

Abstract
Inhaled anesthetics are thought to alter the conformational states of Cys-loop ligand-gated ion channels (LGICs) by binding within discrete cavities that are lined by portions of four α-helical transmembrane domains. Because Cys-loop LGICs are complex molecules that are notoriously difficult to express and purify, scaled-down models have been used to better understand the basic molecular mechanisms of anesthetic action. In this study, stopped-flow fluorescence spectroscopy was used to define the kinetics with which inhaled anesthetics interact with (Aα2-L1M/L38M)2, a four-α-helix bundle protein that was designed to model anesthetic binding sites on Cys-loop LGICs. Stopped-flow fluorescence traces obtained upon mixing (Aα2-L1M/L38M)2 with halothane revealed immediate, fast, and slow components of quenching. The immediate component, which occurred within the mixing time of the spectrofluorimeter, was attributed to direct quenching of tryptophan fluorescence upon halothane binding to (Aα2-L1M/L38M)2. This was followed by a bi-exponential fluorescence decay containing fast and slow components, reflecting anesthetic-induced conformational transitions. Fluorescence traces obtained in studies using sevoflurane, isoflurane, and desflurane, which poorly quench tryptophan fluorescence, did not contain the immediate component. However, these anesthetics did produce the fast and slow components, indicating that they also alter the conformation of (Aα2-L1M/L38M)2. Cyclopropane, an anesthetic which acts with unusually low potency on Cys-loop LGICs, acted with low apparent potency on (Aα2-L1M/L38M)2. These results suggest that four-α-helix bundle proteins may be useful models of in-vivo sites of action that allow use of a wide range of techniques to better understand how anesthetic binding leads to changes in protein structure and function.
Keywords: Anesthesia, Cyclopropane, Desflurane, Fluorescence, Halothane, Isoflurane, Kinetics, Sevoflurane, Spectroscopy, Stopped-flow
Inhaled general anesthetics alter the function of numerous proteins (1-5). Although the receptor site(s) responsible for producing anesthesia are not known with certainty, members of an anesthetic-sensitive superfamily of homologous Cys-loop ligand-gated ion channels (LGICs) are believed to be among the most important targets (6-8). Members of this superfamily include the nicotinic acetylcholine, serotonin type 3, γ-aminobutyric acid type A (GABAA), and glycine receptors (9, 10). Electrophysiological studies using receptor chimeras and site-directed mutagenesis techniques suggest that inhaled anesthetics bind to LGICs within discrete protein cavities that are lined by portions of four α-helical transmembrane domains (M1-M4), producing conformational states with increased sensitivity to agonist (11-14). In the case of the GABAA receptor, inhaled anesthetics can also directly activate the ion channel even in the absence of agonist (15, 16). Both of these actions may occur if general anesthetics stabilize receptors in an open channel state. However because Cys-loop LGICs are complex molecules that cannot be expressed in significant quantities and at high purity, there remain considerable obstacles to applying powerful biophysical and biochemical approaches to gain insight into how inhaled anesthetics act on these targets.
To overcome these obstacles, small, water-soluble four-α-helix bundle proteins have been designed, produced, and used as well-defined model systems to explore how inhaled anesthetics bind to proteins (17-21). Each four-α-helix bundle protein is a dimer of two 27-residue α-helices joined by an eight-residue glycine linker (figure 1). It contains a hydrophobic cavity that is lined by the four α-helices and binds inhaled anesthetics. Recently, it has been shown that such binding can alter the protein's conformational state as evidenced by changes in its fluorescence, circular dichroism and nuclear magnetic resonance spectra following equilibration with anesthetic (22). Such observations suggest that four-α-helix bundle proteins can serve not only as models to explore how anesthetics bind, but also how such binding leads to changes in protein structure and function.
Figure 1.

(A) Modeled structure of the expressed four-α-helix bundle (Aα2-L1M/L38M)2. The cylinders represent the two 27-residue amphiphilic α-helical portions of each 62-residue di-α-helical peptide, joined by an eight-residue glycine linker. Red and white halves of each cylinder represent hydrophilic and hydrophobic residues, respectively. (B) End-on view of a four-α-helix bundle, showing the interaction of the hydrophobic core residues at the heptad a and d positions. The dashed lines indicate how successive hydrophobic core layers are composed of two a and two d residues. (C) An opened-out and flattened representation of the expressed (Aα2-L1M/L38M)2 bundle illustrating the amino acids present at the hydrophobic heptad a and d positions. There are a total of eight hydrophobic core layers, each composed of two a and two d position residues. The heptad W15 residues are highlighted. Equivalent binding sites for anesthetic molecules reside in hydrophobic core layers III and VI where larger leucines were replaced with smaller alanines (17).
The present study was undertaken to examine the kinetics with which inhaled anesthetics interact with the four-α-helix bundle protein (Aα2-L1M/L38M)2. Stopped-flow fluorescence spectroscopic techniques having millisecond time resolution were used to monitor anesthetic-protein interactions. This was possible because each peptide in (Aα2-L1M/L38M)2 contains a tryptophan residue at position 15 (W15) whose fluorescence is reduced upon equilibration with clinically relevant concentrations of anesthetic (22, 23). We focused our studies on two inhaled anesthetics, halothane and sevoflurane. Halothane directly and efficiently quenches tryptophan fluorescence (24) whereas sevoflurane does not (22).
EXPERIMENTAL PROCEDURES
Materials
Halothane (2-bromo-2-chloro-1,1,1-trifluoroethane) was obtained from Halocarbon Laboratories (Hackensack, NJ). Sevoflurane (fluoromethyl 2, 2, 2-trifluoro-1-(trifluoromethyl) ethyl ether) and isoflurane (l-chloro-2,2,2-trifluoroethyl difluoromethyl ether) were obtained from Abbott Laboratories (North Chicago, IL). Desflurane (1,2,2,2,-tetrafluoroethyl difluoromethyl ether) was purchased from Baxter Healthcare Co (Deerfield, IL) and cyclopropane was from Sigma-Aldrich Chemical Co. (St. Louis, MO).
Expression and purification of (Aα2-L1M/L38M)2
The Aα2-L1M/L38M peptide was expressed in Escherichia coli cells and purified to homogeneity as described (23). Peptide identity was confirmed with laser desorption mass spectrometry (Protein Chemistry Laboratory, University of Pennsylvania, Philadelphia, PA). The expected molecular weight was 6863.1 Da, and the experimental value was 6863.0 Da.
Stopped-flow studies of anesthetic-induced quenching of (Aα2-L1M/L38M)2 intrinsic fluorescence
Anesthetic-induced changes in intrinsic protein fluorescence were measured using a SX.18MV stopped-flow spectrofluorimeter (Applied Photophysics, Leatherhead, UK). The four-α-helix bundle protein (Aα2-L1M/L38M)2 at a concentration of 6.8 − 8.0 μM in buffer (130 mM NaCl and 20 mM NaH2PO4 at pH 7.0) was loaded into one of the spectrofluorimeter's gas-tight mixing syringes and anesthetic in buffer was loaded into the other. The contents of the two syringes were rapidly mixed (1:1 vol:vol). Tryptophan was excited at 280 nm (bandwidth 1.5 nm) and the fluorescence intensity was recorded on a logarithmic timebase (1000 points over 10 sec) through a 305 nm cutoff filter. For each experiment, 3−6 individual traces were signal averaged to reduce noise. Unless otherwise indicated, experiments were performed at 25.0 ± 0.2 °C.
Calculation of Q10
The change in the apparent rate of the fast and slow components associated with a 10° C change in temperature, the Q10, was calculated using the following equation (25):
where khigh and klow are the apparent rates at the high and low temperatures, respectively. Thigh and Tlow are the high and low temperatures, respectively. In practice, the values for khigh and klow were obtained by linear regression from the slope of an Arrhenius plot of the data.
Preparation of anesthetic solutions
Volatile anesthetic solutions were prepared by adding an excess of agent to a sealed bottle containing buffer solution, and stirring overnight. These saturated solutions of known concentration were then diluted with buffer within gas-tight syringes to yield the final desired anesthetic concentrations. For cyclopropane, the anesthetic gas was bubbled at a rate of 100 to 120 ml per minute for at least 3 minutes through 150 ml of buffer solution in a 250 ml glass bottle sealed with a Teflon septum. Lines for gas inlet and outlet were introduced through the septum. The resulting solution was sealed and continuously stirred to allow equilibration for at least 30 minutes. The anesthetic gas was then bubbled through the buffer solution again for at least 2 more minutes and allowed to equilibrate. Gas chromatographic studies indicated that the aqueous concentration of cyclopropane produced using this approach are within 10% of that predicted by cyclopropane's aqueous:gas partition coefficient. The saturated solution was subsequently diluted with buffer to the final desired concentration using gas-tight syringes.
RESULTS
Figure 2 shows a representative stopped-flow fluorescence trace obtained upon mixing (Aα2-L1M/L38M)2 with 0.85 mM halothane (in buffer). For comparison, this figure also shows a trace obtained upon mixing (Aα2-L1M/L38M)2 with buffer alone. Upon mixing with halothane, three components of W15 fluorescence quenching were observed. There was an immediate component that occurred within the one millisecond mixing time of the stopped-flow spectrofluorimeter. It was detected as a reduction (i.e. an offset) in fluorescence intensity observed immediately upon mixing (Aα2-L1M/L38M)2 with halothane. This was followed by a bi-exponential fluorescence decay containing fast and slow components that were essentially complete by 500 milliseconds and 10 seconds, respectively. A fit of the fluorescence trace obtained using halothane to a double exponential equation yielded normalized amplitudes and rates of 0.2000 ± 0.0005 and 14.02 ± 0.08 sec−1 for the fast component and 0.0928 ± 0.0005 and 0.479 ± 0.008 sec−1 for the slow component.
Figure 2.

Stopped-flow fluorescence traces obtained upon mixing (Aα2-L1M/L38M)2 with either 0.85 mM halothane in buffer or buffer alone. Mixing (Aα2-L1M/L38M)2 with halothane produced three distinct components of quenching (immediate, fast, and slow). The immediate component was detected as a 36% reduction in fluorescence intensity upon mixing (A). This was followed by a bi-exponential fluorescence decay (A & B). The same fluorescence traces are shown in A & B on different timescales. Trace amplitudes were normalized between 0 (buffer only) and 1 (protein, no anesthetic). The curve is a fit of the decay to a double exponential equation to obtain the normalized amplitudes and rates of the fast and slow components.
Typical stopped-flow fluorescence traces obtained upon mixing (Aα2-L1M/L38M)2 with a range of halothane concentrations are shown in figure 3. For concentrations above 0.026 mM halothane, all three components (immediate, fast, and slow) were measurable. However, the immediate and slow components were typically too small to be quantified in traces obtained using 0.026 mM halothane. Therefore, these traces were fit to a single exponential equation to obtain the normalized amplitude and rate of the fast component. Figure 4A demonstrates that the normalized amplitudes of the three components varied as a function of halothane concentration. The immediate component amplitude increased with halothane concentration in a hyperbolic manner, becoming half maximal at 0.68 ± 0.08 mM (table 1), whereas the fast and slow component amplitudes increased at low concentrations but decreased at high concentrations. The total quenched fluorescence at equilibrium, equal to the sum of the immediate, fast, and slow component amplitudes, increased with halothane concentration before reaching a plateau and was half-maximal at 0.17 ± 0.02 mM. This is similar to the value of 0.12 ± 0.02 mM previously reported to produce half-maximal fluorescence quenching of (Aα2-L1M/L38M)2 by halothane using steady-state fluorescence measurements (22). The rates of the fast and slow components varied little over a 100-fold range of halothane concentrations and averaged 22 ± 7 sec−1 and 0.51 ± 0.01 sec−1, respectively (figure 4B).
Figure 3.

Stopped-flow fluorescence traces obtained upon mixing (Aα2-L1M/L38M)2 with a range of halothane concentrations. The same fluorescence traces are shown in panels A & B on different timescales. Trace amplitudes were normalized between 0 (buffer only) and 1 (protein, no anesthetic). The curves are fits of the decays to a double exponential equation to obtain the normalized amplitudes and rates of the fast and slow components.
Figure 4.

Halothane concentration-dependence of the normalized amplitude (A) and rates (B) of the immediate (■), fast (●), and slow (O) components obtained from stopped-flow fluorescence experiments. The rate of the immediate component was not shown in panel B because it was immeasurably fast. The total (□) component reflects equilibrium fluorescence quenching and is equal to the sum of the immediate, fast, and slow components. No slow component was observed at 0.026 mM halothane. Data points are the means of three experiments and the errors represent the SD. The curves are fits of the immediate and total components to a Hill equation whose parameters are given in table 1. The anesthetizing concentration of halothane is 0.31 mM.
Table 1.
Hill Equation Parameters Defining the Anesthetic Concentration-Dependence of Quenching
| Halothane | Sevoflurane | ||||||
|---|---|---|---|---|---|---|---|
| Component | Immediate | Fasta | Slowa | Total | Fast | Slow | Total |
| EC50 (mM) | 0.68 ± 0.08 | NA | NA | 0.17 ± 0.02 | 0.11 ± 0.01 | 0.52 ± 0.09 | 0.17 ± 0.01 |
| Hill Coefficient | 1.3 ± 0.1 | NA | NA | 1.0 ± 0.1 | 1.2 ± 0.1 | 1.4 ± 0.2 | 1.1 ± 0.1 |
| Maximal Quenching at High Anesthetic Concentration | 0.60 ± 0.03 | NA | NA | 0.74 ± 0.03 | 0.28 ± 0.01 | 0.11 ± 0.01 | 0.39 ± 0.01 |
Plots of the fast and slow components versus halothane concentration were biphasic and not fit to a Hill equation.
In contrast to halothane, sevoflurane produced no immediate fluorescence quenching when rapidly mixed with (Aα2-L1M/L38M)2 even at a concentration that is five-times higher than that required to induce anesthesia (figure 5A). However, sevoflurane did produce a bi-exponential fluorescence decay containing fast and slow components (figures 5A and B). A fit of the decay to a double exponential equation yielded normalized amplitudes and rates of 0.1855 ± 0.0003 and 18.95 ± 0.08 sec−1 for the fast component and 0.0730 ± 0.0004 and 0.453 ± 0.007 sec−1 for the slow component, values that are similar to those obtained using 0.85 mM halothane.
Figure 5.

Stopped-flow fluorescence traces obtained upon mixing (Aα2-L1M/L38M)2 with either 2.85 mM sevoflurane in buffer or buffer alone. The same fluorescence traces are shown in panels A & B on different timescales. Trace amplitudes were normalized between 0 (buffer only) and 1 (protein, no anesthetic). The curve is a fit of the decay to a double exponential equation to obtain the normalized amplitudes and rates of the fast and slow components.
Figure 6A demonstrates that the amplitudes of the fast and slow components increased with sevoflurane concentration before reaching a plateau at high concentrations. As with studies using halothane, only the fast component could be quantified at the lowest concentrations. The rates of the fast and slow components varied little with sevoflurane concentration and averaged 20 ± 8 sec−1 and 0.41 ± 0.04 sec−1, respectively (figure 6B).
Figure 6.

Sevoflurane concentration-dependence of the normalized amplitudes (A) and rates (B) of the fast (●) and slow (O) components obtained from stopped-flow fluorescence experiments. The total (□) component reflects equilibrium fluorescence quenching and is equal to the sum of the fast, and slow components. The slow component was not consistently observed below 0.2 mM sevoflurane. Data points are the means of three experiments and the errors represent the SD. The curves are fits of the components to a Hill equation whose parameters are given in table 1. The anesthetizing concentration of sevoflurane is 0.57 mM.
We also surveyed the effects of three additional anesthetics on (Aα2-L1M/L38M)2 intrinsic fluorescence. Figure 7 shows representative stopped-flow fluorescence traces obtained upon mixing (Aα2-L1M/L38M)2 with isoflurane, desflurane, or cyclopropane. At their in-vivo anesthetizing concentrations, isoflurane (0.31 mM) and desflurane (0.57 mM) caused no immediate fluorescence quenching when rapidly mixed with (Aα2-L1M/L38M)2, but did produce the bi-exponential fluorescence decay containing fast and slow components. The rates of the fast and slow components were 18.34 ± 0.09 sec−1 and 0.48 ± 0.02 sec−1, respectively, for isoflurane and 24.0 ± 0.1 sec−1 and 0.44 ± 0.03 sec−1, respectively, for desflurane. At its anesthetizing concentration (0.81 mM), cyclopropane had no detectable effect on the intrinsic fluorescence intensity of (Aα2-L1M/L38M)2. At a supraphysiological concentration (5.3 mM), cyclopropane produced a small fast component having a rate of 57 ± 1 sec−1 when rapidly mixed with (Aα2-L1M/L38M)2, but no measurable immediate component or slow component.
Figure 7.

Stopped-flow fluorescence traces obtained upon mixing (Aα2-L1M/L38M)2 with isoflurane, desflurane, or cyclopropane at the indicated concentrations. With the exception of 5.3 mM cyclopropane, the concentrations indicated represent the in-vivo anesthetizing concentrations. Trace amplitudes were normalized between 0 (buffer only) and 1 (protein, no anesthetic). For isoflurane and desflurane, the curves are fits of the decays to a double exponential equation. For 5.3 mM cyclopropane, the curve is a fit to a single exponential equation.
The rates of the fast and slow components observed upon mixing (Aα2-L1M/L38M)2 with halothane (0.31 mM) or sevoflurane (0.57 mM) were highly temperature-sensitive (figure 8A). Arrhenius plots for the fast and slow components obtained upon mixing (Aα2-L1M/L38M)2 with either halothane or sevoflurane were linear over the entire temperature range studied (figure 8B). For halothane, the Q10s were 3.1 ± 0.1 and 2.8 ± 0.1 for the fast and slow components, respectively. For sevoflurane, the Q10s were 2.9 ± 0.1 and 2.7 ± 0.2 for the fast and slow components, respectively.
Figure 8.

(A) Stopped-flow fluorescence traces illustrating the effect of temperature on intrinsic W15 fluorescence quenching of (Aα2-L1M/L38M)2 by halothane (top) and sevoflurane (bottom). Note the different time axes for traces obtained at 283 °K versus 308 °K. (B) Arrhenius plot showing the temperature dependence of halothane's fast (●) and slow (■) components of quenching and sevoflurane's fast (O) and slow (□) components of quenching. The lines through the data points were derived by linear regression and used to calculate the Q10s.
DISCUSSION
In the present study, stopped-flow techniques were used to characterize anesthetic interactions with a four-α-helix bundle protein designed to model anesthetic binding sites on Cys-loop LGICs. Anesthetic binding to (Aα2-L1M/L38M)2 produced stopped-flow fluorescence traces containing up to three components of intrinsic fluorescence quenching (immediate, fast, and slow) distinguished by their different timescales. The immediate component of quenching was observed only in studies using halothane and was so fast that it was completed within the solution mixing time of the stopped-flow spectrofluorimeter. A distinctive characteristic of halothane is that it contains a bromine atom that efficiently quenches tryptophan fluorescence. In fact, experiments comparing the abilities of halothane and sevoflurane to quench the fluorescence of N-acetyltryptophanamide indicate that halothane directly quenches indole fluorescence 3600-times more efficiently than sevoflurane (22). This suggests that the immediate component that we observe in studies using halothane represents direct bromine quenching of W15 upon halothane binding within the hydrophobic core of (Aα2-L1M/L38M)2. This interpretation is consistent with nuclear magnetic resonance spectroscopy studies of anesthetic-protein interactions demonstrating that anesthetics bind to proteins with rate constants on the order of ∼107 M−1sec−1 (26, 27) because such binding would occur in less than one millisecond even at the lowest halothane concentration studied. It also implies that the concentration of halothane that produces half-maximal immediate quenching (0.68 ± 0.08 mM) is equivalent to halothane's dissociation constant for binding to this site.
Fast and slow components of quenching occurred on timescales of tens of milliseconds and seconds, respectively, and could be observed even when (Aα2-L1M/L38M)2 was mixed with anesthetics that are highly inefficient quenchers of tryptophan fluorescence. The rates of these components were orders of magnitude slower than those expected for diffusion-limited anesthetic binding and were highly temperature-sensitive. In addition, these rates were insensitive to the particular anesthetic used and the anesthetic concentration chosen. Based on these findings, we conclude that the fast and slow components reflect anesthetic-induced conformational transitions of (Aα2-L1M/L38M)2. Although our studies do not define the precise nature of the anesthetic-induced structural changes, previous spectroscopic studies using equilibrium techniques have shown that inhaled anesthetic binding leads to an increase in the protein's α-helical content and a reorientation of the W15 into a more polar environment (22). Presumably, this reorientation reduces the fluorescence quantum yield of W15 and produces the fast and slow components of quenching. Hydrogen exchange and one-dimensional 1H nuclear magnetic resonance spectroscopic studies indicate that anesthetics also allosterically stabilize and structurally tighten four-α-helix bundle proteins (22, 28). Anesthetics similarly affect the structure of albumin, suggesting that this may represent a fundamental mode of action of anesthetics on proteins (29-31). By producing analogous structural changes in the α-helical transmembrane domains of Cys-loop LGICs, anesthetics may stabilize receptors in their open channel conformational states, a concept supported by mutagenesis studies showing that the open channel stability of Cys-loop LGICs is governed by the structure of these domains (32-35).
In the classical Monod-Wyman-Changeaux allosteric model, proteins exist in an equilibrium between two (or more) conformational states (36). Ligands bind preferentially (i.e. with higher affinity) to particular states, stabilizing them and shifting this equilibrium. Within the context of this model, (Aα2-L1M/L38M)2 exists primarily in a state(s) with relatively low anesthetic affinity, but is stabilized in states having higher anesthetic affinity upon equilibration with anesthetic. Fast and slow components of quenching occur as an anesthetic shifts the conformational equilibrium towards the higher affinity states because the W15 quantum yield in these states is lower. Consistent with this model, the Kd for halothane determined from the immediate component of quenching is six-fold higher than that reported at equilibrium (0.68 ± 0.08 mM vs. 0.12 ± 0.02 mM, respectively) (23). Application of this model to interpret the actions of anesthetics on Cys-loop LGICs leads to the prediction that anesthetics bind with higher affinity to the open channel state as compared to the closed channel state. This agrees with steady-state photoaffinity labeling studies demonstrating increased binding of halothane to cerebellar tissue in the presence of GABA (37) and time-resolved photoaffinity labeling studies showing increased incorporation of an anesthetic-like hydrophobic probe to the cavity formed by the interfaces of the α-helical transmembrane domains when the channel is in the open state (38).
Although the data are not sufficient to define a specific, detailed kinetic scheme to explain all of our results, the observation that anesthetic binding produces a fluorescence decay that is biexponential suggests that anesthetics shift the protein conformational equilibrium from favoring the state having low anesthetic affinity (L) to the state having high anesthetic affinity (H) via an intermediate state (I): Within the framework of scheme 1, the fast component of quenching reflects the conformational transition from L to I and the slow component of quenching reflects the conformational transition from I to H.
Scheme 1.
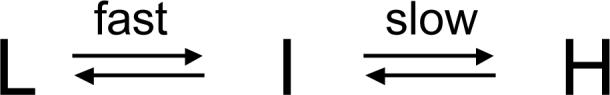
At low halothane concentrations, there is little initial halothane binding as reflected by the small immediate component. Under these conditions, almost all W15 quenching occurs as a consequence of the halothane-induced shift in the conformational equilibrium. At high halothane concentrations, a large fraction of W15 fluorescence is directly quenched by halothane binding as indicated by the large immediate component. This reduces the quantity of W15 fluorescence that can subsequently be quenched as a result of the anesthetic-induced shift in the conformational equilibrium and likely accounts for the reduced amplitudes of the fast and slow components observed with high concentrations of halothane (figure 4). This would also explain why no reduction in the amplitudes of the fast and slow components occurs with high concentrations of sevoflurane (figure 6), as it does not directly quench W15 fluorescence with any appreciable efficiency (22).
The conformational transitions induced by halothane, sevoflurane, desflurane, and isoflurane occurred at clinically relevant anesthetic concentrations as indicated by the presence of fast and slow components of quenching in traces obtained upon rapidly mixing the protein with anesthetics at their anesthetizing concentrations. However at its anesthetizing concentration, cyclopropane failed to produce any detectable conformational transition as it had no effect on (Aα2-L1M/L38M)2 fluorescence. At a concentration that is more than six times greater than that required to cause anesthesia, cyclopropane did produce stopped-flow fluorescence traces containing a relatively small amplitude fast component but no measurable slow component. Such traces were indistinguishable from those obtained using the lowest concentrations of halothane or sevoflurane studied, suggesting that cyclopropane acts on (Aα2-L1M/L38M)2, with low potency. Such anomalously low potency for cyclopropane in this model system would parallel that observed in functional studies using native and heterologously expressed Cys-loop LGICs (16, 39-41). For example, when normalized to their anesthetizing potencies, cyclopropane is an order of magnitude less potent than isoflurane at stabilizing the GABAA receptor in a conformational state that has increased sensitivity to agonist (16). To explain these observations, we have hypothesized that cyclopropane's low potency for altering Cys-loop LGIC conformational states reflects its inability to engage in attractive electrostatic interactions (e.g. hydrogen-bonding or dipolar interactions) with amino acids that contribute to anesthetic binding sites. Such interactions can contribute several kcal/mol to the overall free energy change associated with binding (42). If so, the results of the present studies suggest that electrostatic interactions are also important for anesthetic binding to the hydrophobic core of (Aα2-L1M/L38M)2. This interpretation is supported by the favorable enthalpy changes associated with the binding of halothane, sevoflurane, desflurane, and isoflurane to the four-a-helix bundle (Aα2-L38M)2 (43).
The rates of the fast and slow components were highly temperature sensitive with Q10s near 3. This approximates the values associated with important Cys-loop LGIC conformational transitions such as channel opening and closing (44) and is indicative of significant conformational rearrangements. We also note that experiments performed using halothane and sevoflurane exhibited similar temperature dependence. This suggests that both anesthetics induce (Aα2-L1M/L38M)2 to undergo the same conformational transitions, although we can not rule out the possibility that these two anesthetics induce different (Aα2-L1M/L38M)2 conformations with similar energy barriers between states.
In summary, we have characterized the interactions between inhaled anesthetics and (Aα2-L1M/L38M)2 using stopped-flow fluorescence spectroscopy. Anesthetic binding produces time-dependent quenching of tryptophan fluorescence on the millisecond and second timescales reflecting anesthetic-induced changes in protein structure. Within the context of the Monod-Wyman-Changeaux allosteric model, this occurs because anesthetics bind with higher affinity to and stabilize conformational states having lower tryptophan fluorescence quantum yields. The rates of these conformational transitions are highly temperature-sensitive, indicative of a large change in the protein's structure following anesthetic binding. With the exception of cyclopropane, anesthetics induced conformational transitions in (Aα2-L1M/L38M)2 at concentrations that cause their in-vivo behavioral actions. Cyclopropane, which acts with unusually low potency on Cys-loop LGICs, also acted with low apparent potency on (Aα2-L1M/L38M)2. Inhaled anesthetics may produce similar structural changes in Cys-loop LGICs, leading to stabilization of the open channel state. As Cys-loop LGICs are highly complex proteins that are extremely difficult to express and purify, four-α-helix bundle proteins may be useful scaled-down models that allow a wide range of biochemical and biophysical techniques to be used to better understand how anesthetic binding leads to changes in protein structure and function.
ACKNOWLEDGMENT
Mass spectrometry was performed at the Protein Chemistry Laboratory, University of Pennsylvania, Philadelphia, PA.
Footnotes
This work was supported by grants from the National Institutes of Health GM61927 and GM58448 for DER and GM55876 and GM65218 for JSJ
REFERENCES
- 1.Flood P, Role LW. Neuronal nicotinic acetylcholine receptor modulation by general anesthetics. Toxicol Lett. 1998;100−101:149–153. doi: 10.1016/s0378-4274(98)00179-9. [DOI] [PubMed] [Google Scholar]
- 2.Yost CS, Gray AT, Winegar BD, Leonoudakis D. Baseline K+ channels as targets of general anesthetics: studies of the action of volatile anesthetics on TOK1. Toxicol Lett. 1998;100−101:293–300. doi: 10.1016/s0378-4274(98)00198-2. [DOI] [PubMed] [Google Scholar]
- 3.Gray AT, Zhao BB, Kindler CH, Winegar BD, Mazurek MJ, Xu J, Chavez RA, Forsayeth JR, Yost CS. Volatile anesthetics activate the human tandem pore domain baseline K+ channel KCNK5. Anesthesiology. 2000;92:1722–1730. doi: 10.1097/00000542-200006000-00032. [DOI] [PubMed] [Google Scholar]
- 4.Hollmann MW, Liu HT, Hoenemann CW, Liu WH, Durieux ME. Modulation of NMDA receptor function by ketamine and magnesium. Part II: interactions with volatile anesthetics. Anesth Analg. 2001;92:1182–1191. doi: 10.1097/00000539-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 5.Bru-Mercier G, Hopkins PM, Harrison SM. Halothane and sevoflurane inhibit Na/Ca exchange current in rat ventricular myocytes. Br J Anaesth. 2005;95:305–309. doi: 10.1093/bja/aei185. [DOI] [PubMed] [Google Scholar]
- 6.Franks NP, Lieb WR. Which molecular targets are most relevant to general anaesthesia? Toxicol Lett. 1998;100−101:1–8. doi: 10.1016/s0378-4274(98)00158-1. [DOI] [PubMed] [Google Scholar]
- 7.Campagna JA, Miller KW, Forman SA. Mechanisms of actions of inhaled anesthetics. N Engl J Med. 2003;348:2110–2124. doi: 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- 8.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- 9.Stroud RM, McCarthy MP, Shuster M. Nicotinic acetylcholine receptor superfamily of ligand-gated ion channels. Biochemistry. 1990;29:11009–11023. doi: 10.1021/bi00502a001. [DOI] [PubMed] [Google Scholar]
- 10.Reeves DC, Lummis SC. The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel. Mol Membr Biol. 2002;19:11–26. doi: 10.1080/09687680110110048. [DOI] [PubMed] [Google Scholar]
- 11.Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- 12.Mascia MP, Trudell JR, Harris RA. Specific binding sites for alcohols and anesthetics on ligand-gated ion channels. Proc Natl Acad Sci U S A. 2000;97:9305–9310. doi: 10.1073/pnas.160128797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopreato GF, Phelan R, Borghese CM, Beckstead MJ, Mihic SJ. Inhaled drugs of abuse enhance serotonin-3 receptor function. Drug Alcohol Depend. 2003;70:11–5. doi: 10.1016/s0376-8716(02)00330-7. [DOI] [PubMed] [Google Scholar]
- 15.Krasowski MD, Harrison NL. The actions of ether, alcohol and alkane general anaesthetics on GABAA and glycine receptors and the effects of TM2 and TM3 mutations. Br J Pharmacol. 2000;129:731–743. doi: 10.1038/sj.bjp.0703087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raines DE, Claycomb RJ, Forman SA. Modulation of GABAA receptor function by nonhalogenated alkane anesthetics: the effects on agonist enhancement, direct activation, and inhibition. Anesth Analg. 2003;96:112–118. doi: 10.1097/00000539-200301000-00024. [DOI] [PubMed] [Google Scholar]
- 17.Johansson JS, Gibney BR, Rabanal F, Reddy KS, Dutton PL. A designed cavity in the hydrophobic core of a four-α-helix bundle improves volatile anesthetic binding affinity. Biochemistry. 1998;37:1421–1429. doi: 10.1021/bi9721290. [DOI] [PubMed] [Google Scholar]
- 18.Davies LA, Zhong Q, Klein ML, Scharf D. Molecular dynamics simulation of four-α-helix bundles that bind the anesthetic halothane. FEBS Lett. 2000;478:61–66. doi: 10.1016/s0014-5793(00)01792-0. [DOI] [PubMed] [Google Scholar]
- 19.Johansson JS, Solt K, Reddy KS. Binding of the general anesthetics chloroform and 2,2,2-trichloroethanol to the hydrophobic core of a four-α-helix bundle proteins. Photochem Photobiol. 2003;77:89–95. doi: 10.1562/0031-8655(2003)077<0089:botgac>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 20.Manderson GA, Michalsky SJ, Johansson JS. Effect of four-α-helix bundle cavity size on volatile anesthetic binding energetics. Biochemistry. 2003;42:11203–11213. doi: 10.1021/bi034623b. [DOI] [PubMed] [Google Scholar]
- 21.Zhang T, Johansson JS. A calorimetric study on the binding of six general anesthetics to the hydrophobic core of a model protein. Biophys Chem. 2005;113:169–174. doi: 10.1016/j.bpc.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Pidikiti R, Zhang T, Mallela KM, Shamim M, Reddy KS, Johansson JS. Sevoflurane-induced structural changes in a four-α-helix bundle protein. Biochemistry. 2005;44:12128–12135. doi: 10.1021/bi050896q. [DOI] [PubMed] [Google Scholar]
- 23.Pidikiti R, Shamim M, Mallela KM, Reddy KS, Johansson JS. Expression and characterization of a four-α-helix bundle protein that binds the volatile general anesthetic halothane. Biomacromolecules. 2005;6:1516–1523. doi: 10.1021/bm049226a. [DOI] [PubMed] [Google Scholar]
- 24.Johansson JS, Eckenhoff RG, Dutton PL. Binding of halothane to serum albumin demonstrated using tryptophan fluorescence. Anesthesiology. 1995;83:316–324. doi: 10.1097/00000542-199508000-00012. [DOI] [PubMed] [Google Scholar]
- 25.DeCoursey TE, Cherny VV. Temperature dependence of voltage-gated H+ currents in human neutrophils, rat alveolar epithelial cells, and mammalian phagocytes. J Gen Physiol. 1998;112:503–522. doi: 10.1085/jgp.112.4.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y, Seto T, Tang P, Firestone L. NMR study of volatile anesthetic binding to nicotinic acetylcholine receptors. Biophys J. 2000;78:746–751. doi: 10.1016/S0006-3495(00)76632-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y, Tang P, Firestone L, Zhang TT. 19F nuclear magnetic resonance investigation of stereoselective binding of isoflurane to bovine serum albumin. Biophys J. 1996;70:532–538. doi: 10.1016/S0006-3495(96)79599-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johansson JS, Scharf D, Davies LA, Reddy KS, Eckenhoff RG. A designed four-α-helix bundle that binds the volatile general anesthetic halothane with high affinity. Biophys J. 2000;78:982–993. doi: 10.1016/S0006-3495(00)76656-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanner JW, Eckenhoff RG, Liebman PA. Halothane, an inhalational anesthetic agent, increases folding stability of serum albumin. Biochim Biophys Acta. 1999;1430:46–56. doi: 10.1016/s0167-4838(98)00258-1. [DOI] [PubMed] [Google Scholar]
- 30.Liu R, Pidikiti R, Ha CE, Petersen CE, Bhagavan NV, Eckenhoff RG. The role of electrostatic interactions in human serum albumin binding and stabilization by halothane. J Biol Chem. 2002;277:36373–36379. doi: 10.1074/jbc.M205479200. [DOI] [PubMed] [Google Scholar]
- 31.Johansson JS, Zou H, Tanner JW. Bound volatile general anesthetics alter both local protein dynamics and global protein stability. Anesthesiology. 1999;90:235–245. doi: 10.1097/00000542-199901000-00030. [DOI] [PubMed] [Google Scholar]
- 32.Scheller M, Forman SA. Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABA(A) receptors. J Neurosci. 2002;22:8411–8421. doi: 10.1523/JNEUROSCI.22-19-08411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bianchi MT, Macdonald RL. Mutation of the 9' leucine in the GABAA receptor gamma2L subunit produces an apparent decrease in desensitization by stabilizing open states without altering desensitized states. Neuropharmacology. 2001;41:737–744. doi: 10.1016/s0028-3908(01)00132-0. [DOI] [PubMed] [Google Scholar]
- 34.Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- 35.Chang Y, Weiss DS. Substitutions of the highly conserved M2 leucine create spontaneously opening rho1 gamma-aminobutyric acid receptors. Mol Pharmacol. 1998;53:511–523. doi: 10.1124/mol.53.3.511. [DOI] [PubMed] [Google Scholar]
- 36.Monod J, Wymann J, Changeaux J. On the nature of allosteric transitions: A plausible model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 37.Eckenhoff MF, Eckenhoff RG. Gamma-aminobutyric acid enhancement of halothane binding in rat cerebellum. Neurosci Lett. 2000;286:111–114. doi: 10.1016/s0304-3940(00)01105-8. [DOI] [PubMed] [Google Scholar]
- 38.Arevalo E, Chiara DC, Forman SA, Cohen JB, Miller KW. Gating-enhanced accessibility of hydrophobic sites within the transmembrane region of the nicotinic acetylcholine receptor's δ-subunit. A time-resolved photolabeling study. J Biol Chem. 2005;280:13631–13640. doi: 10.1074/jbc.M413911200. [DOI] [PubMed] [Google Scholar]
- 39.Hara K, Eger EI, 2nd, Laster MJ, Harris RA. Nonhalogenated alkanes cyclopropane and butane affect neurotransmitter-gated ion channel and G-protein-coupled receptors: differential actions on GABAA and glycine receptors. Anesthesiology. 2002;97:1512–1520. doi: 10.1097/00000542-200212000-00025. [DOI] [PubMed] [Google Scholar]
- 40.Raines DE, Claycomb RJ. The role of electrostatic interactions in governing anesthetic action on the torpedo nicotinic acetylcholine receptor. Anesth Analg. 2002;95:356–361. doi: 10.1097/00000539-200208000-00021. [DOI] [PubMed] [Google Scholar]
- 41.Raines DE, Claycomb RJ, Scheller M, Forman SA. Nonhalogenated alkane anesthetics fail to potentiate agonist actions on two ligand-gated ion channels. Anesthesiology. 2001;95:470–477. doi: 10.1097/00000542-200108000-00032. [DOI] [PubMed] [Google Scholar]
- 42.Eckenhoff RG, Johansson JS. Molecular interactions between inhaled anesthetics and proteins. Pharmacol Rev. 1997;49:343–367. [PubMed] [Google Scholar]
- 43.Zhang T, Johansson JS. An isothermal titration calorimetry study on the binding of four volatile general anesthetics to the hydrophobic core of a four-α-helix bundle protein. Biophys J. 2003;85:3279–3285. doi: 10.1016/S0006-3495(03)74746-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dilger JP, Brett RS, Poppers DM, Liu Y. The temperature dependence of some kinetic and conductance properties of acetylcholine receptor channels. Biochim Biophys Acta. 1991;1063:253–258. doi: 10.1016/0005-2736(91)90379-m. [DOI] [PubMed] [Google Scholar]