FR, Schmid RM. IKKalpha controls p52/RelB at the skp2 gene promoter to regulate G1- to S-phase progression. Embo J. 2006;25:3801–12. doi: 10.1038/sj.emboj.7601259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu F, Xia X, Liu B, Shen J, Hu Y, Person M. IKKalpha shields 14-3-3sigma, a G(2)/M cell cycle checkpoint gene, from hypermethylation, preventing its silencing. Mol Cell. 2007;27:214–27. doi: 10.1016/j.molcel.2007.05.042. [DOI] [PubMed] [Google Scholar]
- 20.Dougherty MK, Morrison DK. Unlocking the code of 14-3-3. J Cell Sci. 2004;117:1875–84. doi: 10.1242/jcs.01171. [DOI] [PubMed] [Google Scholar]
- 21.Dellambra E, Golisano O, Bondanza S, Siviero E, Lacal P, Molinari M, D’Atri S, De Luca M. Downregulation of 14-3-3sigma prevents clonal evolution and leads to immortalization of primary human keratinocytes. J Cell Biol. 2000;149:1117–30. doi: 10.1083/jcb.149.5.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki H, Itoh F, Toyota M, Kikuchi T, Kakiuchi H, Imai K. Inactivation of the 14-3-3 sigma gene is associated with 5′ CpG island hypermethylation in human cancers. Cancer Res. 2000;60:4353–7. [PubMed] [Google Scholar]
- 23.Leffers H, Madsen P, Rasmussen HH, Honore B, Andersen AH, Walbum E, Vandekerckhove J, Celis JE. Molecular cloning and expression of the transformation sensitive epithelial marker stratifin. A member of a protein family that has been involved in the protein kinase C signalling pathway. J Mol Biol. 1993;231:982–98. doi: 10.1006/jmbi.1993.1346. [DOI] [PubMed] [Google Scholar]
- 24.Hermeking H, Lengauer C, Polyak K, He TC, Zhang L, Thiagalingam S, Kinzler KW, Vogelstein B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 25.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–20. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 26.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–55. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 27.Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol. 2003;10:913–21. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 28.Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, de Lima RL, Daack-Hirsch S, Sander A, McDonald-McGinn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–9. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moynagh PN. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005;26:469–76. doi: 10.1016/j.it.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 30.Becker MN, Diamond G, Verghese MW, Randell SH. CD14-dependent lipopolysaccharide-induced beta-defensin-2 expression in human tracheobronchial epithelium. J Biol Chem. 2000;275:29731–6. doi: 10.1074/jbc.M000184200. [DOI] [PubMed] [Google Scholar]
- 31.Furrie E, Macfarlane S, Thomson G, Macfarlane GT. Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology. 2005;115:565–74. doi: 10.1111/j.1365-2567.2005.02200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfs TG, Buurman WA, van Schadewijk A, de Vries B, Daemen MA, Hiemstra PS, van ’t Veer C. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. 2002;168:1286–93. doi: 10.4049/jimmunol.168.3.1286. [DOI] [PubMed] [Google Scholar]
- 33.Mempel M, Voelcker V, Kollisch G, Plank C, Rad R, Gerhard M, Schnopp C, Fraunberger P, Walli AK, Ring J, Abeck D, Ollert M. Toll-like receptor expression in human keratinocytes: nuclear factor kappaB controlled gene activation by Staphylococcus aureus is toll-like receptor 2 but not toll-like receptor 4 or platelet activating factor receptor dependent. J Invest Dermatol. 2003;121:1389–96. doi: 10.1111/j.1523-1747.2003.12630.x. [DOI] [PubMed] [Google Scholar]
- 34.Einat M, Resnitzky D, Kimchi A. Close link between reduction of c-myc expression by interferon and, G0/G1 arrest. Nature. 1985;313:597–600. doi: 10.1038/313597a0. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Y, Wang S, Gobl A, Oberg K. Inhibition of CDK2, CDK4 and cyclin E and increased expression of p27Kip1 during treatment with interferon-alpha in carcinoid tumor cells. J Biol Regul Homeost Agents. 1999;13:207–15. [PubMed] [Google Scholar]
- 36.Hasan UA, Trinchieri G, Vlach J. Toll-like receptor signaling stimulates cell cycle entry and progression in fibroblasts. J Biol Chem. 2005;280:20620–7. doi: 10.1074/jbc.M500877200. [DOI] [PubMed] [Google Scholar]
- 37.Hasan UA, Caux C, Perrot I, Doffin AC, Menetrier-Caux C, Trinchieri G, Tommasino M, Vlach J. Cell proliferation and survival induced by Toll-like receptors is antagonized by type I IFNs. Proc Natl Acad Sci U S A. 2007;104:8047–52. doi: 10.1073/pnas.0700664104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheng S, Carey J, Seftor EA, Dias L, Hendrix MJ, Sager R. Maspin acts at the cell membrane to inhibit invasion and motility of mammary and prostatic cancer cells. Proc Natl Acad Sci U S A. 1996;93:11669–74. doi: 10.1073/pnas.93.21.11669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang M, Volpert O, Shi YH, Bouck N. Maspin is an angiogenesis inhibitor. Nat Med. 2000;6:196–9. doi: 10.1038/72303. [DOI] [PubMed] [Google Scholar]
- 40.Jiang N, Meng Y, Zhang S, Mensah-Osman E, Sheng S. Maspin sensitizes breast carcinoma cells to induced apoptosis. Oncogene. 2002;21:4089–98. doi: 10.1038/sj.onc.1205507. [DOI] [PubMed] [Google Scholar]
- 41.Bailey CM, Khalkhali-Ellis Z, Seftor EA, Hendrix MJ. Biological functions of maspin. J Cell Physiol. 2006;209:617–24. doi: 10.1002/jcp.20782. [DOI] [PubMed] [Google Scholar]
- 42.Chen EI, Florens L, Axelrod FT, Monosov E, Barbas CF, 3rd, Yates JR, 3rd, Felding-Habermann B, Smith JW. Maspin alters the carcinoma proteome. Faseb J. 2005;19:1123–4. doi: 10.1096/fj.04-2970fje. [DOI] [PubMed] [Google Scholar]
- 43.Odero-Marah VA, Khalkhali-Ellis Z, Chunthapong J, Amir S, Seftor RE, Seftor EA, Hendrix MJ. Maspin regulates different signaling pathways for motility and adhesion in aggressive breast cancer cells. Cancer Biol Ther. 2003;2:398–403. doi: 10.4161/cbt.2.4.471. [DOI] [PubMed] [Google Scholar]
- 44.Cui JQ, Wang H, Reddy ES, Rao VN. Differential transcriptional activation by the N-terminal region of BRCA1 splice variants BRCA1a and BRCA1b. Oncol Rep. 1998;5:585–9. doi: 10.3892/or.5.3.585. [DOI] [PubMed] [Google Scholar]
- 45.Chiba H, Muramatsu M, Nomoto A, Kato H. Two human homologues of Saccharomyces cerevisiae SWI2/SNF2 and Drosophila brahma are transcriptional coactivators cooperating with the estrogen receptor and the retinoic acid receptor. Nucleic Acids Res. 1994;22:1815–20. doi: 10.1093/nar/22.10.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coisy-Quivy M, Disson O, Roure V, Muchardt C, Blanchard JM, Dantonel JC. Role for Brm in cell growth control. Cancer Res. 2006;66:5069–76. doi: 10.1158/0008-5472.CAN-05-0596. [DOI] [PubMed] [Google Scholar]
- 47.Muchardt C, Bourachot B, Reyes JC, Yaniv M. ras transformation is associated with decreased expression of the brm/SNF2alpha ATPase from the mammalian SWI-SNF complex. Embo J. 1998;17:223–31. doi: 10.1093/emboj/17.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.LeGouy E, Thompson EM, Muchardt C, Renard JP. Differential preimplantation regulation of two mouse homologues of the yeast SWI2 protein. Dev Dyn. 1998;212:38–48. doi: 10.1002/(SICI)1097-0177(199805)212:1<38::AID-AJA4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 49.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 50.Barnes BJ, Kellum MJ, Pinder KE, Frisancho JA, Pitha PM. Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res. 2003;63:6424–31. [PubMed] [Google Scholar]
- 51.Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW, Taniguchi T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell. 1994;77:829–39. doi: 10.1016/0092-8674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 52.Kim TY, Lee KH, Chang S, Chung C, Lee HW, Yim J, Kim TK. Oncogenic potential of a dominant negative mutant of interferon regulatory factor 3. J Biol Chem. 2003;278:15272–8. doi: 10.1074/jbc.M205792200. [DOI] [PubMed] [Google Scholar]