

Abstract
Experimental allergic encephalomyelitis (EAE) is a T cell-mediated autoimmune disease model of multiple sclerosis (MS). Signal transducer and activator of transcription 4 (Stat4) is a transcription factor activated by interleukin 12 (IL-12) and IL-23, two cytokines known to play important roles in the pathogenesis of EAE by inducing T cells to secrete IFN-_ and IL-17 respectively. We and others have shown earlier that therapeutic intervention or targeted disruption of Stat4 was effective in ameliorating EAE. Recently, a splice variant of Stat4 termed Stat4β has been characterized that lacks 44 amino acids at the C-terminus of the full length Stat4α. In this study we examined whether T cells expressing either isoform could impact the pathogenesis of EAE. We found that transgenic mice expressing Stat4βon a Stat4-deficient background develop an exacerbated EAE compared to wild-type mice following immunization with MOGp35–55 peptide, while Stat4α transgenic mice have greatly attenuated disease. The differential development of EAE in transgenic mice correlates with increased IFNγ and IL-17 in Stat4β-expressing cells in situ, contrasting increased IL-10 production by Stat4α-expressing cells. This study demonstrates that Stat4 isoforms differentially regulate inflammatory cytokines in association with distinct effects on the onset and severity of EAE.
Keywords: Autoimmunity, EAE/MS, Inflammation, Cytokine, Stat4 signaling
Introduction
Multiple Sclerosis (MS) is an inflammatory demyelinating disease of the central nervous system (CNS) that afflicts more than one million people worldwide (1, 2). The disease usually begins in young adults and affects women more frequently than men (3). About 30% of MS patients develop clinical paralysis and become wheel chair-bound for the rest of their lives (1, 2). There is currently no medical treatment available that can cure MS. The destruction of the oligodendrocyte myelin sheath and axonal loss in the CNS are the pathological hall-marks of MS (4). Although the etiology of MS remains unknown, it is generally viewed as an organ-specific autoimmune disease, mediated by myelin-reactive T cells in the CNS. Activation of immune cells, secretion of inflammatory cytokines and differentiation of encephalitogenic T cells are key processes associated with the pathogenesis of MS (1–4). Experimental allergic encephalomyelitis (EAE) is a CD4+ Th1/Th17 cell-mediated inflammatory demyelinating autoimmune disease of the CNS (5). EAE can be induced in susceptible animals by immunization with whole brain homogenate and purified neural antigens such as myelin basic protein (MBP), proteolipid protein (PLP) and myelin oligodendrocyte glycoprotein (MOG) or adoptive transfer of neural antigen-specific T cells. The clinical and pathological features of EAE show close similarity to human MS and therefore it has commonly been used as an animal model to study the mechanisms of MS pathogenesis and to test the efficacy of potential therapeutic agents for the treatment of MS (5, 6).
The pathogenesis of EAE/MS is a complex process involving activation of antigen presenting cells (APCs), differentiation of neural antigen-specific T cells and secretion of inflammatory cytokines in the CNS (4–6). Interleukin-12 is a 70 kD heterodimeric cytokine composed of IL-12p40 and IL-12p35 subunits (7). IL-23 is a related heterodimeric cytokine composed of IL-12p40 and IL-23p19 subunits (8). IL-12 and IL-23 are produced by APCs which play critical roles in the pathogenesis of EAE/MS (9–11). Signaling through specific receptors that share the IL-12Rβ1 chain, IL-12 and IL-23 induce tyrosine phosphorylation and activation of Janus kinases, Jak-2 and Tyk-2, and Signal Transducer and Activator of Transcription, including Stat3, Stat4 and Stat5, resulting in the generation of effector T cells and production of inflammatory cytokines (12–14). We and others have shown earlier that the inhibition of IL-12/IL-23 production or IL-12/IL-23 signaling was effective in preventing the differentiation of Th1/Th17 cells and pathogenesis of EAE (9–11, 15–21). Gene targeting studies have demonstrated an essential role for Stat4 in mediating IL-12/23-induced differentiation of Th1/Th17 cells and pathogenesis of EAE (22–24). Thus Stat4 remains a central target for altering the downstream effects of IL-12 and IL-23 and the effector functions of Th1 and Th17 cells in autoimmune diseases.
Many of the Stat proteins exist as alternatively spliced isoforms which vary at the C-terminal domain (25). The full-length proteins are referred to as _, while the forms lacking the putative transactivation domain are designated _. While the _ isoforms of Stat1 and Stat5 have been shown to function as dominant-negative factors, Stat3β and Stat4β are capable of activating gene transcription (26–29). In a recent study, we generated mice that express either Stat4α or Stat4_, an isoform that lacks 44 amino acids at the C-terminus, as a T cell specific transgene on a Stat4-deficient genetic background. Purified CD4+ T cells from these mice were capable of generating IFNγ-secreting Th1 cells in vitro (26). However, the role of Stat4 isoforms in the pathogenesis of organ-specific autoimmune diseases has not been examined.
In this study we used Stat4−/− transgenic mice expressing Stat4α or Stat4β to study their distinct roles in the pathogenesis of EAE. We found that the Stat4β transgenic mice develop an exacerbated EAE in association with an increased expression of inflammatory cytokines. The Stat4α transgenic mice remain resistant to EAE, suggesting that Stat4β is more efficient than Stat4α in mediating the pathogenesis of EAE.
Materials and Methods
Animals
The C57BL/6 mice were purchased from Harlan (Indianapolis, IN). The Stat4−/− mouse in C57BL/6 background was generated as described earlier (26, 30). The transgenic mice expressing Stat4α or Stat4β genes were generated as described earlier (26). The mice were maintained in the animal care facility at Methodist Research Institute. All animal protocols used in the experiments were approved by the Institutional Animal Care and Use Committee.
Reagents
The 21 amino acid peptide [MEVGWYRSPFSRVVHLYRNGK] corresponding to mouse MOGp35–55 was obtained from Genemed Synthesis. Inc. (San Francisco, CA). Murine recombinant IL-17, IFNγ and IL-10 were purchased from R&D Systems, Inc. (Minneapolis, MN). The biotin/FITC-conjugated anti-IL-17, anti-IFNγ and anti-IL-10 antibodies were purchased from e-Bioscience. All reagents for qRT-PCR were purchased from Applied Biosystems (Foster City, CA).
Induction of EAE
To induce EAE, 4 to 6 weeks old female mice (5 per group) were immunized (s.c.) with 100 μg of MOGp35–55 peptide antigen in 150 μl emulsion of Incomplete Freund’s Adjuvant containing 50 μg/ml heat-killed Mycobacterium tuberculosis (H37Ra, Difco Laboratories, Detroit, MI) in the lower dorsum on days 0 and 7. The mice also received (i.p) 100 ng of pertussis toxin (Sigma Chemicals, St Louis, MO) on days 0 and 2. The clinical symptoms were scored every day from day 0 to 30 in a blinded manner as follows; 0, normal; 0.5, stiff tail; 1, limp tail; 1.5, limp tail with inability to right; 2, paralysis of one limb; 2.5, paralysis of one limb and weakness of one other limb; 3, complete paralysis of both hind limbs; 4, moribund; 5, death. Mean clinical score (MCS) was calculated by adding every day clinical score for all mice in a group and then divided by total number of mice. Mean maximum clinical score (MMCS) was the MCS at the peak of disease. Average mean clinical score (AMCS) was calculated by adding the MCS for all days (from day 0 to 30) and then divided by number of days. The mean clinical score more than one (MCS>1) was obtained by counting the number of days with MCS more than one (20). The area under the curve (AUC) was calculated using GraphPad Prism 5.0 Software.
Histological analysis
The mice induced to develop EAE were euthanized on day 30 by CO2 asphyxiation and perfused by intracardiac infusion of 4% paraformaldehyde and 1% glutaraldehyde in PBS. Brain and spinal cord samples were removed and fixed in 10% formalin in PBS. Tissues were processed and transverse sections from cervical, upper thoracic, lower thoracic and lumbar regions of the spinal cord were stained with Luxol Fast Blue or hematoxylin and eosin. Inflammation and demyelination in the CNS were assessed under microscope in a blinded manner. The spinal cord sections were viewed as anterior, posterior and two lateral columns (4 quadrants). Each quadrant displaying the infiltration of mononuclear cells or loss of myelin was assigned a score of one inflammation or one demyelination, respectively. Thus, each animal has a potential maximum score of 16 and this study represents the analysis of spinal cords from 5 mice per group. The pathological score from each group is expressed as percent positive over total number of quadrants examined (20).
Quantitative real-time polymerase chain reaction
The quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) was performed using the ABI Prism 7900 Fast Sequence Detection System (Applied Biosystems, Foster City, CA) according to manufacturer’s instructions. The brain and spleen samples were isolated on day 14 following induction of EAE. Spleen cells were cultured in 24 well tissue culture plates in RPMI medium with 10 μg/ml MOGp35–55 antigen or 5 μg/ml Con A for 24 hrs. Total RNA was extracted from brain, spleen, and cultured spleen cells using TRIzol reagent (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. The RNA samples (5 μg/100 μl reaction) were reverse transcribed into cDNA (RT-PCR) using random hexamer primers and TaqMan reverse transcription kit (Applied Biosystems, Foster City, CA). The cDNA (2 _l/sample) was subjected to qPCR analysis in triplicate using forward and reverse primers, TaqMan Universal Master Mix and probe (10μl/reaction) in fast optical 96-well plate. Controls include RT-PCR in the absence of RNA and real-time PCR in the absence of cDNA. The data were analyzed using the ABI Prism 7900 relative quantification (delta-delta-Ct) study software (Applied Biosystems, Foster City, CA). In this study we have used primer sets for 10 selected inflammatory genes with GAPDH (Applied Biosystems, Foster City, CA) as internal controls. The expression levels of inflammatory genes normalized to GAPDH are presented as arbitrary fold changes compared to control samples.
T cell proliferation assay
T cell proliferation was measured by WST-1 assay (Roche, Indianapolis, IN). The spleen cells were isolated on day 14 following induction of EAE and cultured in 96 well tissue culture plates in RPMI medium (1×105/200 μl/well) with 0, 1, 5 and 10 μg/ml MOGp35–55 peptide. WST-1 reagent (10 μl/well) was added after 72 hrs of culture and the absorbance was measured at 450 nm using 2100 microplate reader (Alpha Diagnostics Inc., San Antonio, TX) as a measure of viable cell count.
Intracellular cytokine staining
Spleen, lymph node and brain cells isolated on day 14 following induction of EAE were cultured in 24 well tissue culture plates in RPMI medium (1×106/ml) in the presence of 10 μg/ml MOGp35–55 antigen or 5 μg/ml Con A for 24 hrs. Monensin (2 μM) was added during the last 4 hrs to block protein secretion. The cells were isolated fixed and permeabilized by incubating in PBS containing 1% paraformaldehyde and 0.02% Triton X-100 at 4°C for 15 min. After washing in PBS, the cells were stained with fluorochrome conjugated IL-17 and IFNγ antibodies at 4°C for 30 min and analyzed using a three color FACSCanto flow cytometer to determine the percentage of cells expressing cytokines.
ELISA for IFNγ, IL-17 and IL-10
To determine the cytokine response, spleen cells from MOGp35–55-sensitized mice were cultured in 24 well plates in RPMI medium (1×106/ml) in the presence of 0 and 10 μg/ml MOGp35–55 or 5 μg/ml Con A. The culture supernatants were collected after 48 hrs and the levels of IFNγ, IL-17 and IL-10 measured by ELISA. Briefly, 96-well ELISA plates were coated with 2 μg/ml of anti-IL-17 or anti-IFNγ capture antibody in 100 μl/well of 0.06 M Carbonate buffer, pH 9.6. After overnight incubation at 4°C, the excess Abs were washed off and the residual binding sites blocked by the addition of 100 μl/well of 1% BSA in PBS for 1 h. The test samples (culture supernatants) and standards (rIL-17, rIFNγ, rIL-10) were added and incubated at 4°C overnight. Plates were washed with PBS containing 0.05% Tween-20 and 0.2 μg/ml of biotin-conjugated anti-IL-17, anti-IFNγ or anti-IL-10 added as detection antibody. After incubation at room temperature for 1 h, the plates were washed three times and avidin-alkaline phosphatase added followed by 1 mg/ml of p-nitrophenyl phosphate. After 30 min incubation at room temperature, the absorbance was read at 405 nm and the concentrations of IL-17, IFNγ and IL-10 in the culture supernatants were calculated from the standard curve. For some experiments, CD4+ T cells were cultured under Th1 conditions as described earlier (23).
Statistical analysis
All the experiments were repeated two or three times and the values are expressed as mean ± SD. The differences between groups were analyzed by ANOVA and the values of p<0.05 were considered significant.
Results
Stat4β transgenic mice develop an exacerbated EAE
To study the distinct roles played by Stat4 isoforms in autoimmune disease, we examined the development of EAE in transgenic mice that express Stat4α and Stat4β directed by the CD2 locus control region backcrossed to a Stat4-deficient background (26) and compared with wild type and Stat4−/− mice. As shown in figure 1A, Stat4β transgenic mice developed an exacerbated EAE compared to wild type mice. The day of onset and MCS in Stat4β transgenic mice was similar to wild type mice in the early phase of EAE, but the MCS continued to worsen in Stat4β transgenic mice in the later phase of EAE. In contrast, Stat4 deficient mice remained resistant to EAE and Stat4α transgenic mice developed mild EAE with delayed onset and earlier remission than the Stat4β transgenic and wild type mice (Fig. 1A). The Stat4β transgenic mice also showed a significant increase in AUC, MMCS and AMCS than the wild type and Stat4α transgenic mice (Table 1). These results show that Stat4β transgenic mice develop an exacerbated EAE compared to Stat4α transgenic or wild type mice and suggest the distinct abilities of Stat4 isoforms to mediate the pathogenesis of EAE.
FIGURE 1.
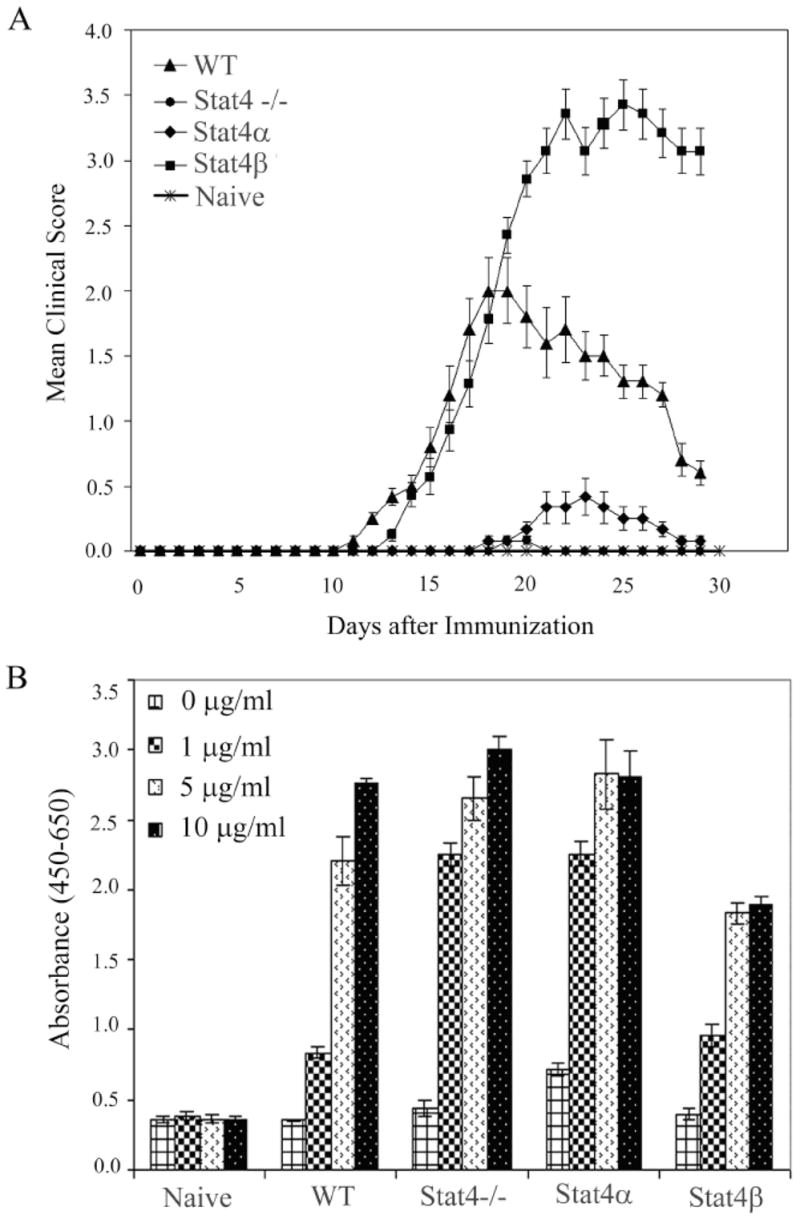
Development of EAE in Stat4α and Stat4β transgenic mice. A, C57BL/6 wild type (WT), Stat4 deficient (Stat4−/−), Stat4α transgenic (Stat4α) and Stat4β transgenic (Stat4β) mice were induced to develop EAE by immunization with MOGp35–55 antigen. The clinical symptoms were scored every day in a blinded manner. The mean clinical score of all 10 mice from two different experiments is shown. The figure is representative of three independent experiments. B, Neural antigen-induced proliferation of spleen T cells from Stat4α and β transgenic mice in vitro. Spleen cells were isolated from C57BL/6 wild type (WT), Stat4 deficient (Stat4−/−), Stat4α transgenic (Stat4α), and Stat4β transgenic (Stat4β) mice on day 14 following induction of EAE. The cells were cultured with MOGp35–55 for 48 hrs and the proliferation measured by WST-1 assay.
Table 1.
Clinical analysis of EAE in Stat4 transgenic mice
| Analyses | Naïve | WT | Stat4−/− | Stat4α | Stat4β |
|---|---|---|---|---|---|
| MMCS | 0.0 (0.0) | 2.00 (100) | 0.08 (4.0) | 0.42 (21.0) | 3.36 (168) |
| AMCS | 0.0 (0.0) | 0.89 (100) | 0.0064 (0.72) | 0.083 (9.3) | 1.31 (147) |
| MCS>1/days | 0.0 (0.0) | 12.00 (100) | 0.0 (0.0) | 0.0 (0.0) | 13.0 (108) |
| MCS/Day30 | 0.0 (0.0) | 0.60 (100) | 0.0 (0.0) | 0.08 (13.3) | 3.07 (512) |
| AUC | 0.0 (0.0) | 21.85 (100) | 0.16 (0.73) | 2.53 (11.6) | 37.83 (173) |
The mean clinical score (MCS) of EAE was calculated by adding every day clinical score for all mice in a group and then divided by total number of mice. The mean maximum clinical score (MMCS) is the MCS at the peak of the disease. Average mean clinical score (AMCS) was calculated by adding the MCS for all days (from day 0 to 30) and then divided by 30. The mean clinical score more than one (MCS>1) was obtained by counting the number of days with MCS more than one. The values in parenthesis are percent change based on clinical scores in WT mice with EAE. Area under the curve (AUC) was calculated using the GraphPad Prism 5.0 software.
To confirm that Stat4α transgenic and Stat4−/− mice were sensitized to MOGp35–55 peptide, we measured the antigen-induced T cell proliferation ex vivo. As shown in figure 1B, in vitro culture of spleen cells from wild type, Stat4-deficient, Stat4α transgenic and Stat4β transgenic mice showed a dose-dependent proliferation in response to MOGp35–55 antigen with the Stat4β transgenic mice displaying a slightly decreased proliferation compared to cells from mice of the other genotypes. These results suggest that the lack of disease in Stat4α transgenic and Stat4−/− mice is not due to the lack of development of MOG-specific T cell responses.
Stat4β transgenic mice develop severe inflammation and demyelination in the CNS
To further establish the differential regulation of EAE by Stat4 isoforms, we examined the pathology of CNS inflammation and demyelination. As shown in figure 2, the wild type mice with EAE showed extensive myelin loss (demyelination) associated with infiltration of immune cells (inflammation) in the spinal cord. When compared with wild type, the Stat4β transgenic mice with EAE showed a significant increase in the extent of inflammation and demyelination inthe spinal cord. However, the Stat4−/− and Stat4α transgenic mice induced to develop EAE showed no sign of inflammation or demyelination in the CNS. Therefore, T cells expressing Stat4β caused more CNS pathology compared to T cells lacking Stat4 or those expressing Stat4α.
FIGURE 2.

Histology of CNS inflammation and demyelination in Stat4α and Stat4β transgenic mice with EAE. The spinal cord samples were isolated from C57BL/6 wild type (WT), Stat4 deficient (Stat4−/−), Stat4α transgenic (Stat4α) and Stat4β transgenic (Stat4β) mice on day 30 following induction of EAE. The transverse sections of cervical, upper thoracic, lower thoracic and lumbar regions of spinal cord were obtained and stained with LFB/PAS (luxol fast blue/periodic acid scriff) along with H&E (hematoxylin and eosin). The pathology of demyelination (left) and inflammation (right) in the spinal cord sections were visualized by microscopy and the representative 10X pictures are shown. The number of positive quadrants with inflammation and demyelination were scored and expressed as percentage over the total number of quadrants examined in the histogram.
Histological analyses revealed that wild type mice with EAE display 18% and 21% spinal cord quadrants positive for demyelination and inflammation, respectively (Fig. 2). When compared with wild type mice, the Stat4β transgenic mice developed severe pathology with 71% (4 fold increase; p<0.01) and 61% (3 fold increase; p<0.01) spinal cord quadrants positive for demyelination and inflammation, respectively. The Stat4α transgenic mice developed very mild CNS pathology with 3% and 1.47% spinal cord quadrants positive for demyelination and inflammation respectively. The Stat4−/− mice failed to show any symptoms of CNS pathology (Fig. 2). These results suggest that the clinical symptoms of EAE correlate with the pathology of CNS inflammation and demyelination in Stat4α and Stat4β transgenic mice.
Stat4β transgenic mice with EAE express elevated levels of effector T cell-derived inflammatory cytokines in the CNS and lymphoid organs
We next wanted to define the mechanism in the differential regulation of EAE in Stat4α and Stat4β transgenic mice. As mice that are deficient in Stat4 have multiple defects in Th1 differentiation (22), Th17 function (23), migration and adhesion of T cells to inflamed sites (31–33), we focused our analysis on comparing the Stat4α and Stat4β transgenic immune cells where differences likely reflect specific effects of the isoforms. We have previously shown that Th1 differentiation in vitro is largely similar in Stat4α and Stat4β transgenic cells (26), and we did not observe consistent differences between these cells on the expression of Ccr5, Ccr7, Gcnt1, St3gal6, or α4 or β1 integrin (data not shown). We then examined the expression of effector T cell-derived inflammatory cytokines in the CNS, spleen and spleen cells cultured with antigen. The levels of IFNγ and IL-17 mRNA detected in the brain and spleen of mice with EAE were significantly increased over unimmunized naïve mice and largely correlated to disease severity with tissues from wild type or Stat4β transgenic mice having the highest levels (Fig. 3). Expression of the Th1 transcription factor T-bet also correlated with IFNγ expression in tissues from Stat4β transgenic mice though less well in tissues from wild type mice. The mRNA levels of cytokines from antigen stimulated spleen cells were somewhat different with higher levels of IFNγ observed in wild type and Stat4β transgenic cultures but higher IL-17 in Stat4α transgenic cultures (Fig. 3). Thus, while the expression of IFNγ and IL-17 in the CNS correlated with disease severity, differences in cytokine profile between Stat4α and Stat4β transgenic mice suggest that Stat4 isoforms may differentially regulate cytokine production in EAE.
FIGURE 3.

Expression of effector T cell-derived inflammatory cytokines in the CNS, spleen and cultured spleen cells from Stat4α and Stat4β transgenic mice with EAE. Brain and spleen samples were isolated from C57BL/6 wild type (WT), Stat4α transgenic (Stat4α), and Stat4β transgenic (Stat4β) mice on day 14 following induction of EAE by immunization with MOGp35–55 antigen. Total RNA was extracted from brain, spleen or spleen cells cultured with neural antigens and the expression of IFNγ, IL-17 and T-bet analyzed by qRT-PCR using GAPDH as internal control. The fold changes in the expression of cytokines in EAE mice were calculated based on naïve mice as control.
Stat4β transgenic mice with EAE express elevated levels of APC-derived inflammatory cytokines in the CNS and lymphoid organs
To further define the mechanism in the differential regulation of EAE in Stat4α and Stat4β transgenic mice, we examined the expression of antigen presenting cell-derived inflammatory cytokines in the CNS and lymphoid organs. As shown in Figure 4, the wild type and Stat4β transgenic mice with EAE showed an increased expression of IL-12p35, IL-12p40 and IL-23p19 in the brain and spleen compared to naïve mice. Stat4α transgenic mice with EAE showed little or no increase in the expression of IL-12p35, IL-12p40 or IL-23p19 in the brain and spleen. Interestingly, the levels of IL-12p35 mRNA correlated well with IFNγ mRNA levels and the levels of IL-12p40 and IL-23p19 mRNA correlated with IL-17 mRNA levels in both brain and spleen (compare Fig. 4 to Fig. 3) and with the clinical and pathological symptoms of EAE in Stat4α and Stat4β transgenic mice.
FIGURE 4.

Expression of APC-derived inflammatory cytokines in the CNS and spleen of Stat4α and β transgenic mice with EAE. Brain and spleen were isolated from C57BL/6 wild type (WT), Stat4α transgenic (Stat4α) and Stat4β transgenic (Stat4β) mice on day 14 following induction of EAE by immunization with MOGp35–55 antigen. Total RNA was extracted from brain and spleen and the expression of IL-12p35, IL-12p40 and IL-23p19 analyzed by qRT-PCR using GAPDH as internal control. The fold changes in the expression of cytokines in EAE mice were calculated based on naïve mice as control.
IFNγ production in the periphery correlates with the severity of EAE in Stat4β transgenic mice
To determine if the differences observed in mRNA expression in figure 3 and 4 results in differential cytokine production, we examined IFNγ and IL-17 production by intracellular cytokine staining (Fig. 5) and ELISA (Fig. 6). Cells isolated from CNS, spleen or draining LN were stimulated with PMA and ionomycin before intracellular staining with anti-IL-17 and anti-IFNγ antibodies in CD4+ cells. Early cytokine production in WT, Stat4α and Stat4β transgenic cells were not dramatically different, with Stat4β transgenic cells having a slightly greater propensity for IFNγ production (Fig. 5). Moreover, while there was decreased inflammation in the Stat4α transgenic CNS (Fig. 2), Stat4α transgenic T cells in the CNS were capable of producing IL-17 and IFNγ at levels similar to wild-type cells (Fig. 5). In response to antigen stimulation, spleen cells from wild type and Stat4β transgenic mice produced higher levels of IFNγ than Stat4α transgenic cells, while, Stat4α transgenic cells produced more IL-17 than either wild type or Stat4β transgenic cells (Fig. 6). We did not detect IL-12 or IL-23 production from antigen-stimulated spleen cells (data not shown). These results highlight that the decreased disease in Stat4α transgenic mice is not due to an inability to develop inflammatory cell types in vivo.
FIGURE 5.

Intracellular IFNγ and IL-17 in immune cells from Stat4α and β transgenic mice. Spleen, lymph node and brain cells were isolated from C57BL/6 wild type (WT), Stat4α transgenic (Stat4α) and Stat4β transgenic (Stat4β) mice on day 14 following induction of EAE. The cells were cultured with PMA + ionomycin for 6 hours before staining with IFNγ and IL-17 specific antibodies and analyzed by flow cytometry.
FIGURE 6.

Neural antigen-induced secretion of IFNγ and IL-17 from Stat4α and β transgenic spleen cells in culture. Spleen cells were isolated from C57BL/6 wild type (WT), Stat4α transgenic (Stat4α) and Stat4β transgenic (Stat4β) mice on day 14 following induction of EAE. The cells were cultured with MOGp35–55 or Con A for 36 h, and the release of IFNγ (A) and IL-17 (B) was analyzed by ELISA.
To identify other genes that demonstrate Stat4-dependence, we performed a Stat4 ChIP-on-chip experiment that will be described in detail elsewhere.5 Il10 was identified in this analysis and bound by Stat4 in the second and third introns (Fig. 7A). Wild type and Stat4−/− Th1 culture stimulated with either IL-12 or anti-CD3 demonstrated Stat4-dependence in the induction of IL-10 production (Fig. 7B). As IL-10 is an important regulatory cytokine that inhibits the development of EAE (34), we examined the regulation of IL-10 by Stat4 isoforms. Wild type, Stat4α transgenic and Stat4β transgenic cells re-stimulated with anti-CD3 after culture under Th1 conditions demonstrated similar production of IFNγ (Fig. 7C). In contrast, Stat4β transgenic cells had decreased IL-10 production in these cultures (Fig. 7C). To test if this phenotype was reflected in vivo during disease, we tested RNA from wild type, Stat4α transgenic and Stat4β transgenic mice for Il10 expression in situ. Three-five fold more Il10 mRNA was detected in Stat4α transgenic samples than in wild type or Stat4β transgenic samples (Fig. 7D). We further examined splenic cultures from wild type, Stat4α transgenic and Stat4β transgenic to assay for IL-10 production following stimulation as in Fig. 6. While wild type and Stat4β transgenic cells had similar IL-10 production, Stat4α transgenic cells produced 2-3-fold higher levels of IL-10 (Fig. 7E). Thus, Stat4α-expressing T cells have an increased propensity for IL-10 production and this is associated with decreased CNS inflammation and pathology in Stat4α transgenic mice, compared to wild type or Stat4β transgenic mice.
FIGURE 7.

Differential regulation of IL-10 by Stat4 isoforms. A, Affymetrix Integrated Genome Browser analysis of Stat4 binding across the Il10 locus using data from a Stat4 ChIP-on-chip dataset. Bars indicate the intensity of Stat4-bound DNA hybridizing to oligonucleotides spanning −7.5 kb to +2.5 kb relative to the Il10 transcriptional start. The exon-intron structure of Il10 is indicated below the graph. B, Naïve, wild type and Stat4−/− CD4+ T cells were cultured under Th1 conditions for five days before re-stimulation with IL-12 (left) or anti-CD3 (right) for 24 hrs. IL-10 levels were examined in supernatants using ELISA. C, Naïve, wild type, Stat4α transgenic and Stat4β transgenic CD4+ T cells were cultured under Th1 conditions for five days before re-stimulation with anti-CD3 for 24 hours. IL-10 and IFNγ levels were examined in supernatants using ELISA. D, Il10 mRNA levels were assessed in total RNA from spleens isolated from C57BL/6 wild type (WT), Stat4α transgenic (Stat4α) and Stat4β transgenic (Stat4β) mice on day 14 following induction of EAE by immunization with MOGp35–55 antigen by qRT-PCR using GAPDH as internal control. The fold changes in the expression levels were calculated based on naïve spleen. E, Spleen cells were isolated from C57BL/6 wild type (WT), Stat4α transgenic (Stat4α) and Stat4β transgenic (Stat4β) mice on day 14 following induction of EAE. The cells were cultured as in Fig. 6 and the production of IL-10 was analyzed using ELISA.
Discussion
Stat proteins are expressed as multiple isoforms; alpha forms that are full length and beta forms that lack the C-terminal transactivation domain of the alpha form and rather have a novel C-terminal domain resulting from the lack of splicing of the last exon. Although the isoform phenomenon is well documented, the biological role of these isoforms is not entirely clear. For Stat1 and Stat5, the beta isoforms are dominant negatives (35, 36). The functions of Stat3 are more context dependent, where the beta isoform may interfere with transcription of some genes but activate others (28, 29, 37). Indeed, Stat3β can mediate some aspects of liver inflammation and rescue the embryonic lethality of Stat3-deficiency (28, 29). We have described that Stat4 also exists as alpha and beta isoforms and that each is able to mediate Th1 differentiation in vitro (26). In this report we have used Stat4α and Stat4β transgenic mice to define the ability of Stat4 isoforms to mediate CNS inflammation and demyelination in the EAE model of MS. We found that the Stat4β transgenic mice develop exacerbated EAE compared to wild type mice, while the Stat4α transgenic mice develop mild EAE. The exacerbation of EAE in Stat4β transgenic mice associates with lower levels of IL-10 production and increased expression of inflammatory cytokines including IFNγ and IL-17 compared to Stat4α transgenic mice. These findings highlight the fact that Stat4 isoforms play distinct roles in the pathogenesis of EAE.
EAE is an extensively studied T cell- and Stat4-dependent model of inflammatory disease (24). Our results indicate that T cells expressing Stat4β are much more efficient in mediating inflammation than T cells expressing the Stat4α isoform. Stat4β transgenic mice develop much more severe disease with greater levels of demyelination than those observed in Stat4α transgenic mice or wild type mice. The mechanism for this increased disease likely includes the altered cytokine environments observed in the transgenic mice. While Stat4α and Stat4β transgenic cells are equally capable of becoming IFNγ- or IL-17-secreting cells in vitro, Stat4α transgenic cells have increased levels of IL-10 production (Fig. 7 and data not shown) (26). We similarly observed increased IL-10 production in Stat4α transgenic mice with EAE in vivo and ex vivo (Fig. 7). The lower levels of IL-10 produced in Stat4β transgenic mice are associated with increased IL-12 and IL-23 mRNA levels in CNS and spleen tissue, and increased IFNγ expression in tissue and from antigen-stimulated Stat4β-expressing cells compared to those observed in Stat4α-expressing cells (Fig. 3–7). The lower levels of IFNγ produced by Stat4α transgenic cells compared to Stat4β transgenic cells may, at least in part, be responsible for the observed increases in IL-17 from Stat4α transgenic cells in the periphery (Fig. 4 and 6) as has been described (38, 39). The ability of Stat4 to promote IL-10 production is consistent with other reports of Stat4-dependent IL-10 production in Th1 and NK cell cultures (40–42). We provide data from a ChIP-on-chip assay that Stat4 directly binds Il10, and show that acute stimulation of Th1 cells with IL-12 results in IL-10 production from wild type but not Stat4−/− cells (Fig. 7). Moreover, Stat4α, but not Stat4β, can mediate the programming of the Il10 gene for increased expression in Th1 cultures. Thus, while Stat4α can rescue Stat4-deficiency in vitro and compensates for some in vivo Stat4 functions (30), altered cytokine profiles from these cells limit their ability to promote the development of EAE. As IL-10 is critical regulator of inflammation in EAE (34), the increased IL-10 production in Stat4α transgenic mice (Fig. 7) provides a mechanism how Stat4 isoforms differentially regulate the pathogenesis of EAE. These results also suggest that modulating the splicing between the alpha and beta isoforms of Stat4 could be therapeutic for inflammatory diseases.
It is also possible that other Stat4α- or Stat4β-specific functions might be important for the pathogenesis of EAE. While we previously observed that both isoforms could mediate Th1 development in vitro, we did notice that Stat4β-expressing cells produced slightly less IFNγ in response to IL-12 (26). Since there was more severe disease in Stat4β transgenic mice, it seems unlikely this contributes to the level of disease. We also observed that Stat4β transgenic cells had much higher proliferation than Stat4α or wild type cells in a pattern that paralleled the severity of disease. However, MOGp35–55-specific proliferation suggests that there is no significant increase in the overall number of antigen-reactive T cells in EAE (Fig. 1). Similarly, intracellular cytokine staining did not show dramatic differences among the percentages of cytokine-positive cells, though IFNγ was increased in the Stat4β transgenic cells. As noted above, this is more likely to result from changes in the balance of IL-10 and IFNγ production, and their resulting effects on IL-17 production. However, there may be additional genes that are differentially regulated by Stat4 isoforms which may also contribute to the development of inflammatory diseases.
The importance of Stat4 in mediating inflammation has been extensively studied in mouse models of infectious and inflammatory diseases (22). We have recently shown that Stat4 is also required for IL-12 responses in human cells (43), suggesting that much of what we have learned will be applicable to understanding human inflammatory responses. Our model further demonstrates that Stat4 expression in T cells is sufficient to mediate inflammatory immunity. The Stat4α and Stat4β transgenes are expressed from a CD2 locus control region that promotes transcription primarily in T cells, with considerably lower expression in other lymphoid cells (26). The transgenic mice have been backcrossed to the Stat4−/− background so that the Stat4 isoforms are expressed in T cells but not other cells in the mouse. Previous work has shown that Stat4 can be expressed in myeloid cells following an appropriate stimulus (44). However, as the transgenic mice in this study lack Stat4 in any myeloid compartment, our results indicate that Stat4 expression in non-lymphoid cells is not required for the development of EAE. While targeting Stat4 using small molecule inhibitors has proven difficult, it may be possible to alter Stat4 function by modulating the splicing of Stat4 isoforms and thus altering the ability of immune cells to mediate disease.
Footnotes
Abbreviations used in this paper: MS, multiple sclerosis; EAE, experimental allergic encephalomyelitis; MBP, myelin basic protein; MOG, myelin oligodendrocyte glycoprotein; PLP, proteolipid protein; qRT-PCR, quantitative reverse transcription polymerase chain reaction; MCS, mean clinical score; MMCS, mean maximum clinical score; AMCS, average mean clinical score; AUC, area under the curve.
Good, S. R., Thieu, V. T., Mathur, A. N., O’Malley, J. T., Perumal, N. B. and Kaplan, M. H. Temporal induction patterns of STAT4 target genes defines potential for Th1 lineage-specific programming. In preparation.
Funding support: This work was supported in part by grants from the National Institutes of Health, R01 NS42257 (to JJB) and AI045515 (to MHK).
References
- 1.Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- 2.Bitsch A, Bruck W. Differentiation of multiple sclerosis subtypes: implications for treatment. CNS Drugs. 2002;6:405–418. doi: 10.2165/00023210-200216060-00004. [DOI] [PubMed] [Google Scholar]
- 3.Whitacre CC, Reingold SC, O’Looney PA. A gender gap in autoimmunity. Science. 1999;283:1277–1278. doi: 10.1126/science.283.5406.1277. [DOI] [PubMed] [Google Scholar]
- 4.Steinman L, Martin M, Bernard C, Conlon P, Oksenberg JR. Multiple sclerosis: deeper understanding of its pathogenesis reveals new targets for therapy. Annu Rev Neurosci. 2002;25:491–505. doi: 10.1146/annurev.neuro.25.112701.142913. [DOI] [PubMed] [Google Scholar]
- 5.Gold R, Hartung HP, Toyka KV. Animals model for auto immune demyelinating disorders of the nervous system. Mol Med Today. 2000;6:88–91. doi: 10.1016/s1357-4310(99)01639-1. [DOI] [PubMed] [Google Scholar]
- 6.Steinman L, Zamvil SS. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann Neurol. 2006;60:12–21. doi: 10.1002/ana.20913. [DOI] [PubMed] [Google Scholar]
- 7.Trinchieri G. IL-12: A proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen specific adaptive immunity. Ann Rev Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 8.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 9.Leonard JP, Waldburger KE, Goldman SJ. Prevention of EAE by antibodies against interleukin-12. J Exp Med. 1995;181:381–385. doi: 10.1084/jem.181.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bright JJ, Musuro BF, Du C, Sriram S. Expression of IL-12 in CNS and lymphoid organs of mice with experimental allergic encephalomyelitis. J Neuroimmunol. 1998;82:22–26. doi: 10.1016/S0165-5728(97)00184-7. [DOI] [PubMed] [Google Scholar]
- 11.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, Murphy KM. Interleukin-12 signaling in Th1 cells involves tyrosine phosphorylation of Stat 3 and Stat 4. J Exp Med. 1995;181:1755–1762. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacon CM, Petricoin EM, 3rd, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci USA. 1995;92:7307–7311. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, To W, Wagner J, O’Farrell AM, McClanahan T, Zurawski S, Hannum C, Gorman D, Rennick DM, Kastelein RA, de Waal Malefyt R, Moore KW. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 15.Bright JJ, Du C, Coon M, Sriram S, Klaus SJ. Prevention of experimental allergic encephalomyelitis via inhibition of IL-12 signaling and IL-12-mediated Th1 differentiation: an effect of the novel anti-inflammatory drug lisofylline. J Immunol. 1998;161:7015–7022. [PubMed] [Google Scholar]
- 16.Bright JJ, Du C, Sriram S. Tyrphostin B42 inhibits IL-12-induced tyrosine phosphorylation and activation of Janus kinase-2 and prevents experimental allergic encephalomyelitis. J Immunol. 1999;162:6255–6262. [PubMed] [Google Scholar]
- 17.Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Gene Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 18.Natarajan C, Bright JJ. Curcumin inhibits experimental allergic encephalomyelitis by blocking IL-12 signaling through JAK-STAT pathway in T cells and differentiation of neural antigen specific Th1 cells. J Immunol. 2002;169:6506–6513. doi: 10.4049/jimmunol.168.12.6506. [DOI] [PubMed] [Google Scholar]
- 19.Muthian G, Bright JJ. Quercetin ameliorates experimental allergic encephalomyelitis by blocking IL-12 signaling through JAK-STAT pathway in T lymphocyte. J Clin Immunol. 2003;24:541–551. doi: 10.1023/B:JOCI.0000040925.55682.a5. [DOI] [PubMed] [Google Scholar]
- 20.Raikwar HP, Muthian G, Rajasingh J, Johnson C, Bright JJ. PPARγ antagonists exacerbate neural antigen-specific Th1 response and experimental allergic encephalomyelitis. J Neuroimmunol. 2005;167:99–107. doi: 10.1016/j.jneuroim.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 21.Muthian G, Raikwar HP, Rajasingh J, Bright JJ. 1, 25 Dihydroxyvitamin-D3 modulates JAK-STAT pathway in IL-12/IFNγ axis leading to Th1 response in experimental allergic encephalomyelitis. J Neurosci Res. 2006;83:1299–1309. doi: 10.1002/jnr.20826. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan MH. STAT4: a critical regulator of inflammation in vivo. Immunol Res. 2005;31:231–242. doi: 10.1385/IR:31:3:231. [DOI] [PubMed] [Google Scholar]
- 23.Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, Kapur R, Levy DE, Kansas GS, Kaplan MH. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–4907. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 24.Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest. 2001;108:739–747. doi: 10.1172/JCI12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darnell JEJ. STATs and gene regulation. Science. 1997;277:1630–1636. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 26.Hoey T, Zhang S, Schmidt N, Yu Q, Ramchandani S, Xu X, Naeger LK, Sun YL, Kaplan MH. Distinct requirements for the naturally occurring splice forms Stat4alpha and Stat4beta in IL-12 responses. EMBO J. 2003;22:4237–4248. doi: 10.1093/emboj/cdg393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Sullivan A, Chang HC, Yu Q, Kaplan MH. STAT4 is required for interleukin-12-induced chromatin remodeling of the CD25 locus. J Biol Chem. 2004;279:7339–7345. doi: 10.1074/jbc.M309979200. [DOI] [PubMed] [Google Scholar]
- 28.Yoo JY, Huso DL, Nathans D, Desiderio S. Specific ablation of Stat3beta distorts the pattern of Stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell. 2002;108:331–344. doi: 10.1016/s0092-8674(02)00636-0. [DOI] [PubMed] [Google Scholar]
- 29.Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X, Murray-Tait V, Chiarle R, Poli V. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5:401–409. doi: 10.1038/ni1052. [DOI] [PubMed] [Google Scholar]
- 30.Broxmeyer HE, Bruns HA, Zhang S, Cooper S, Hangoc G, McKenzie AN, Dent AL, Schindler U, Naeger LK, Hoey T, Kaplan MH. Th1 cells regulate hematopoietic progenitor cell homeostasis by production of oncostatin M. Immunity. 2002;16:815–825. doi: 10.1016/s1074-7613(02)00319-9. [DOI] [PubMed] [Google Scholar]
- 31.White SJ, Underhill GH, Kaplan MH, Kansas GS. Cutting edge: differential requirements for Stat4 in expression of glycosyltransferases responsible for selectin ligand formation in Th1 cells. J Immunol. 2001;167:628–631. doi: 10.4049/jimmunol.167.2.628. [DOI] [PubMed] [Google Scholar]
- 32.Underhill GH, Zisoulis DG, Kolli KP, Ellies LG, Marth JD, Kansas GS. A crucial role for T-bet in selectin ligand expression in T helper 1 (Th1) cells. Blood. 2005;106:3867–3873. doi: 10.1182/blood-2005-03-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwasaki M, Mukai T, Nakajima C, Yang YF, Gao P, Yamaguchi N, Tomura M, Fujiwara H, Hamaoka T. A mandatory role for STAT4 in IL-12 induction of mouse T cell CCR5. J Immunol. 2001;167:6877–6883. doi: 10.4049/jimmunol.167.12.6877. [DOI] [PubMed] [Google Scholar]
- 34.Bettelli E, Das MP, Howard ED, Weiner HL, Sobel RA, Kuchroo VK. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol. 1998;161:3299–3306. [PubMed] [Google Scholar]
- 35.Vinkemeier U, Cohen SL, Moarefi I, Chait BT, Kuriyan J, Darnell JE., Jr DNA binding of in vitro activated Stat1_, Stat1_ and truncated Stat1: interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J. 1996;15:5616–5626. [PMC free article] [PubMed] [Google Scholar]
- 36.Caldenhoven E, van Dijk TB, Solari R, Armstrong J, Raaijmakers JA, Lammers JW, Koenderman L, de Groot RP. STAT3_, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J Biol Chem. 1996;271:13221–13227. doi: 10.1074/jbc.271.22.13221. [DOI] [PubMed] [Google Scholar]
- 37.Yoo JY, Wang W, Desiderio S, Nathans D. Synergistic activity of STAT3 and c-Jun at a specific array of DNA elements in the alpha 2-macroglobulin promoter. J Biol Chem. 2001;276:26421–2649. doi: 10.1074/jbc.M009935200. [DOI] [PubMed] [Google Scholar]
- 38.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 40.Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O’Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 41.Rutz S, Janke M, Kassner N, Hohnstein T, Krueger M, Scheffold A. Notch regulates IL-10 production by T helper 1 cells. Proc Natl Acad Sci USA. 2008;105:3497–3502. doi: 10.1073/pnas.0712102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grant LR, Yao ZJ, Hedrich CM, Wang F, Moorthy A, Wilson K, Ranatunga D, Bream JH. Stat4-dependent, T-bet-independent regulation of IL-10 in NK cells. Genes Immun. 2008;9:316–327. doi: 10.1038/gene.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robertson MJ, Chang HC, Pelloso D, Kaplan MH. Impaired interferon-gamma production as a consequence of STAT4 deficiency after autologous hematopoietic stem cell transplantation for lymphoma. Blood. 2005;106:963–970. doi: 10.1182/blood-2005-01-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frucht DM, Aringer M, Galon J, Danning C, Brown M, Fan S, Centola M, Wu CY, Yamada N, El Gabalawy H, O’Shea JJ. Stat4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1-mediated inflammation. J Immunol. 2002;164:4659–4664. doi: 10.4049/jimmunol.164.9.4659. [DOI] [PubMed] [Google Scholar]